Article Text

Abstract
Given the high prevalence and rising incidence of non-alcoholic fatty liver disease (NAFLD), the absence of approved therapies is striking. Although the mainstay of treatment of NAFLD is weight loss, it is hard to maintain, prompting the need for pharmacotherapy as well. A greater understanding of disease pathogenesis in recent years was followed by development of new classes of medications, as well as potential repurposing of currently available agents. NAFLD therapies target four main pathways. The dominant approach is targeting hepatic fat accumulation and the resultant metabolic stress. Medications in this group include peroxisome proliferator-activator receptor agonists (eg, pioglitazone, elafibranor, saroglitazar), medications targeting the bile acid-farnesoid X receptor axis (obeticholic acid), inhibitors of de novo lipogenesis (aramchol, NDI-010976), incretins (liraglutide) and fibroblast growth factor (FGF)-21 or FGF-19 analogues. A second approach is targeting the oxidative stress, inflammation and injury that follow the metabolic stress. Medications from this group include antioxidants (vitamin E), medications with a target in the tumour necrosis factor α pathway (emricasan, pentoxifylline) and immune modulators (amlexanox, cenicriviroc). A third group has a target in the gut, including antiobesity agents such as orlistat or gut microbiome modulators (IMM-124e, faecal microbial transplant, solithromycin). Finally, as the ongoing injury leads to fibrosis, the harbinger of liver-related morbidity and mortality, antifibrotics (simtuzumab and GR-MD-02) will be an important element of therapy. It is very likely that in the next few years several medications will be available to clinicians treating patients with NAFLD across the entire spectrum of disease.
- NONALCOHOLIC STEATOHEPATITIS
- FATTY LIVER
- PHARMACOTHERAPY
Statistics from Altmetric.com
Introduction
Non-alcoholic fatty liver disease (NAFLD), defined as excess accumulation of fat in the liver, has become the most common cause for chronic liver disease in the Western world and is estimated to impact at least 30% of Americans1 ,2 or Chinese3 with the prevalence appearing to rise in recent years.4 ,5 Non-alcoholic steatohepatitis (NASH) is a subset of NAFLD, estimated to affect 2–5% of Americans, in which increased liver fat is accompanied by cellular injury, inflammatory infiltrate and, subsequently, liver fibrosis, which can progress to cirrhosis with its associated complications.6 Although fatty liver by itself is associated with other features of the metabolic syndrome such as obesity, diabetes mellitus type 2, hypertension and dyslipidemia, increased liver-related mortality is essentially limited to patients with NASH.7
Increased triglyceride deposition in the liver reflects an input/output imbalance of hepatic free fatty acid (FFA) metabolism. In obesity-associated NAFLD, there is an increase of FFA delivery to the liver, especially during the fed state, due to adipose tissue insulin resistance.8 ,9 In addition, de novo lipogenesis is increased,10 driven by the hyperinsulinemia as well as excess availability of carbohydrates. Compensatory increase in very low density lipoprotein (VLDL) secretion is not sufficient to overcome the excess formation of triglycerides11 while it is unclear whether β-oxidation is increased or decreased in these subjects.12
The accumulated triglycerides in steatosis appear to be relatively inert with benign outcome; hepatocellular injury is driven by lipotoxicity from FFAs and their derivatives,13 as well as overloading of mitochondrial capacity. This initial metabolic stress activates multiple cell stress pathways, including generation of reactive oxygen species, endoplasmic reticulum stress and apoptosis. Injury signals from stressed or dying hepatocytes, lipids and chemokines activate an immune response, including recruitment and activation of variety of immune cells, further increasing cellular injury. Key mediators are the Kupffer cells and macrophages, which are further activated by bacterial products from the gut microbiome.
Hepatocellular injury and immune cell activity converge to activate hepatic stellate cells, causing a change in their phenotype and deposition of collagen, resulting in increased fibrosis and hepatic architectural distortion.
Although the injury patterns are common and conserved, there is variability between patients with NAFLD in the degree of activation of each individual pathway, likely accounting for the heterogeneity of clinical phenotypes and severity. This may be secondary to different external stimuli (ie, dietary composition), genetic components and modulation by the gut microbiome, among other factors.
The remarkable progress that has been made in previous years in understanding disease pathogenesis has led to an explosion of medical therapies targeting various aspects of the fat accumulation and injury pathways. These can be grouped into four general classes (figure 1), according to their intended targets: (1) Medications with a primary metabolic target, geared to reduce hepatic fat accumulation and metabolic stress. (2) Medications addressing oxidative stress or the inflammation and injury components of NASH. (3) Medications with a primary gut target, modulating the interaction between the gut and the liver in NAFLD. (4) Antifibrotics, aiming to decrease the progressive fibrosis and resultant complications.
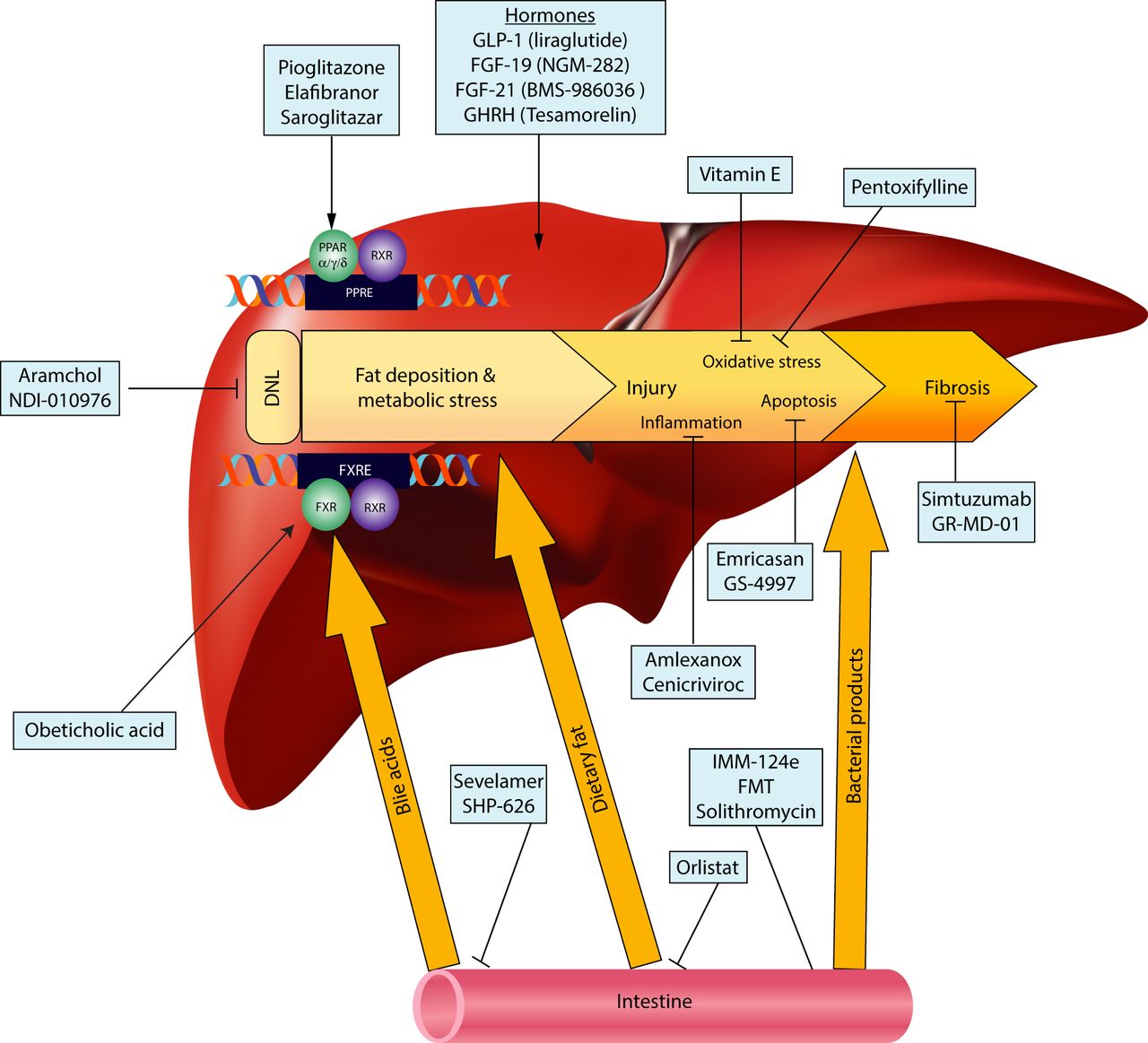
{kind=link}
Targets of upcoming therapies for non-alcoholic fatty liver disease (NAFLD). DNL, de novo lipogenesis; FGF, fibroblast growth factor; FMT, faecal microbial transplant; FXR, farnesoid X receptor; FXRE, FXR response element; GHRH, growth hormone-releasing hormone; GLP-1, glucagon-like peptide-1; PPAR, peroxisome proliferator-activated receptor; PPRE, PPAR response element; RXR, retinoid X receptor.
Medications with a primary metabolic target
PPAR agonists
The peroxisome proliferator-activator receptors (PPARs) are a family of nuclear receptors that bind a wide range of fatty acids and fatty acid derivatives and transcriptionally regulate a wide variety of metabolic processes (table 1). There are three PPARs—α, β/δ and γ—that share the same target DNA sequence but differ in ligand selectivity and tissue distribution.14
Clinical trials of medications for non-alcoholic fatty liver disease (NAFLD)
PPARα is expressed extensively in the liver, adipose tissue, heart, skeletal muscle and kidney;15 its activity increases in the fasting state and transcriptionally drives the expression of a large number of genes, including those regulating gluconeogenesis, β-oxidation, lipid transport and the hormone fibroblast growth factor (FGF)-21. In various animal models, PPARα deletion, either at germline level16 or in hepatocytes only,17 is associated with worsening of hepatic steatosis. Fibrates, which are synthetic agonists of PPARα, are used extensively for clinical treatment of hypertriglyceridemia, but have not shown a consistent beneficial effect for the treatment of NAFLD,18 possibly related to their effect on PPARα outside the liver. PPARδ, another member of the PPAR family, has a wider expression distribution pattern, and beyond hepatocytes is also expressed in high levels in skeletal muscle and macrophages19 and its activation improves insulin sensitivity, decreases hepatic glucose production, increases fatty acid oxidation and decreases macrophage and Kupffer cell activation. PPARδ activation has also been shown to decrease atherosclerotic disease in animal models.20 Treatment with a PPARδ agonist in a pilot study decreased hepatic fat content, likely through an increase in fatty acid oxidation,21 but development has been halted due to safety concerns. Elafibranor (GFT-505) is a dual PPARα/δ agonist, aiming to combine the beneficial effects of activating the two receptors. Animal data demonstrate that a beneficial effect of elafibranor on serum triglycerides, cholesterol and high density lipoprotein (HDL), and a reduction in hepatic fat that is mediated, at least in part, by non-PPARα-dependent mechanism.22 ,23 Post hoc analysis of short-term (4–12 weeks) phase II clinical trials using elafibranor for the treatment of metabolic syndrome demonstrated a significant reduction in ALT in subjects in the top two quartiles at baseline22 ,24 and has shown an improvement in liver, adipose and peripheral tissue insulin sensitivity,24 making it a potentially attractive therapeutic agent for NASH. Recently, a phase IIb randomized double-blind placebo controlled trial (RDBPCT), GOLDEN-505, assessed the effectiveness of elafibranor (80 or 120 mg/day) or placebo for 1 year to treat biopsy-proven NASH.25 The study included 276 patients with mild–severe NASH and allowed for inclusion of diabetics, but excluded patients with cirrhosis. The primary end point of the study was ambitiously selected as histological resolution of NASH without worsening of fibrosis, based on recommendations from a recent US Food and Drug Administration/American Association for the Study of Liver Diseases (AASLD) workshop.26 The primary end point was achieved in 23% and 21% of patients in the 80 mg and 120 mg/day groups, respectively, and in 17% of controls; the difference between the groups was not statistically significant. A more stringent definition of NASH resolution was assessed post hoc, and using that criteria, NASH resolution was achieved in 19% of the 120 mg/day group compared with 12% of placebo-treated subjects (p=0.045). The failure to show benefit appears to be primarily due to a high response rate in the placebo groups of mild–moderate (NAFLD activity score (NAS)27 3–5) disease and in fact, when subjects with mild disease at baseline (NAS=3) were excluded from the analysis, the 120 mg/day dose was significantly superior than placebo across both response definitions. The 120 mg/day dose had a modest effect on alanine aminotransferase (ALT) (decrease of 9.5 U/L compared with placebo) and in patients with diabetes improved insulin sensitivity. A phase III trial (NCT02704403) is currently recruiting subjects with NASH and NAS≥4, aiming to determine the effects of 72 weeks of treatment with 120 mg/day on NASH resolution without worsening of fibrosis. For the first time in therapeutic trials of NAFLD, a clinical co-primary end point is included, assessing the effect of elafibranor on mortality, cirrhosis and liver-related clinical outcomes.
PPARγ, another member of the PPAR family, is predominantly expressed in the adipose tissue, controlling lipogenesis, glucose metabolism and adipose tissue differentiation. Thiazolidinediones (TZDs), synthetic PPARγ agonists, are insulin sensitisers with proven efficacy for treatment of diabetes and have been shown in multiple trials to be effective for the treatment of NASH.28–30 In the PIVENS RDBPCT, the largest trial of a PPARγ agonist to date, 80 patients with NASH but not diabetes or cirrhosis received pioglitazone 30 mg/day for 96 weeks and were compared with 83 placebo-treated subjects.30 Pioglitazone treatment was associated with histological improvement in 34% of subjects compared with 19% of controls. The significance level of p=0.04 did not meet the prespecified cut-off, despite apparent effectiveness, mainly due to discrepancies in the interpretation of entry liver biopsies in this group. Unfortunately, concerns with the safety profile of TZDs (especially regarding cardiovascular safety of rosiglitazone) and a side effect profile that includes weight gain due to redistribution of body fat have led to poor acceptance of these agents for the treatment of NASH in clinical practice.31
The glitazars are a class of medications designed as dual PPARα/γ agonists, aiming to synergise the beneficial effects of PPARα and PPARγ agonism. However, development of most compounds in this class has been halted due to safety concerns. The only glitazar in clinical use, saroglitazar, is currently approved in India for the treatment of diabetic dyslipidemia.32 ,33 In a mouse model of NASH, treatment with saroglitazar induced histological improvement as well as a decrease in liver fat content and ALT.34 A retrospective analysis of patients with NAFLD treated with saroglitazar for dyslipidemia demonstrated significant and marked decrease in ALT (from 64±6 to 28±3, p<0.01) after 24 weeks of treatment.35 In PRESS VIII, a phase II open-label study, the efficacy of saroglitazar was evaluated in 32 patients with biopsy-proven NASH and a 52% reduction in ALT was demonstrated after 12 weeks of treatment.36 A phase III RDBPCT of saroglitazar for 52 weeks in non-cirrhotic patients with biopsy-proven NASH is currently ongoing in India, with the primary end point defined as improvement in NASH histology with no worsening of fibrosis (Clinical Trials Registry—India CTRI/2015/10/006236).
Novel selective modulators of PPARα (pemafibrate, K-877) and PPARγ (INT-131), a PPARδ agonist (HPP-593) and a PPARα/γ agonist (DSP-8658), are currently in clinical trials for other indications (mainly diabetic dyslipidemia). Whether these agents will prove to have a beneficial effect on NAFLD is yet unknown.
Although TZDs are typically considered to act through PPARγ agonism, there is evidence to suggest other mechanisms as well. In an animal model, MSDC-0602, an experimental TZD with poor affinity to PPARγ, was able to suppress hepatic glucose production and de novo lipogenesis in mice with hepatocyte-specific PPARγ knockout,37 likely through interaction with mitochondrial proteins Mcp1 and Mcp2.38 Clinical trials in human NASH are planned but have not begun to date.
Targeting the FXR-bile acid axis
The interaction of bile acids with the farnesoid X receptor (FXR), their intracellular receptor, negatively regulates bile acid synthesis and acts transcriptionally to decrease hepatic lipogenesis and steatosis,39 as well as decrease hepatic gluconeogenesis and improve peripheral insulin sensitivity.40 Obeticholic acid (6-ethylchenodeoxycholic acid (OCA)) is a synthetic lipophilic bile acid acting as FXR agonist and was recently evaluated as potential treatment for NASH in a phase IIb multicentre clinical trial. In the RDBPCT FLINT study,41 283 non-cirrhotic patients with biopsy-proven NASH (NAS ≥4) were randomised to receive OCA (25 mg/day) or placebo for 72 weeks. The primary end point of the study was histological improvement, defined as a decrease of two points in the NAS with no worsening of fibrosis. Histological improvement was seen in 45% of patients treated with OCA compared with 21% of those treated with placebo (p=0.0002). Resolution of NASH was seen in 22% of patients versus 13% of controls (p=0.08) and improvement in fibrosis score was detected in 35% versus 19% in controls (p=0.004). A concomitant significant decrease in liver enzymes was noted. Interestingly, patients treated with OCA had a significant decrease in body mass index (BMI) (decrease of 0.7 vs gain of 0.1 kg/m2 in the placebo group, p=0.01); whether this was responsible, at least in part, for the histological improvement is unclear. However, patients treated with OCA had a reversible significant increase in total and low density lipoprotein (LDL)-cholesterol and decrease in HDL-cholesterol. These changes were of low magnitude, appeared predominantly at treatment initiation and improved with treatment continuation, suggesting adaptation or initiation of cholesterol-reducing medications. Whether these changes will be sustained with prolonged treatment and whether they will be associated with an increase in cardiovascular risk remains to be shown. The main side effect of OCA was pruritus, noted in 23% of OCA-treated patients and requiring intervention or treatment discontinuation in several patients. Thus, although OCA appears effective for the treatment of NASH, its long-term safety and tolerability is still unclear. A phase III trial (NCT02548351) is currently recruiting patients with non-cirrhotic NASH and will compare the effects of OCA or placebo for 72 weeks on two co-primary histological outcomes—resolution of NASH without worsening of fibrosis or improvement of fibrosis without worsening of NASH. Similar to the elafibranor phase III trial, a clinical end point is included to assess the effect of treatment on mortality and liver-related outcomes at 6 years.
Other FXR agonists are being tested in clinical trials. GS-9674, a synthetic non-steroidal FXR agonist, is currently in a phase I study. In contrast to OCA, GS-9674 and similar agents are less likely to undergo enterohepatic circulation and may have more predictable pharmacokinetics.42 INT-767 is a bile acid analogue that acts as a dual agonist on FXR and on TGR5, the transmembrane G-protein bile acid receptor. In an animal model, INT-767 improved histological features of NASH and modulated the activation of hepatic monocytes.43 A phase I trial in human subjects is scheduled to start. In contrast to OCA, treatment with ursodeoxycholic acid (UDCA), a naturally occurring secondary bile acid, has not been shown to improve histological features of NASH,44 ,45 despite lowering liver enzymes. The difference between the effects of UDCA and OCA may be related to the poor affinity of UDCA to FXR or even its ability to antagonise FXR activity.46 Other UDCA derivatives, such as nor-UDCA or tauroursodeoxycholic acid, were not tested in humans for the treatment of NAFLD.
An alternative approach to direct targeting of FXR has been the use of bile acid sequestrants that disrupt the enterohepatic circulation and result in reduction in serum lipids. An RDBPCT assessing the effectiveness of 24 weeks of colesevelam treatment in patients with biopsy-proven NASH47 failed to show histological improvement and in fact demonstrated a rise in liver enzymes and liver fat content with colesevelam treatment. These findings are consistent with a reduction in FXR and liver X receptor (LXR)48 activation, as seen in animal models as well.48 All sequestrants may not be the same, as sevelamer, a phosphate-binding medication with bile salt binding capacity,49 has been shown in an animal model of steatohepatitis to improve liver fat, inflammation and fibrosis50 in an FXR-independent effect.51 Similarly to the effect of sequestration, enterohepatic circulation of bile acids can be disrupted by inhibiting the ileal apical sodium-dependent bile acid transporter (ASBT), the major route of reabsorption of bile acids in the terminal ileum. ASBT inhibitors cause an increase in faecal bile acids and improve glycaemic control in animal models.52 An oral inhibitor of ASBT, volixibat (SHP-626), recently concluded phase I studies and a phase II trial in patients with NASH is enrolling (NCT02787304).
Inhibition of de novo lipogenesis
Aramchol is a conjugate of cholic and arachidic acid that was shown to inhibit stearoyl CoA desaturase (SCD) and de novo lipogenesis in cell and animal models.53 ,54 In a recent phase II RDBPCT, 300 mg/day of aramchol given for 3 months significantly decreased hepatic fat content by 12.5%.55 The study enrolled predominantly subjects with NAFLD but not NASH and did not use histological end points. Treatment was not associated with improvement in liver enzymes, raising a concern that the reduction in hepatic fat was not accompanied by an improvement in inflammation or cellular injury. A phase IIb study (NCT02279524) is currently evaluating the effects of higher doses (400 and 600 mg/day) for 1 year in patients with non-cirrhotic biopsy-proven NASH (NAS≥4). The primary end point is decrease in liver fat content by MRI, while histological end points such as improvement or resolution of NASH are defined as secondary end points.
Malonyl-CoA acts as a key gatekeeper of fatty acid metabolism, controlling the balance between de novo lipogenesis and fatty acid oxidation.56 It serves as the building block for fatty acid synthesis and elongation, while inhibiting the entry of fatty acids to the mitochondria for β-oxidation. Acetyl-CoA carboxylase (ACC) is the key enzyme generating malonyl-CoA from acetyl-CoA and regulating this process. Inhibition of ACC by antisense oligonucleotides in a murine model of NAFLD increases fatty acid oxidation, decreases lipogenesis, decreased hepatic fat content and improved insulin sensitivity.56 An allosteric inhibitor of ACC, NDI-010976, was recently tested in obese subjects in a phase I trial57 and demonstrated dose-dependent inhibition of de novo lipogenesis, reaching up to 98% decrease from baseline following a single dose, making it a potential treatment for NAFLD.
Dur-928 is an endogenous sulfated oxysterol that has been shown to decrease hepatic fat content in animal models via inhibition of LXR and SREBP58 and is being developed as an oral agent for the treatment of NASH. A phase Ib study (ACTRN12615001355561) to evaluate its safety in patients with NASH compared with controls is currently ongoing.
Incretins and DPP-4 inhibitors
Glucagon-like peptide 1 (GLP-1) is an incretin hormone derived from the proglucagon polyprotein that is also the precursor to glucagon.59 GLP-1 is secreted by intestinal L-cells in response to meal ingestion and acts on the pancreas to improve glycaemic control by stimulating insulin secretion from pancreatic β-cells and inhibiting α-cell glucagon secretion. Beyond the pancreas, GLP-1 improves peripheral insulin sensitivity, increases hepatic glucose uptake and glycogen synthesis, delays gastric emptying and decreases appetite.60 Several long-acting GLP-1 receptor agonists (GLP-1RAs) are approved for the treatment of type 2 diabetes mellitus. In retrospective analyses of GLP-1RA trials in diabetic subjects, beneficial effects on liver enzymes and hepatic fat content were shown. For example, liraglutide treatment for 26 weeks was associated with an average decrease of 8.2 U/L in ALT activity in subjects with baseline elevated ALT, but this appeared to be fully explained by the concomitant decrease in weight and HbA1c.61 Similar findings were reported for exenatide62 and lixisenatide.62 Recently, the LEAN study63 was an RDBPCT specifically designed to examine the utility of liraglutide to treat NASH. Fifty-two subjects with histologically proven NASH (only 33% of whom were diabetics) were randomised to receive liraglutide (1.8 mg/day) or placebo for 48 weeks. At the end of the study, histological resolution of NASH without worsening of fibrosis, the primary end point, was seen in 39% of the patients assigned to liraglutide versus 9% of the placebo group (p=0.02). Mechanistically, liraglutide treatment improved hepatic insulin sensitivity, with resultant reduction in hepatic endogenous glucose production, decreased hepatic de novo lipogenesis and promoted an improvement in adipose tissue insulin sensitivity with reduction of lipolysis and delivery of FFAs to the liver.64 As expected, treatment with liraglutide was associated with weight loss; histological responders lost on average 2.1 kg more than non-responders. Unfortunately, the study was not powered to show whether the beneficial effect on the liver was independent from the effect of weight loss alone.
A potentially alternative approach for augmentation of endogenous incretin effects is by use of small-molecule inhibitors of dipeptidyl peptidase 4 (DPP-4), the enzyme responsible for rapid degradation of GLP-1. Studies of sitagliptin, a DPP-4 inhibitor, were small and limited to diabetic patients with fatty liver. Only one study used histological end points.65 In this small, uncontrolled trial with 15 patients, treatment with 100 mg/day of sitagliptin for 1 year was associated with improvement in liver enzymes, hepatocyte ballooning, histological activity scores and steatosis. A modest decrease in liver fat content by magnetic resonance spectroscopy (MRS) was also shown after 24 weeks of sitagliptin66 or vildagliptin.66 Other studies, on the other hand, failed to show an effect of sitagliptin treatment on liver fat content67 or liver enzymes.68 Recently, Cui et al69 performed the largest study to date, enrolling 50 subjects with NAFLD and pre-diabetes or early diabetes in an RDBPCT. In that study, 24 weeks of treatment with sitagliptin 100 mg/day were not associated with improvement in liver fat, liver enzymes or liver stiffness. Thus, DPP-4 inhibition does not seem to be highly effective for the treatment of NASH.
Lipid-lowering agents
As metabolic syndrome is closely associated with fatty liver disease, many of the patients have dyslipidemia and increased risk for cardiovascular disease. Furthermore, in patients with NASH, there is evidence for excess accumulation of free cholesterol in the liver, which could play a role in disease pathogenesis.70 ,71 Statin use is generally safe in patients with chronic liver disease, including those with NAFLD.72 In a retrospective analysis, statin use was associated with decreased risk of NASH and advanced fibrosis in a large cohort of patients who underwent a liver biopsy for possible NASH.73 However, this could also reflect hesitancy of providers to prescribe statins to patients with advanced liver disease and in fact there is clear evidence for underuse of statins in patients with NAFLD.74 Prospective clinical trials using statins to treat the liver disease are few and limited but generally demonstrated a beneficial effect of statins on liver fat content (reviewed in ref.72). In a recent prospective uncontrolled trial, 20 patients with biopsy-proven NASH (baseline NAS score of 8) and dyslipidemia were treated for 12 months with 10 mg/day of rosuvastatin. In total, 19 of the 20 patients reportedly had a complete resolution of NASH including complete resolution of steatosis, inflammation and ballooning, despite experiencing no weight loss.75 Whether such phenomenal findings can be replicated in a larger controlled trial remains to be seen.
In a pilot RDBPCT,76 ezetimibe, an inhibitor of intestinal absorption of cholesterol, was given for 24 weeks to patients with biopsy-proven NASH. Treatment had no effect on liver fat content, histology or liver enzymes.
Targeting hormonal signalling
FGF-21 is a peptide hormone, secreted predominantly from the liver. FGF-21 levels increase in the fasting state and regulate the fasting response in the adipose tissue and other organs.77 BMS-986036 (previously ARX-618) is a pegylated FGF-21 analogue that in animal models improved insulin sensitivity, hepatic fat content and de novo lipogenesis.78 A phase II RDBPCT is evaluating the effectiveness of 16 weeks of BMS-986036 treatment on hepatic fat content (by MRS) in patients with NASH (NCT02413372).
FXR activation in the terminal ileum by reabsorbed bile acids results in transcription and secretion of FGF-19, a peptide hormone that acts on the liver and leads to decreased bile acid synthesis and decreased gluconeogenesis in an insulin independent manner.77 It also has a proliferative effect on hepatocytes, predominantly through activation of the FGFR4 receptor, raising concern for potential tumourigenesis.79 NGM-282 is a recombinant variant FGF-19 that reportedly retains its beneficial metabolic effect but not the tumorigenic effects. In a mouse model of NASH, treatment with NGM-282 for 3 weeks resulted in a marked reduction of hepatic fat, histological features of NASH and decrease in ALT.80 NGM-282 is currently in a phase II study (NCT02443116) to determine the effects of 12 weeks of treatment on liver fat content in patients with biopsy-proven NASH.
VK2809 (previously MB07811) is a liver-directed agonist of the thyroid β receptor. In murine models, VK2809 was shown to increase fatty acid oxidation and decrease hepatic fat content and plasma triglycerides.81 A phase II trial in patients with NAFLD and hypercholesterolaemia is scheduled to begin enrolling soon.
Tesamorelin, a growth hormone-releasing hormone (GHRH) analogue, is approved for treating HIV-associated lipodystrophy. In an RDBPCT on subjects with HIV and increased abdominal fat, tesamorelin treatment was associated with a mild decrease of 2% in liver fat content (by MRS) but not with any change in liver enzymes.82 However, given that baseline liver fat content and liver enzymes were normal or near-normal for many of the subjects, a specific effect on NAFLD could not be determined. A prospective trial in HIV-infected subjects with fatty liver and with histological and radiological end points is underway (NCT02196831). Although relative growth hormone (GH) deficiency has been associated with NAFLD and GH treatment may improve NASH,83 no studies in non-HIV NAFLD with tesamorelin or other GH-axis agents have been conducted so far.
MicroRNA-based treatment
The microRNAs miR-103 and miR-107 are upregulated in the liver of obese animals84 and in the serum of human NAFLD patients.85 In animal models, miR-103/7 have been shown to modulate insulin sensitivity through direct interaction with caveolin-1.84 RG-125/AZD4076, an anti-Mir directed against miR-103/107 is being developed for the treatment of NASH and is currently in phase I (NCT02612662).
Medications affecting oxidative stress and inflammation
Antioxidants
Vitamin E, a fat-soluble antioxidant, has been used in multiple studies to treat fatty liver disease (table 1). In the PIVENS phase III RDBPCT,30 vitamin E at a dose of 800 IU/day for 96 weeks was superior to placebo, achieving histological response in 43% of subjects (compared with 19% of placebo, p=0.001) and resolution of NASH in 36%. Vitamin E treatment resulted in reduction in hepatocyte ballooning and lobular inflammation in approximately half of the patients, reflecting its expected effect as an antioxidant decreasing oxidative stress-mediated injury. Interestingly, it was also associated with a decrease in hepatic steatosis scores in 54% of subjects, with an average decline of 0.7 units in the steatosis score, through an unclear mechanism. No effect on fibrosis was seen. The effects of vitamin E were further demonstrated in a recent analysis86 that pooled together data from the PIVENS study and from patients receiving vitamin E on the placebo arm of the FLINT trial.41
Vitamin E is a mixture of eight different tocopherols and tocotrienols, with α-tocopherol thought to be the main active component in humans due to its greater affinity to tocopherol transfer protein, the main transporter of vitamin E from the gut to the liver.87 Within the α-tocopherol molecule, there are three chiral centres with eight possible enantiomers. The synthetic form of vitamin is a racemic mixture of different enantiomers and is approximately 50% as potent as the natural form, RRR-α-tocopherol. Vitamin E formulations (considered a food supplement) markedly differ in their components and their racemic structure. Thus, when comparing results from different clinical trials or prescribing vitamin E treatment to patients, the specific formulation and doses need to be clarified. Currently, the best available data for the benefit of vitamin E in NASH, as discussed above, come from the PIVENS trial,30 which used natural, RRR-α-tocopherol at a dose of 800 IU/day.
Although vitamin E use was not associated with any major safety signals in clinical trials for NASH, data from other studies suggest it may not be a completely risk-free intervention as it is sometimes perceived. A large meta-analysis of 135 000 patients from 19 studies that used vitamin E demonstrated an increase in all-cause mortality,88 although this has not been shown by other meta-analyses.89 Treatment with vitamin E was associated with an increased risk of haemorrhagic stroke, attributed to an antiplatelet effect of high-dose treatment.90 In addition, the SELECT trial demonstrated an increased risk of prostate cancer in elderly men, receiving long-term vitamin E for cancer prevention.91 Although these studies suggest potential risks with vitamin E treatment, the overall magnitude of risk is relatively small and was seen in trials where the clinical benefit (for cancer prevention or prevention of cardiovascular disease) was low or non-existent. In the context of NASH, where the efficacy of vitamin E is well proven, these risks should be taken into consideration when selecting patients for treatment and compared against the risks of other possible medications or the risk of no treatment. Finally, the PIVENS trial excluded diabetics and patients with cirrhosis; thus, there is lack of data regarding vitamin E efficacy and safety in these patients.
Cysteamine is an aminothiol that can act as scavenger for reactive oxygen species and can replenish glutathione stores, and thus has potential benefit in NASH. In a small pilot study, Dohil et al67 treated 11 children with biopsy-proven NAFLD with enteric-coated cysteamine bitartarate for 24 weeks. There was a significant improvement in liver enzymes without an effect on BMI and an increase in serum adiponectin levels. Based on the results of this preliminary study, the NIDDK NASH-CRN conducted CyNCh, a phase IIb multicentre RDBPCT of cysteamine bitartarate. In total, 169 children with biopsy-proven NAFLD (NAS≥4) were randomised to weight-based cysteamine or placebo for 1 year.92 Although cysteamine therapy was associated with significant improvement in ALT (decrease of 53 U/L vs 8 U/L in controls, p=0.02), there was no histological benefit, either in overall improvement of NASH (by NAS score) or in individual histological parameters.
Targeting apoptosis and TNFα pathway
A major component of steatohepatitis is hepatocyte injury and apoptosis, driven by both the intrinsic and extrinsic pathways, predominantly through tumour necrosis factor (TNF)α signalling.93 ,94 Both pathways converge to a shared mechanism via the enzymatic cascade and activation of caspases, controlling apoptosis and inflammation. Emricasan is an oral irreversible pan-caspase inhibitor with high first-pass metabolism,95 making it a potentially attractive agent for the treatment of liver diseases associated with apoptosis. Emricasan has been shown to decrease liver enzymes in patients with chronic hepatitis C96 ,97 and to markedly improve inflammation, hepatocyte injury and fibrosis in mice fed a high-fat diet (HFD), without having an effect on hepatic fat accumulation or features of the metabolic syndrome.98 Recently, in a short-term phase II RDBPCT, 38 subjects with non-cirrhotic NAFLD and elevated transaminases were randomised to receive emricasan 25 mg twice daily or placebo for 28 days.99 The emricasan-treated group demonstrated marked decrease in liver enzymes (median ALT decrease 25.8 vs 9.4 U/L for placebo, p<0.05) and, as expected, a decrease in serum cytokeratin 18 fragments, a marker of liver apoptosis. Whether this improvement in liver enzymes is associated with a histological improvement is yet to be seen. ENCORE-NF, an ongoing phase IIb trial (NCT02686762), is evaluating the efficacy of two doses of emricasan (10 mg and 100 mg/day) for 72 weeks in patients with biopsy-proven NASH and fibrosis (but not cirrhosis). The primary end point is improvement in fibrosis without worsening of NASH, with secondary end points aiming to demonstrate histological improvement or resolution of NASH.
Pentoxifylline has been suggested as a potentially beneficial therapy for NASH due to its putative effects on TNFα, reduction of oxidative stress and possible antifibrotic effects. Furthermore, it was initially thought to be beneficial for the treatment of severe acute alcoholic hepatitis,100 although this has not been replicated in later, larger trials.101 ,102 In a small, two-centre RDBPCT, Zein et al103 treated 55 patients with biopsy-proven NASH with pentoxifylline 400 mg three times daily (or placebo) for 1 year. Histological improvement, defined as a decrease in NAS by ≥2 points, occurred in 38.5% of patients treated with pentoxifylline compared with 13.8% of placebo-treated ones (p=0.036). The main factors driving the improvement were a reduction in steatosis and inflammation, while hepatocyte ballooning did not change significantly from baseline. There was also a modest (0.2 unit) but significant decrease in fibrosis scores. Resolution of NASH was seen in 25% of patients treated with pentoxifylline (p=0.03 for the comparison with placebo). No effect was seen on markers of insulin sensitivity or on serum TNFα levels. Histological response to pentoxifylline was associated with a decrease in plasma levels of several oxidised lipids,104 suggesting that a reduction in lipid peroxidation may underlie the beneficial effect. Two recent meta-analyses105 ,106 included that study as well as several other smaller, lower-quality ones and concluded that pentoxifylline has a beneficial effect on liver enzymes and histology.
Apoptosis signal-regulating kinase 1 (ASK1) is a MAP3 kinase that upon activation by extracellular TNFα or intracellular oxidative or endoplasmic reticulum stress activates the p38/JNK pathway, resulting in hepatocyte apoptosis and fibrosis. In murine models, it has also been shown to contribute to TNFα-mediated insulin resistance and steatosis107 and inhibition of ASK1 ameliorates diet-induced steatosis and fibrosis.108 In an ongoing randomised open-label phase II trial, GS-4997, an oral ASK1 inhibitor, is studied for 24 weeks with or without simtuzumab in patients with NASH and stage 2–3 fibrosis (NCT 02466516).
Immune modulators
Two IκB kinases (IKKs)—IKKε and TBK1—appear to be important in linking obesity and inflammation. Both are upregulated in adipose tissue in animals with diet-induced obesity and deletion protects these animals from insulin resistance, obesity and steatosis.109 Amlexanox, a medication used to treat asthma and aphthous ulcers, was found in a screening assay to be an inhibitor of both IKKε and TBK1 and in animal models of diet-induced or genetic obesity caused weight loss through increased energy expenditure.110 In these animals, amlexanox treatment improved insulin sensitivity and decreased steatosis and hepatic expression of inflammatory genes. A phase II RDBPCT is currently assessing the effects of 12 weeks of amlexanox in patients with diabetes, obesity and fatty liver on hepatic fat content by imaging, HbA1c and weight (NCT01975935).
In NASH, there is overexpression of inflammatory chemokines, including CCL2 (MCP1) and CCL5 (RANTES),111 and these play an important role in the activation and migration of inflammatory cells into the liver, as well as the progression of fibrosis.112 ,113 Cenicriviroc is an oral antagonist of CCR2/CCR5, the chemokine receptors for MCP1 and RANTES, respectively. In clinical trials of HIV-infected patients, cenicriviroc was associated with improvement in serum markers of fibrosis, although in patients with no apparent liver disease.114 Cenicriviroc is currently studied in patients with obesity, insulin resistance and suspected NAFLD in a phase IIa study (ORION) aiming to assess the effects of 24-week treatment on insulin sensitivity, liver enzymes and liver imaging. Simultaneously, CENTAUR, an ongoing phase IIb trial, is assessing the histological effects of up to 2 years of cenicriviroc or placebo on patients with NASH and fibrosis (but not cirrhosis).115 The results of both studies are expected later this year.
Vascular adhesion protein 1 (VAP-1, also known as SSAO) is a membrane sialoglycoprotein expressed on hepatic sinusoidal endothelial cells. VAP-1 functions as a receptor to support adhesion and recruitment of lymphocytes to the liver.116 VAP-1 also has amine oxidase activity, which can result in the generation of reactive oxygen species and subsequent injury. In patients with NAFLD, serum levels of the soluble form of VAP-1 are increased and hepatic VAP-1 expression is upregulated; furthermore, VAP-1 is also detected in these patients on activated stellate cells, the major mediators of fibrosis.117 Inhibition or knockdown of VAP-1 decreased hepatic inflammation, injury and fibrosis in murine models of NASH.117 PXS-4728A, a selective irreversible inhibitor of the VAP-1 enzymatic activity, improved histological features of NASH in the streptozotocin (STZ)/HFD mouse model and completed phase I in healthy volunteers.118
Medications with a primary gut target
Antiobesity medications
Lifestyle modification has been consistently shown to improve fatty liver disease119 ,120 and should be considered the backbone of any therapeutic intervention (table 1).121 However, sustained (as opposed to short term) weight loss is difficult to achieve and maintain. Orlistat is a gut lipase inhibitor, decreasing the absorption of dietary fats and approved for the treatment of obesity. In a small prospective RDBPCT, orlistat treatment for 6 months was associated with a significant decrease in ALT and liver fat by ultrasound compared with the control, diet-alone, group, despite a similar degree of weight loss.122 However, in another prospective open-label study, 9 months of orlistat, vitamin E and diet were compared with vitamin E and diet alone. Weight loss in both groups was associated with histological improvement as well as decrease in liver enzymes, but there was no difference between the orlistat-treated subjects and controls.123 Interestingly, there was no significant difference in the degree of weight loss between the subjects treated with orlistat and controls, but this may reflect the small sample size and lack of sufficient power. Furthermore, since vitamin E (which was subsequently shown to have beneficial effects on NAFLD) was included in both treatment groups, and since it is fat-soluble, impaired absorption of the vitamin E supplementation in the orlistat group could have offset the benefits from this intervention. Thus, although orlistat is not likely to provide benefit independently of weight loss, it can be considered as an adjunct to assist in weight loss in patients with NAFLD.
Targeting gut microbiome
The gut microbiome is involved in the pathogenesis of NAFLD and NASH,124 at least in part through exposure of the liver to bacterial products such as lipopolysaccharide (LPS). IMM-124e is an IgG-rich extract of bovine colostrum obtained from cows immunised against LPS. In ob/ob mice, oral IMM-124e treatment is associated with improved liver fat content, liver enzymes and insulin sensitivity,125 thought to be due to a reduction of delivery of LPS and bacterial products from the gut to the liver and subsequent activation of Kupffer cells. In a small pilot study on 10 patients with biopsy-proven NASH, 30 days of IMM-124e treatment improved insulin sensitivity and glycaemic control, with a small effect on liver enzymes.126 A phase II RDBPCT is currently evaluating the effects of 24 weeks of IMM-124e in patients with biopsy-proven NASH (NCT02316717). The study end points include changes in liver fat content (by MRS) and in liver enzymes.
The effects of the gut on the liver can also be addressed by modulating the gut microbiome itself, through a faecal microbiota transplantation (FMT). FMT is a proven effective therapy for recurrent Clostridium difficile infection and transplantation of gut microbiota from lean subjects was shown to improve insulin sensitivity in subjects with the metabolic syndrome.127 In an ongoing open-label pilot study, investigators are performing FMT from lean donors to patients with biopsy-proven non-cirrhotic NASH to assess the effects on liver fat content and markers of injury (NCT02469272). Although FMT is unlikely to become a main therapeutic option for NAFLD, the results of the study will hopefully shed light on an important element of the disease pathogenesis.
Solithromycin is a new generation macrolide antibiotic in clinical trials for the treatment of bacterial infections. In a murine model of diet and STZ-induced NASH, solithromycin has demonstrated reduction in hepatocyte ballooning and inflammation, without an effect on liver fat content128 as well as a decrease in blood glucose levels and downregulation of hepatic gluconeogenic enzymes.129 The mechanism of response may not be related to solithromycin's antibacterial activity as it is not active against gut Gram-negative bacteria. Based on the preclinical results, a phase II open-label study is currently ongoing to determine the effect of 13 weeks of solithromycin treatment on liver histology in patients with non-cirrhotic NASH (NCT02510599).
Antifibrotic medications
Given the clear association between hepatic fibrosis and clinical outcomes in liver disease in general and in NASH in particular,7 reduction in fibrosis is a main goal of treatment (table 1). Beyond targeting NASH-specific disease mechanisms, as detailed above, several medications focus on the fibrotic process itself in a NASH-independent manner. Simtuzumab is a monoclonal antibody against lysyl oxidase-like 2 (LOXL2), a matrix enzyme responsible for cross-linking of collagen chains that is expressed extensively in fibrotic regions in the liver.130 Simtuzumab is currently evaluated in a long-term phase II study (NCT01672866) to determine whether it can decrease hepatic collagen in non-cirrhotic subjects with NASH and advanced fibrosis. In cirrhotic patients with NASH, simtuzumab treatment for 2 years is being studied (NCT01672879) with the goal of decreasing hepatic venous pressure gradient (HVPG), as well as a reduction in liver-related mortality, transplant or decompensation. Both studies have completed enrolment but results are not available so far.
Galectin-3 is a protein expressed predominantly in immune cells that recognises and binds to galactose residues. Galectin-3 is essential to the development of liver fibrotic process in NASH and GR-MD-02, a galectin-3 inhibitor, decreased NASH disease activity and fibrosis in an animal model.131 GR-MD-02 is currently evaluated in two phase II clinical trials; one trial is recruiting patients with NASH cirrhosis and portal HTN to evaluate the ability of 1-year treatment to reduce HVPG (NCT02462967), while the other study is evaluating the reduction of hepatic fibrosis by MRI after 16 weeks of treatment in patients with NASH and advanced fibrosis (NCT02421094).
Summary
In recent years, remarkable progress has been made in understanding the pathogenesis of NAFLD/NASH and, subsequently, in developing medications to target the disease. Interestingly, despite this progress, histological improvement of NASH has not exceeded 50% and NASH resolution rates are remarkably lower. This may reflect ineffectiveness of therapies, inability to overcome continuing caloric excess, or the heterogeneity in pathway activation between subjects. It is possible that no one-medication-fits-all strategy will be successful and future research should assess predictors of response to specific therapies.
It is becoming apparent from clinical trials that the short-term results of therapeutic intervention will depend on the target for therapy. Agents aimed at metabolic targets are likely to impact steatosis first, and then inflammation and ballooning followed by fibrosis, whereas agents targeting the interface of inflammation and fibrosis or fibrosis alone will impact their target. While prevention of progression to cirrhosis is a critical end point and a marker of therapeutic benefit, it should be noted that therapies based simply on pure antifibrotic effects do not address the drivers of disease progression such as cell stress, apoptosis and inflammation. The long-term implications of impacting fibrosis without affecting disease activity remain to be determined but may set the stage for combination therapies along with drugs targeting the metabolic components of the disease. Conversely, the benefits of drugs targeting metabolic targets may be amplified by adding agents with anti-inflammatory and antifibrotic effects. The current understanding of the pathophysiology of NASH provides a strong rationale for combination therapeutics for NASH and it is hoped that current agents in pivotal trials will be used to achieve high rates of response in use as combination therapy.
Currently, no pharmacological therapy is approved for NAFLD or NASH. Clinicians treating these patients should emphasise the importance of lifestyle changes, weight loss and exercise as the mainstay of treatment. Orlistat can be safely used as an adjunct to weigh loss and may provide additional benefit. Clinicians should also address and treat other components of the metabolic syndrome, including hypertension, dyslipidemia and insulin resistance or diabetes. Statins can be safely used to treat hypercholesterolaemia in patients with NAFLD or NASH. For patients with biopsy-proven NASH, currently available medications with proven benefit, although for off-label use, include liraglutide, pioglitazone, vitamin E and pentoxifylline. It should be noted that the vast majority of trials excluded patients with cirrhosis and these are best treated in the context of clinical studies.
Given the breadth of the pipeline of new agents and its dynamicity, in the coming years the arsenal of medications available to the hepatologist treating NASH is likely to significantly expand.
References
Footnotes
Contributors YR: manuscript design, writing, revision and approval of final version. AJS: manuscript design, revision and approval of final version.
Funding NIDDK Intramural Research Program.
Competing interests YR is funded by the NIDDK intramural Research Program.
AJS has stock options in Genfit. He has served as a consultant to AbbVie, Astra Zeneca, Nitto Denko, Nimbus, Salix, Tobira, Takeda, Fibrogen, Immuron, Exhalenz and Genfit. He has been an unpaid consultant to Intercept and Echosens. His institution has received grant support from Gilead, Salix, Tobira and Novartis. None of these are related to the current study. He is the president and CMO of Sanyal Biotechnologies.
Provenance and peer review Commissioned; externally peer reviewed.