Article Text

Abstract
Objective We aimed to characterise the liver tissue bacterial metataxonomic signature in two independent cohorts of patients with biopsy-proven non-alcoholic fatty liver disease (NAFLD) diagnosis, as differences in the host phenotypic features—from moderate to severe obesity—may be associated with significant changes in the microbial DNA profile.
Design and methods Liver tissue samples from 116 individuals, comprising of 47 NAFLD overweight or moderately obese patients, 50 NAFLD morbidly obese patients elected for bariatric surgery and 19 controls, were analysed using high-throughput 16S rRNA gene sequencing.
Results Liver bacterial DNA profile significantly differs between morbidly obese and non-morbidly obese patients with NAFLD. Bacteroidetes (p=1.8e-18) and Firmicutes (p=0.0044) were over-represented in morbidly obese patients and Proteobacteria (p=5.2e-10)—specifically Gammaproteobacteria and Alphaproteobacteria, and Deinococcus-Thermus (p=0.00012)—were over-represented in the non-morbidly obese cohort. Cohort-specific analysis of liver microbial DNA signatures shows patterns linked to obesity. The imbalance in Proteobacteria (Alpha or Gamma) among non-morbidly obese patients, and Peptostreptococcaceae, Verrucomicrobia, Actinobacteria and Gamma Proteobacteria DNA among morbidly obese patients was associated with histological severity. Decreased amounts of bacterial DNA from the Lachnospiraceae family were associated with more severe histological features. Proteobacteria DNA was consistently associated with lobular and portal inflammation scores. Microbial DNA composition corresponded to predicted functional differences.
Conclusion This is the first comprehensive study showing that the liver tissue of NAFLD patients contains a diverse repertoire of bacterial DNA (up to 2.5×104 read counts). The liver metataxonomic signature may explain differences in the NAFLD pathogenic mechanisms as well as physiological functions of the host.
- nonalcoholic steatohepatitis
- intestinal microbiology
- liver biopsy
- fatty liver
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The natural history of non-alcoholic fatty liver disease (NAFLD) is modulated by genetic and environmental factors.
Recent discoveries revealed the role of the gut microbiota in human health and disease, including NAFLD. However, the impact of the liver tissue microbial DNA profiling on the disease biology remains unknown.
What are the new findings?
The liver tissue contains a diverse repertoire of bacterial DNA (up to 2.5×104 read counts).
Obesity is an important driver of liver microbial DNA composition.
Liver samples of non-morbidly obese patients presented a dominant expansion of Proteobacteria DNA, which was linked to non-alcoholic steatohepatitis, ballooning degeneration, lobular and portal inflammation and liver fibrosis.
In contrast, in morbidly obese patients, the potentially detrimental association between liver histology and Proteobacteria DNA was shared with bacterial DNA derived from other taxa, including Verrucomicrobiae, Actinobacteria, Nitrospira and Bacterioidia.
Depletion of DNA from the Lachnospiraceae family members was associated with severe histological features.
Lipopolysaccharides (LPS) derived from Gram-negative bacteria localise in the portal tract.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
NAFLD subtypes might be distinguished by the microbial composition of the liver 16S rRNA gene profile.
The correlation of disease phenotypes and host phenotypic features with tissue bacterial metataxonomic profiling will enable better understanding of NAFLD pathogenesis, as well as provide a platform on which to evaluate interventions.
Liver bacterial metataxonomic profiling suggests that therapeutic options, including probiotic selection, should be precisely defined according to specific clinical scenarios, including the host phenome features.
Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a major burden on health systems around the world, as it has become the most prevalent chronic liver disease.1 The NAFLD natural history is modulated by genetic and environmental factors,2 including the gut microbiota that can explain a fraction of the disease heritability.2 3 Ample body of evidence indicates that the magnitude and severity of obesity and type 2 diabetes play a significant role in the disease prognosis.2 4 5 In fact, NAFLD exhibits high degree of comorbidity with disorders of the metabolic syndrome and shares with them pathogenic mechanisms of disease, including a state of chronic systemic inflammation.6
Much progress has been made in understanding NAFLD biology and pathogenesis. However, answers to some fundamental questions regarding mechanistic basis of disease remain elusive. For example, while epidemiological studies have robustly demonstrated that obesity is a major risk factor for NAFLD development,1 4 5 7 8 how and to what extent NAFLD interacts with the host phenome is only partially understood. Results yielded by a meta-analysis on hepatic histology in obese patients undergoing bariatric surgery (BS) suggest that pathogenic mechanisms of NAFLD in severely obese individuals are linked to the obesity status.9 Likewise, findings yielded by large studies exploring NAFLD histological features in morbidly obese patients demonstrate that severe obesity exerts strong influence on the liver disease phenotype.10
Advances in molecular technologies have revolutionised our understanding of mechanisms of complex diseases. Likewise, high-throughput deep sequencing has allowed researchers to elucidate the impact of the gut microbiome on human health, including NAFLD11–16 and non-alcoholic steatohepatitis (NASH)15 17–20—the severe histological form of the disease. Pioneering studies21–23 have consistently shown that obesity alters the gut microbiota. It has also been shown that severe—morbid—obesity per se imposes major changes in the gut microbiota, along with considerable systemic metabolic impairment.24
Earlier explorations of the biogeography of bacterial communities on the human body have demonstrated that microbiota varies systematically across body habitats.25 Consequently, studying the tissue microbiota of affected organs may open new research avenues, including the understanding of pathogenic mechanisms of human diseases.26 However, the impact of the liver microbiome on NAFLD biology and its potential interactions with the host phenome are largely unknown, which greatly limits our mechanistic understanding of microbial associations with the disease. In conducting the present study, we hypothesised that the liver microbial DNA signature in NAFLD may explain a portion of the variance in the disease manifestation, including differences in disease mechanisms and histological phenotypes. Hence, we aimed to demonstrate that a diverse and dynamic repertoire of bacterial DNA inhabits the liver of patients with NAFLD. We reasoned that examining the basic biology of the hepatic metataxonomic profile in patients with NAFLD and diverse host phenotypic features—from moderate to severe obesity—may provide valuable insight into the disease mechanisms as well as physiological functions of the host. The goal of this investigation was thus to characterise the liver tissue 16S rRNA gene signature in two unique and independent cohorts—overweight or moderately obese, and morbidly obese patients—with biopsy-proven NAFLD diagnosis.
Methods
Study design, patient selection and phenotypic characterisation
Using high-throughput 16S rRNA gene sequencing, we obtained pertinent liver metataxonomic information from the tissue samples of 116 individuals, namely 47 overweight or moderately obese patients with NAFLD, 50 NAFLD severely—morbidly—obese patients (body mass index (BMI) >40 kg/m2) elected for BS and 19 control subjects (non-NAFLD). Adopting a case–control design, we ensured that the control liver samples matched patients’ features in each cohort (morbidly obese and non-morbidly obese). Complete details of the study design, patient recruitment strategy, physical, anthropometric and biochemical evaluation, as well as histological assessment are shown in the online supplementary material.
Supplemental material
All investigations performed as a part of the present study were conducted in accordance with the guidelines of the 1975 Declaration of Helsinki, as revised in 1993.
16S rRNA gene metagenomic sequencing, sequence data analysis and operational taxonomy unit (OTU) clustering
DNA was isolated from liver specimens by a manual protocol as explained in the online supplementary material. Extracted bulk DNA samples were amplified with barcoded primers; DNA libraries were constructed, and high-throughput sequencing was performed on an IlluminaMiSeq platform by Macrogen Inc (NGS Division) Seoul, South Korea. A negative (blank) control was used to test the potential presence of contaminant DNA and/or cross-contamination; the sample showed no product and did not pass the quality control analysis (online supplementary figure 1). We used qPCR based on a SYBR Green assay27 and universal primers28 to measure the 16S rRNA gene absolute abundance within each sample. Details of metagenomic sequencing, including Linear Discriminant Analysis (LDA) Effect Size (LEfSe) and functional prediction can be found in the online supplementary material.
Supplemental material
Liver immunohistochemistry (IHQ) for LPS staining
Analysis of LPS abundance and localisation was performed by IHQ using an antibody that recognises the lipid A or endotoxin region of LPS from Escherichia coli. This antibody cross-reacts with Pseudomonas aeruginosa, Klebsiella pneumoniae, Salmonella enteritidis, Enterobacter aerogenes, Escherichia hermannii, Yersinia enterocolitica and Shigella sonnei. Further details can be found in the online supplementary material.
Results
Clinical, biochemical, and histological features of NAFLD patients and controls are presented in table 1.
Clinical and biochemical features of patients with NAFLD and the control group
Liver microbial DNA diversity is related to the host phenome
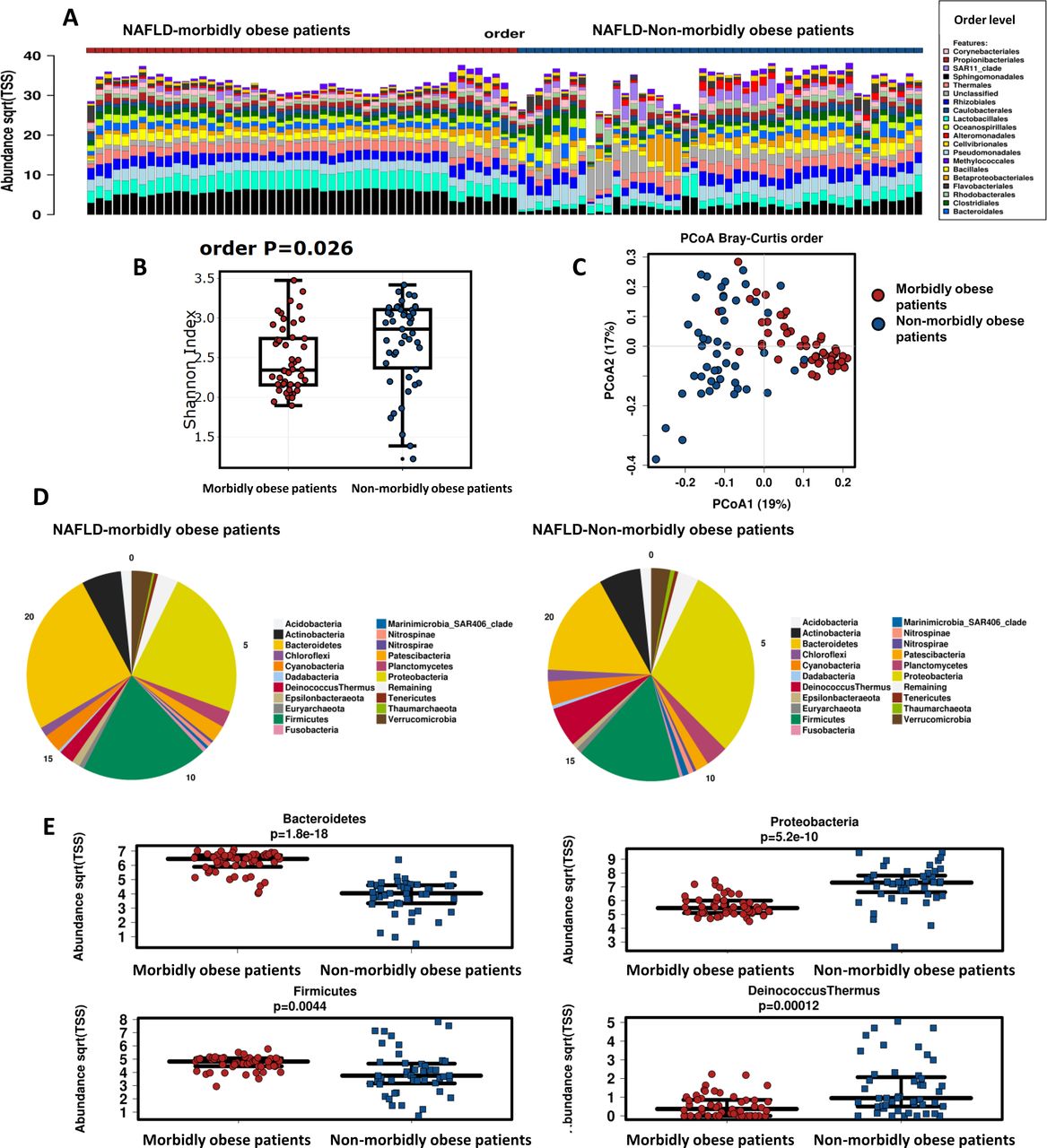
The rarefaction curves obtained for the dataset (OTU based) flatten to the right after ~4000 reads sampled, indicating that the underlying microbial profiling’ richness is well covered by the sequence data, which was higher than ~7500 reads in all samples (online supplementary figure 2). As indicated by the plots of microbial DNA composition in figure 1A, significant differences exist in the microbial diversity between the two NAFLD patient cohorts. The liver 16S rRNA gene signature of morbidly obese patients with NAFLD exhibited a significantly (p=0.026, non-parametric analysis of variance one-way test) decreased microbial diversity at the order taxonomy level (Shannon’s diversity, H index: 2.34±0.34) compared with that of overweight or moderately obese patients with NAFLD (Shannon’s diversity, H index: 2.86±0.5) (figure 1B).
Supplemental material

Liver metataxonomic signature in the two NAFLD cohorts: morbidly obese versus non-morbidly obese patients. (A) Stacked column bar graph depicting within-cohort liver microbial DNA composition. The microbial composition of each patient enrolled into the study is shown at the order level. (B) Bacterial DNA alpha diversity within subjects grouped by cohort, as measured by Shannon’s diversity index H at the order level. Box plot shows decreased microbial diversity (Shannon index) in morbidly obese patients with NAFLD. Horizontal line denotes mean, while box boundaries show SD. (C) Principal coordinate analysis (PcoA) plot showing Bray-Curtis beta diversity among patients by cohort at the order level. (D) Abundance profiling at the phylum taxonomy level in each cohort of patients (morbidly obese vs non-morbidly obese individuals). (E) Box plot showing the level (median) of differential bacterial DNA abundance (taxonomy: phylum level) between groups; box boundaries show SD. P value denotes non-parametric one-way analysis of variance (Wilcoxon rank test) statistics corrected by false discovery rate. NAFLD, non-alcoholic fatty liver disease.
Principal coordinate analysis (PcoA) of Bray-Curtis beta diversities revealed significant differences between the two groups across all taxonomic levels. Permutational analysis of variance of Bray-Curtis beta diversities indicated that intersubject differences between the two cohorts were explained by microbial taxonomic variation (PERMDIS2 p=1.19e-9) (figure 1C) at the order level. Phylogenetic beta diversity calculated using weighted UniFrac by PcoA also showed differences between groups (PERMDIS2 p=2.86e-6) at the order level.
Liver bacterial DNA profile significantly differs between morbidly obese and non-morbidly obese patients with NAFLD
While bacterial phyla Bacteroidetes, Firmicutes and Proteobacteria accounted for up to 90% of sequences on average, distribution of phyla and other taxon levels between the groups was uneven (figure 1D). Significant differences in the intercohort analysis results across most of the taxonomic levels seem to be driven by the differential abundance of the dominant phyla. Specifically, Bacteroidetes (false discovery rate (FDR): 1.8e-18) and Firmicutes (FDR: 0.0044) were significantly over-represented in the NAFLD-morbidly obese cohort, whereas Proteobacteria (FDR: 5.2e-10) and Deinococcus-Thermus (FDR: 0.00012) were over-represented among overweight or moderately obese patients with NAFLD (figure 1E). At the order level, Bacteroidia and Clostridia were over-represented in the NAFLD morbidly obese cohort, while Gamma, Alphaproteobacteria, Bacilli and Deinococci were over-represented in the NAFLD non-morbidly obese cohort (figure 2A). The dendrogram displayed in online supplementary figure 3 shows the phylogenetic relationships among these taxa.
Supplemental material

Liver bacterial DNA profiling is influenced by the host phenome. (A) Box plot showing the level (median and SD) of differential bacterial DNA abundance (taxonomy: class) between groups. P value denotes non-parametric one-way analysis of variance (Wilcoxon rank test) statistics corrected by false discovery rate. (B) Cladogram representing the LDA-LEfSe results, comparing taxonomic composition of liver bacterial DNA in the samples obtained from morbidly obese and non-morbidly obese cohorts, including controls, NAFL and NASH patients. (C) sPLS-DA classification performance of liver microbial DNA composition in morbidly obese versus non-morbidly obese patients with NAFLD based on the relative abundance of bacterial taxa at the class level. Graph shows the first two components with 95% confidence level ellipse plots. LDA, Linear Discriminant Analysis; LEfSe, Linear Discriminant Analysis (LDA) Effect Size; NAFL, non-alcoholic fatty liver; NASH, non-alcoholic steatohepatitis.
To identify the biologically informative features that help in differentiating the two NAFLD cohorts, we applied the LEfSe method. LEfSe findings confirmed that Bacteroidetes, Clostridia, Alpha and Gammaproteobacteria were the features most likely to explain the differences between the two groups (figure 2B).
Statistical comparison of taxon abundances across subsamples at the order, family and genus taxonomic levels, respectively, are shown in online supplementary tables 1-3. online supplementary figure 4 shows 25 of the top 100 features, some of which may assist in classifying patients into the NAFLD non-morbidly obese or NAFLD morbidly obese cohort based on area under the receiver operating characteristic (AUROC) analysis.
Supplemental material
To gain further insight into the contribution of specific bacterial DNA profiles that explain the differences between the two patient cohorts, we used sparse partial least squares linear discriminant analysis (sPLS-DA), which takes into account the sparsity and compositionality of microbiome data. The multivariable method identified similar bacterial DNA profiles as responsible for major differences, including component one that characterised the NAFLD morbidly obese cohort with Bacteroidales and Clostridiales, and to a lesser extent Verrucomicrobia, all of which accounted for ~8% of the variance, and component two that characterised the NAFLD non-morbidly obese cohort with Gram-negative bacteria, including species of the Phylum Proteobacteria and Deinococcus-Thermus, accounting for ~16% of the variance (figure 2C).
Results of the association between the liver tissue bacterial metataxonomic profiling and BMI as continuous variable are shown in the online supplementary material, online supplementary table 4 and online supplementary figure 5.
Supplemental material
Quantitative detection of total bacterial DNA
Total 16S rRNA gene load did not differ between NAFL and NASH patients in any of the cohorts; however, the liver of patients with NASH presented a modest (although non-significant) increase in the abundance of 16S rRNA gene. Complete details are provided in the online supplementary material and online supplementary figure 6.
Supplemental material
Cohort-specific analysis of liver microbial DNA signatures and liver histology
Analysis of the 16S rRNA gene signature in each individual cohort revealed that the liver bacterial metataxonomic signature was significantly influenced by the host phenome (morbid obesity vs non-morbid obesity).
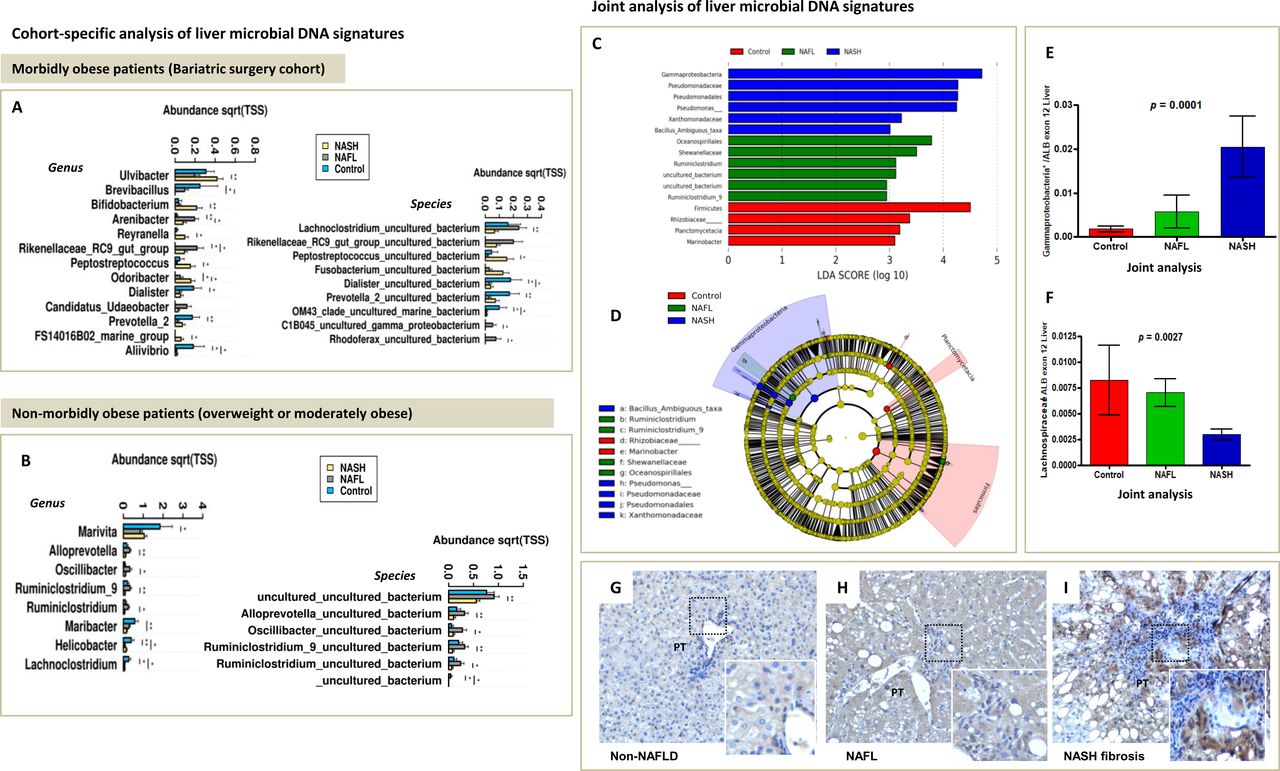
When controls (non-NAFLD) were compared with patients with NAFLD, we found that the hepatic microbial DNA profile varied greatly in taxa composition, indicating that some taxa may be associated with a protective effect against fatty liver. Nevertheless, a distinctive pattern was observed in severely obese individuals (figure 3A,B). For example, cohort-specific analysis showed that some bacterial DNA was seen most frequently in the liver specimens of non-NAFLD morbidly obese subjects (figure 3A), including Brevibacillus, Dialister, Prevotella two and Aliivibrio. Conversely, in the liver of non-NAFLD non-morbidly obese subjects, the most abundant taxa included Marivita (order Rhodobacterales) and Lachnoclostridium (order Clostridiales).

{kind=link}
{kind=link}
{kind=link}
Cohort-specific and joint analysis of liver microbial DNA signatures. (A) Analysis of liver microbial DNA signatures in morbidly obese patients (bariatric surgery cohort). (B) Analysis of liver microbial DNA signatures in non-morbidly obese patients. Measurements across sample groups are compared by the non-parametric Kruskal-Wallis statistical test. Significantly different taxa are shown as bar chart. SE is depicted by error bars. pairwise comparisons are performed by Mann-Whitney U test and are annotated as *p<0.05 and **p<0.01. Filter: include top 300 most abundant taxa. Control: non-NAFLD subjects. (C) LDA score in the joint analysis of liver microbial DNA signatures: visualisation of differential features with statistical and biological significance, ranked according to the effect size. (D) Cladogram representing the LDA LEfSe results, comparing taxonomic composition of control, NAFL and NASH liver tissue. Main features in the cladogram are represented on phylogenetic trees. The cladogram reports the taxa (highlighted by small circles and by shading) showing different abundance values (according to the LEfSe results) in the three groups as described in the results section; for each taxon, the colour denotes the class with higher median for both the small circles and the shading. These graphical outputs were generated by the LEfSe visualisation modules publicly available at the Galaxy website. (E and F) Validation of results by qPCR in the joint analysis. A non-parametric Kruskal-Wallis test was used for pairwise comparison of the subject groups. Bars represent mean±SEM. Data were analysed using GraphPad Prism version 4.00 for Windows (GraphPad software, San Diego, California, USA). The results were considered statistically significant at p< 0.05. *In silico PCR on the Silva database (https://www.arb-silva.de/search/testprime) shows that sequence primers used to validate Gammaproteobacteria DNA abundance have 96% coverage and 82% specificity for Pseudomonadaceae and 89% coverage and 80% specificity for Xanthomonadaceae/Stenotrophomonas. (G–I) Lipopolysaccharides (LPS) from Gram-negative Proteobacteria (Enterobacterales) localise in the portal tracts. Immunohistochemistry analysis was performed to assess the abundance and localisation of LPS in the liver. (A) Representative sample of non-NAFLD liver; (B and C) representative samples of NAFLD (NAFL and NASH fibrosis, respectively). LPS expression was assessed in five non-NAFLD samples and 16 samples of patients with NAFLD (NAFL n=8; NASH n=8). Original magnification 200×. LDA, Linear Discriminant Analysis; LEfSe, Linear Discriminant Analysis Effect Size; NAFL, non-alcoholic fatty liver; NASH, non-alcoholic steatohepatitis; Pt, portal tract.
The within-cohort analysis of bacterial DNA composition aimed at elucidating the NAFLD severity (NAFL vs NASH) showed associations with histological traits across the entire spectrum, including features of more aggressive stages such as ballooning degeneration and fibrosis. Detailed metataxonomic analysis at the genus and family levels and their relationship with main histological traits are shown in online supplementary tables 5 and 6.
Specifically, we observed that the presence of bacterial DNA of Roseibacillus (Verrucomicrobia/Verrucomicrobiae), Peptostreptococcus (Firmicutes/Clostridia), Bifidobacterium (Actinobacteria/Actinobacteria) and Streptomyces (Actinobacteria/Actinobacteria) was particularly dominant in the NAFLD morbidly obese cohort, and the more dramatic increases were associated with NASH (online supplementary table 5). Likewise, bacterial DNA of Proteobacteria—predominantly in the Gamma class—was associated with a more aggressive disease phenotype (online supplementary table 5).
Conversely, in the liver of NAFLD non-morbidly obese patients, significant differences were seen in the level of bacterial DNA of members of the Firmicutes/Clostridia genus and family, respectively, and the taxa Alloprevotella, all of which were associated with a benign disease course. Similarly, bacterial DNA from the Prevotellaceae family (Prevotellaceae_UCG001 and Alloprevotella genus) was found in association with less severe histological features, including lower lobular and portal inflammation scores and fibrosis. In addition, in the liver of NAFLD non-morbidly obese patients, DNA from Proteobacteria in the Gamma, Alpha and Epsilon classes was associated with severe histological features, including NASH, ballooning degeneration, lobular and portal inflammation, and liver fibrosis (online supplementary table 6).
Analyses of the similarities of bacterial DNA from the liver specimens of both NAFLD groups (morbidly obese and non-morbidly obese) yielded remarkable findings in association with members of the Lachnospiraceae family, including the Lachnospiraceae_NK4A136_group, and absence of liver inflammation (online supplementary tables 5,6). Likewise, while overabundance of bacterial DNA from the Gram-positive Lachnoclostridium and Ruminiclostridium was associated with benign histological features in the morbidly obese and non-morbidly obese cohorts, respectively, bacterial DNA from the Xanthomonadaceae family members was detected across samples in both groups and was associated with the presence of NASH (online supplementary tables 5,6).
Joint analysis of metataxonomic profiles suggests that major differences in bacterial DNA composition of the liver tissue in patients with NASH are driven by enrichment with Proteobacteria taxa in the Gamma class
Next, we focused on the differences in liver bacterial DNA composition across the histological stages of disease severity, from the control (non-NAFLD) liver to NASH in the whole population, which was normalised per sample and cohort values (morbid obesity and non-morbid obesity) according to the LEfSe method. All features were analysed by Kruskal-Wallis test, including establishing whether the values in different classes were differentially distributed.
The LEfSe approach was used to identify the taxa that most likely explain differences in the disease severity. The pairwise Wilcoxon test was therefore conducted to assess whether all pairwise comparisons between groups significantly agree with the class-level trend. Finally, an LDA model was developed, yielding relative differences among classes that were used to rank the features. It revealed that the most differentially abundant bacterial taxa in control liver specimens belong to phylum Firmicutes. In contrast, over-represented taxa in NASH specimens included Gammaproteobacteria and Pseudomonadales (figure 3C, online supplementary figure 7). Taxonomic representation of statistically and biologically consistent differences among control liver, NAFL and NASH is shown in figure 3D. Overabundance of Gammaproteobacteria DNA in the liver of patients with NASH was further validated by an independent quantitative molecular approach (RT-qPCR) using primer sequences with high coverage and specificity for Pseudomonadaceae, including Xanthomonadaceae/Stenotrophomonas (figure 3E). Depletion of Lachnospiraceae DNA in the liver of NASH patients was further validated by qPCR (figure 3F).
Supplemental material
Validation of main findings by an independent molecular approach
We validated NGS bacterial DNA abundances of eight representative taxa (Alpha and Gamma Proteobacteria, Actinobacteria, Peptostreptococcus, Verrucomicrobia, Veillonella, Lachnospiraceae and Bacteroides/Prevotella) that showed the most significant differences in the analysis of association with the disease severity and related histological traits. Validation was performed by RT-qPCR using taxon-specific primers listed in the online supplementary material.
Results of copy number abundances of taxa associated with severe histological traits were consistent with the findings of 16S rRNA gene metagenomic sequencing, as all effects remained in the same direction as those yielded by the NGS analysis. There was no detectable amplification in non-template controls in any of the assays. A summary of cohort-specific validation results can be found in online supplementary figures 8,9.
Supplemental material
Supplemental material
LPS derived from Gram-negative bacteria localise in the portal tract
We hypothesised that Gammaproteobacteria DNA found in tissue samples—particularly Enterobacteriales—originates in the gut and translocates to the liver through the portal system. Therefore, the liver tissue was examined for the presence of another bacterial constituent, bacterial LPS. As positive control tissue, we included a tissue specimen of necrotic colon cancer in which we observed marked immunoreactivity of LPS (online supplementary figure 10). Immunohistochemical analysis revealed that LPS immunostaining localised in the portal tracts with absent or minimal immunoreactivity in liver zones 2 or 3 (figure 3H/I). Clusters of LPS particles were more frequently seen in areas of portal inflammation. In addition, compared with non-NAFLD liver specimens (0.2±0.27 mean, SD) and liver samples of patients with NAFL (0.75±0.27), the liver of patients with NASH (2.05±0.8) presented significantly higher number of portal tracts stained positive for antibodies against LPS (p=0.0004) (figure 3 H/I).
Supplemental material
Liver bacterial DNA profiling shows distinctive predicted functional signatures
Specialised pathways were observed in each patient subsample, which were modulated by the host phenome-related factors (severe obesity vs non-severe obesity). Details can be found in the online supplementary material and online supplementary figure 11−15.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Discussion
NAFLD specialists have long relied on liver biopsy to infer the clinical impact of histological findings, but revolutionary advances in molecular and sequencing techniques have provided new avenues for enhancing our understanding of the disease pathogenesis. Here, we extended the analysis of the disease beyond risk factors and histological features to include largely unknown features of the liver bacterial metataxonomic profile. We focused on severe (morbid) obesity and non-morbid obesity and confirmed that the liver microbial DNA signature of NAFLD is dramatically modulated by the host phenome. Although the liver microbiota composition in terms of main phyla was comparable between the morbidly obese and non-morbidly obese NAFLD cohorts, we observed significant differences in the abundance profiling and diversity measurements and in the predicted functionality of bacterial DNA.
Specifically, we found that, compared with non-morbidly obese patients, the liver of morbidly obese patients with NAFLD showed decreased Alpha diversity. In addition, we noted significant differences in the Beta diversity between the two cohorts, as measured by the non-phylogeny-based Bray-Curtis method and the weighted UniFrac, which uses the OTU’s phylogeny. Analysis of other relevant clinical covariates indicated that there were no associations with sex, type 2 diabetes or arterial hypertension.
Furthermore, we found that bacterial DNA profile of the liver of morbidly obese patients with NAFLD is characterised by overabundance of species of the phyla Bacteroidetes and Firmicutes, more specifically the classes Bacteroidia and Clostridia, respectively, that represented ~8% of the diversity variance in the PCoA 1. In contrast, in the liver of non-morbidly obese NAFLD patients, we noted overabundance of species of the phyla Proteobacteria and Deinococcus-Thermus—specifically, Alpha and Gammaproteobacteria, Bacillii and Deinococci, which represented ~16% of the diversity variance in the PCoA 2.
Assessment of differences in the liver bacterial DNA composition across the full disease severity spectrum, including the control liver, and the liver of patients with NAFL and NASH in the whole population normalised per sample and cohort values (BMI) according to the LEfSe method, showed that the disease severity is associated with Proteobacteria in both Alpha and Gamma classes. Specifically, the most pronounced fold changes in microbial DNA associated with NASH were seen for Gammaproteobacteria, Pseudomonadaceae and Pseudomonadales. These findings are in agreement with those yielded by previous explorations of human NAFLD microbiota signatures in non-liver tissues, particularly the gut lumen microbiome,18 19 and the circulating microbiota of patients with NASH29 or alcoholic liver disease.30
Assessment of liver bacterial DNA abundance and histological traits highlighted the specific impact of the architectural metataxonomic signature in each patient cohort, indicating that the uneven distribution of bacterial DNA species differentially influenced the histological phenotypes. Ruminiclostridium and Alloprevotella depletion was associated with NASH in non-morbidly obese patients, whereas increased abundance of Xanthomonadaceae family, as well as some particular members of the Clostridia, Actinobacteria and Verrucomicrobiae classes—including Peptostreptococcus, Bifidobacterium and Roseibacillus—was associated with NASH among morbidly obese patients. Significantly greater proportion of the anaerobes Peptostreptococcus was noted in the samples of morbidly obese patients with NASH relative to NAFL. This genus is recognised as GI inhabitant,31 and recent studies have shown increased levels of Peptostreptococcus sp. in faecal samples of patients with ulcerative colitis32 and colorectal cancer.33 In addition, marked expansion of Verrucomicrobia DNA was seen in the liver specimens of morbidly obese patients with NASH—a finding that was validated by qPCR using a primer set with high specificity (99.9%) for Rosseibacilus (Verrucomicrobiaceae) (online supplementary figure 8). The implication of Rosseibacilus in the context of human diseases remains largely unknown; these Gram-negative taxa have been isolated in aquatic and terrestrial environments.34
The overall analysis showed consistent association of Proteobacteria (Alpha or Gamma) DNA with features of the disease severity, including lobular and portal inflammation, liver fibrosis and the NAFLD Activity Score (NAS) score. This is highly relevant in light of available evidence suggesting that Gram-negative bacteria, particularly Proteobacteria, are involved in the genesis of endotoxaemia—also described as metabolic endotoxemia—35 36 and inflammation,36 37 high levels of ethanol production,14 20 evasion from immunity system surveillance and defence mechanisms or molecular mimicry38 and predisposition to cardiovascular (CV) disease.39
We investigated the putative presence of bacterial LPS in the liver and found that Gram-negative-derived LPS staining was more frequently observed in the liver of NASH patients with severe fibrosis than in NAFL or non-NAFLD samples and we demonstrated that LPS predominantly localised in the portal tracts. These results suggest that Gram-negative-derived DNA (and other constituents) found in the liver may also enter the liver trough the portal system. Hence, it is highly plausible to assume that gut represents the main source of Gram-negative Gammaproteobacteria, particularly Enterobacterales.
We identified some individual bacterial DNA that might be associated with protection against liver inflammation. This beneficial effect was observed for members of the Lachnospiraceae family, the Lachnospiraceae NK4A136 and Lachnospiraceae FCS020, as well as a member of the Bacteroidia class—the Alloprevotella genus that was significantly depleted in the liver of NAFLD-non-morbidly obese patients with more severe histological phenotypes. The validation analysis showed significantly reduced levels of Alloprevotella in patients with ballooning degeneration and fibrosis as well. In agreement with our observation, recent evidence indicates that administration of Lachnospiraceae attenuates obesity, inflammation and dysbiosis in mice fed high fat diet.40 Capsular faecal transplant of beneficial taxa such as Ruminococcaceae and Lachnospiraceae was correlated with cognitive improvement and decrease in inflammation in patients with cirrhosis.41 Likewise, the taxa Alloprevotella has been associated with decreased lifetime CV risk.42 We have found, however, that different genera within the family Prevotellaceae—for example Prevotella_9 and Alloprevotella—had opposite effects on liver histological features. This observation is in agreement with available CV research findings.42
Nevertheless, it is worth noting that the presence of bacterial DNA in the liver tissue does not necessarily imply existence of live bacteria. It is highly plausible that bacterial DNA could be present as debris in immune cells, specifically in Kupffer cells.43
Limitations and possible implications
The main limitation of our study is its cross-sectional design, which does not permit any causality inferences. Specifically, we cannot confirm that the disease severity and progression is caused by the observed microbial signature (bacterial DNA). Similarly, we cannot confirm whether the injured—diseased—host tissue, which is characterised by an environment of metabolic stress,1 7 44 45 is the driving factor behind a specific bacterial profile. However, while, as mentioned above, some bacterial species are representative of the microbial communities residing in the human gut—including Proteobacteria, Bacteroidetes and Firmicutes—other bacterial specimens are specifically abundant in either the oral cavity (http://www.homd.org/)—for example Veillonella, Negativicutes and Ignavibacteria—or the gastric microbiome, for example, Deinococcus-Thermus. Therefore, we cannot identify the source of the liver microbial DNA or assume that it is exclusively colonised by the continuing small leakage of bacteria from the gut. Nevertheless, it is tempting to speculate that alterations in the epithelial gut barrier may play a role in explaining our findings46 and would be amenable to pharmacological or BS intervention.47 48
In conclusion, this is the first comprehensive study showing that the liver tissue of NAFLD patients contains a diverse repertoire of bacterial DNA (up to 2.5×104 read counts after exhaustive filtering). Measurement of 16S rRNA gene content by qPCR using universal primers showed that total bacterial DNA of the liver tissue is not negligible. More importantly, the most pronounced differences within cohorts between mild and severe histological features were independently validated by RT-qPCR.
The liver microbial DNA signature may explain differences in NAFLD pathogenic mechanisms, as well as physiological functions of the host. NAFLD subtypes might be distinguished by the microbial composition of the liver microbiome. Our results may have important implications for unravelling the molecular basis of the disease and for expanding therapeutic options, including probiotic selection or BS that should be more precisely defined according to specific clinical scenarios, such as the obesity status and/or, for instance, treatment and prevention of infections of the mouth.
Footnotes
SS and CJP are joint senior authors.
Twitter @SSookoian, @cjpirola, @panat
Contributors SS: study concept and design; data acquisition; performed liver biopsies and collected biological material; histological evaluation; immunohistochemistry (IHQ); RT-PCR; data analysis and interpretation; general study supervision; manuscript drafting; and securing funding. AS: bioinformatics analysis of sequencing data and data processing and presentation. MSL and CF: sample processing; MG and GOC: performed liver biopsies and collected biological samples. CJP: study concept and design, biological material collection and data acquisition; RT-PCR; data analysis and interpretation; statistical analysis; IHQ; manuscript drafting; and general study supervision and securing funding.
Funding This study was partially supported by grants PID-C2012-0061, PICT 2014-0432, PICT 2014-1816, PICT 2015-0551 and PICT 2016-0135 (Agencia Nacional de Promoción Científica y Tecnológica, FONCyT), CONICET Proyectos Unidades Ejecutoras 2017, PUE 0055.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Biological specimens, including blood samples and liver biopsies from all subjects included in this study, were obtained with written, informed consent in accordance with the Institutional Review Board-approved protocols (protocol numbers: 104/HGAZ/09, 89/100 and 1204/2012).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.