Article Text

Abstract
Objective HCV is a major cause of chronic liver disease worldwide, but the role of neutralising antibodies (nAbs) in its natural history remains poorly defined. We analysed the in vivo role of hypervariable region 1 (HVR1) for HCV virion properties, including nAb susceptibility.
Design Analysis of HCV from human liver chimeric mice infected with cell-culture-derived prototype genotype 2a recombinant J6/JFH1 or HVR1-deleted variant J6/JFH1ΔHVR1 identified adaptive mutations, which were analysed by reverse genetics in Huh7.5 and CD81-deficient S29 cells. The increased in vivo genomic stability of the adapted viruses facilitated ex vivo density analysis by ultracentrifugation and in vivo neutralisation experiments addressing the role of HVR1.
Results In vivo, J6/JFH1 and J6/JFH1ΔHVR1 depended on single substitutions within amino acids 867–876 in non-structural protein, NS2. The identified A876P-substitution resulted in a 4.7-fold increase in genomic stability. In vitro, NS2 substitutions enhanced infectivity 5–10-fold by increasing virus assembly. Mouse-derived mJ6/JFH1A876P and mJ6/JFH1ΔHVR1/A876P viruses displayed similar heterogeneous densities of 1.02–1.1 g/mL. Human liver chimeric mice loaded with heterologous patient H (genotype 1a) immunoglobulin had partial protection against mJ6/JFH1A876P and complete protection against mJ6/JFH1ΔHVR1/A876P. Interestingly, we identified a putative escape mutation, D476G, in mJ6/JFH1A876P. This mutation in hypervariable region 2 conferred 6.6-fold resistance against H06 IgG in vitro.
Conclusions The A876P-substitution bridges in vitro and in vivo studies using J6/JFH1-based recombinants. We provide the first in vivo evidence that HVR1 protects cross-genotype conserved HCV neutralisation epitopes, which advocates the possibility of using HVR1-deleted viruses as vaccine antigens to boost broadly reactive protective nAb responses.
- HCV
- HEPATITIS C
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Little is known about the comparable fitness of HCV recombinants in vitro (cell culture) versus in vivo (mouse models).
Plasma-derived HCV particles display low buoyant density through association with lipoproteins and the virus is able to persist in patients in the presence of potentially neutralising antibodies.
Hypervariable region 1 (HVR1)-deleted HCV has altered physiochemical properties (ie, high density) in vitro.
HVR1 of HCV protects the virus against neutralising antibodies in vitro.
What are the new findings?
Mutations within the HCV gene, NS2, confer in vivo stability of a cell culture infectious HCV recombinant (J6/JFH1), thus bridging the gap between in vitro and in vivo studies.
Identification of adaptive NS2 mutations that might aid in the production of sufficient quantities of culture derived HCV for whole-virus vaccine approaches.
HVR1 protects cross-genotype reactive HCV epitopes in vivo, thus indicating that HVR1-mediated antibody resistance is a contributor to viral persistence in infected patients.
In vivo, HVR1-deleted HCV particles have the same low-density profile as unmodified particles.
Hypervariable region 2 of HCV may contribute to persistence by providing escape from neutralising antibodies.
How might it impact on clinical practice in the foreseeable future?
The study provides much needed in vivo data on the virological determinants of HCV neutralisation and control, with implications for vaccine development.
The development of an HCV vaccine presents major challenges, but is the only effective way to curtail the HCV global heath impact in the foreseeable future.
Introduction
Approximately 180 million people worldwide are chronically infected with HCV, with increased risk of developing liver cirrhosis and hepatocellular carcinoma.1 ,2 Despite recent introduction of direct-acting antivirals, a vaccine is urgently needed to control HCV worldwide3; there are ∼4 million new infections annually. HCV is a positive single-stranded RNA virus of the Flaviviridae family, encoding the structural proteins Core and envelope proteins 1 and 2 (E1 and E2), p7 and non-structural (NS) proteins 2, 3, 4A, 4B, 5A and 5B.4 HCV is stratified into seven major genotypes with many clinically relevant subtypes,5 and it circulates in infected individuals as a heterogeneous quasispecies with implications for effectiveness of immune responses.6
The genotype 2a HCV isolate, JFH1, shown to infect Huh7 hepatoma cell lines,7 has allowed in-depth probing of the virus life cycle.8 Low in vitro infectivity of JFH1 was initially overcome by development of the JFH1-based intragenotypic recombinant, J6/JFH1, in which core-NS2 had been replaced with corresponding genes of another genotype 2a isolate, J6.9 ,10 Additional in vivo studies were made possible by the small animal model, urokinase-type plasminogen activator-severe combined immunodeficiency mice with human liver xenografts (human liver chimeric mice), which is a robust HCV infection model.11–13 Culture-derived J6/JFH1 infected human liver chimeric mice and chimpanzees;14 ,15 we confirmed genomic stability of J6/JFH1 in vitro.16 ,17 However, in vivo genomic stability of this first high-titre HCV recombinant remains unexplored and could represent a virus proliferation bottleneck complicating the interpretation of in vivo studies.
The role of neutralising antibodies (nAbs) in resolving acute HCV infection remains incompletely understood.18 Although some patient studies suggest that the rapid induction of broadly reactive nAbs is associated with viral clearance,19 ,20 the implications for immunotherapy and vaccine development remain unclear. Specifically, the envelope proteins of HCV could be immunologically distinct across genotypes, or even subtypes, resulting in limited antibody cross-reactivity.21 Although cross-reactivity of human sera from patients with chronic HCV has been reported in vitro17 and in vivo,22 no in vivo neutralisation data on hard-to-neutralise strains, such as 2a strain J6,17 ,23 was reported. We previously showed that the E2 27 amino acid motif hypervariable region 1 (HVR1), found to be non-essential for in vivo infectivity,24 ,25 protected the virus from neutralisation at cross-genotype conserved epitopes in vitro.25 However, differences in HCV properties between culture-derived and in vivo derived HCV particles could greatly influence such findings.14 In vitro, HVR1-deleted infectious particles displayed a shift in density from a 1.01 to 1.1 g/mL range to a single peak at ∼1.1 g/mL.25
To address the importance of HVR1 for HCV neutralisation protection in vivo, we adapted the HCV recombinant, J6/JFH1, and the HVR1-deleted variant J6/JFH1ΔHVR1, to yield consistent high titres during in vivo infections of human liver chimeric mice and assayed their physiochemical properties. These in vivo derived, fully adapted and physiochemically similar viruses with and without HVR1 were used to address whether HVR1 protects HCV against cross-genotype reactive nAbs in vivo.
Materials and methods
Plasmids
We used plasmids encoding the genotype 2a recombinant J6/JFH1 and the HVR1-deleted variant J6/JFH1ΔHVR1.9 ,25 Point mutations were inserted by conventional cloning techniques, and the HCV sequence of final maxipreps was confirmed (Macrogen).
Cell culturing
Huh7.5 human hepatoma cells and Huh7-derived S29 cells26 (with low CD81 expression conferring non-susceptibility to HCV) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and antibiotics. Culturing and infections of cells with HCV were done as described.16 Infected cultures were evaluated every second day by NS5A-specific immunostaining. Transfections of cells with in vitro transcribed HCV RNA using Lipofectamine2000 (Life Technologies) were carried out for 4 h in OPTIMEM, trypsinised and reseeded at 100 000 cells/well in 24-well plates, while also plating four wells in slides for HCV-specific immunostaining to monitor viral spread. Supernatant collection and HCV-specific cell staining was carried out at days 1, 2, 3 and 4 post transfection.16 S29 cell transfections and evaluation of intracellular Core and infectivity as well as extracellular Core and infectivity were carried out as described.27 Briefly, intracellular HCV was released by five freeze/thaw cycles and intracellular Core was extracted in cold RIPA buffer supplemented with protease inhibitors. Intracellular and extracellular samples were analysed for Core (Abbott ARCHITECT HCV Ag assay) and HCV infectivity titres. Direct HCV open reading frame (ORF) sequencing of viruses recovered from culture supernatants was done by RT-nested PCR procedures.16
Human liver chimeric mouse infections
Animal studies using human liver chimeric mice engrafted with human hepatocytes were performed at Ghent University, with protocols approved by the local Animal Ethics Committee. Mice were prepared as described,12 ,28 and inoculated by intraperitoneal injection of 104 50% tissue culture infectious dose (TCID50) or focus forming units (FFUs) of culture-derived HCV, or 104 IU HCV RNA of mouse-derived HCV. Mouse liver repopulation by human hepatocytes was confirmed 2–3 days pre-inoculation by measuring human albumin plasma levels. Human polyclonal IgG levels in the animals were measured by an in-house ELISA as described.29 Sequencing the HCV ORF of viruses recovered from plasma or serum was performed as above, except that random hexamers were used in reverse transcription with Superscript II (Invitrogen). Analysing the entire HCV envelope protein sequence of HCV from in vivo-derived serum pools was done by RT-PCR and clonal analysis, and subsequent direct sequencing of any positions with >20% sequence variation. Cell culture experiments using mouse plasma were done with 5 IU/mL heparin.
Equilibrium density gradient centrifugation
Step gradients were made as described.25 Briefly, iodixanol at 40%, 30%, 20% and 10% (Optiprep, Axis-Shield) was layered and left upright for 24 h at 4°C for formation of semi-continuous gradients. Samples were added to the top of the gradients and these were spun at 35 000 rpm (∼151 000×the Relative Centrifugal Force) for 18 h at 4°C. Subsequently, 1 mL gradient fractions were harvested from the bottom and 700 µL were weighed to calculate fraction densities. Fractions were titrated for infectivity and HCV RNA at 1:10 dilutions as described below.
HCV infectivity and RNA titration of mouse and cell culture-derived samples
Initially cell culture supernatant infectivity titres were determined using limiting dilution (10-fold) TCID50 assays with six replicates.16 Subsequently, titrations were performed in triplicates using an assay allowing automated counting of FFU using ImmunoSpot Series 5 UV Analyzer.30 HCV RNA titres of mouse and cell culture samples were measured either by Roche COBAS AmpliPrep/COBAS TaqMan HCV test or in house qPCR.16
In vitro neutralisation
We used purified polyclonal IgG antibodies (H06 IgG) isolated from genotype 1a chronic-phase patient H plasma collected in 2006,22 ,31 as well as monoclonal antibodies AR3A, AR4A and AR5A with the b6 control antibody.32 ,33 We plated 6×103 Huh7.5 cells/well into poly-d-lysine-coated 96-well plates (96 Well Optical Bottom Plates, Nunc). The following day four replicates of culture-derived HCV (yielding FFU/well of between 45 and 170 counts in the virus-only controls) were incubated 1 h at 37°C with an IgG dilution series and subsequently incubated with cells along with eight replicates of virus without IgG. The infected cells were washed after 3 h incubation with the virus/IgG or virus only, and following an additional 45 h the cells were fixed and HCV-specific immunostaining was performed.16 The percent neutralisation was calculated by relating FFU counts to the mean of eight replicates incubated with virus only. Neutralisation data were analysed as dose–response curves using GraphPad Prism V.4.03.
In vivo neutralisation
The methodology was previously described in detail.34 Here, all mice were loaded with 1 mg/g body weight of purified H06 IgG or with purified control IgG isolated from HCV-negative healthy volunteers (Irrelevant IgG). In prior homologous challenge experiments, the 1 mg/g IgG dose appeared to give better protection than 0.2 mg/g34; due to the limited availability of H06 IgG, it was not feasible to use higher doses. Three days after infusion with the antibodies, the animals were injected with a 100% infectious dose of in vivo derived HCV (104 IU/mouse). Both the antibody and the virus were injected intraperitoneally. We previously found that 3 days after injection an insignificant amount of antibody was retained in the peritoneal cavity.34
Results
Human liver chimeric mouse inoculations with culture-derived prototype HCV recombinants J6/JFH1 and J6/JFH1ΔHVR1
Lindenbach et al14 demonstrated that the genotype 2a intragenotypic HCV recombinant, J6/JFH1, infected human liver chimeric mice and chimpanzees. We subsequently showed that J6/JFH1 and J6/JFH1ΔHVR1 infected human liver chimeric mice (B156 and B150R, respectively) without acquiring envelope mutations.25
HCV infection of B156 and B150R were studied among seven inoculations in which three and four mice were inoculated with 104 TCID50 of culture-derived J6/JFH1 (B156, B139 and K888) or J6/JFH1ΔHVR1 (B150R, K826, K890R and B139L), respectively. During a 4-week follow-up, two and three mice infected with J6/JFH1 and J6/JFH1ΔHVR1, respectively, developed infections with HCV genome titres >105 IU/mL (figure 1A, B). Mouse B139L had HCV RNA titres below the assay detection limit of <103 IU/mL (figure 1B) and K888 displayed HCV RNA titres between 104 and 105 IU/mL (figure 1A).
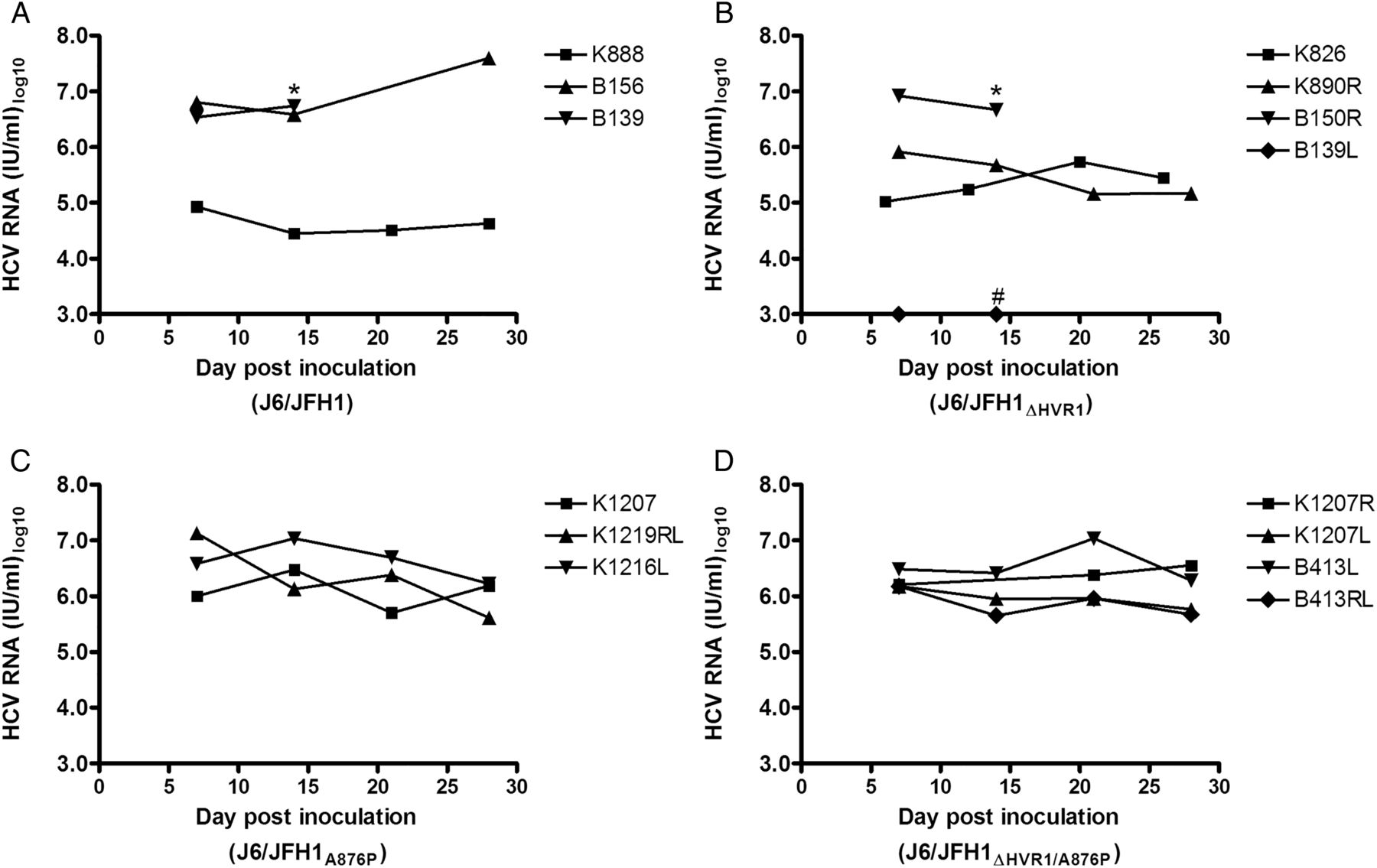
Inoculation of human liver chimeric mice with culture-derived HCV recombinant J6/JFH1 with and without hypervariable region 1 (HVR1). Mice were inoculated with similar doses of culture-derived HCV recombinants (A) J6/JFH1, (B) J6/JFH1ΔHVR1, (C) J6/JFH1A876P and (D) J6/JFH1ΔHVR1/A876P. HCV RNA titres were monitored on animal plasma or serum samples for 4 weeks post inoculation. Limit of HCV RNA detection was 1000 IU/mL.*The mouse died following sampling at the indicated timepoint. # No HCV RNA analyses were performed on later samples from this mouse.
Sequence analysis of HCV recovered from robust in vivo infections identified coding mutations within a short region of NS2
We extracted HCV RNA from high-titre mouse samples (week 2, K826 and B139; week 3, K890R and B150R; and week 4, B156) and directly sequenced the entire genome ORF (table 1). All viruses had developed dominant coding mutations within a short region in or near the third putative transmembrane domain of NS2: R867L, G868D, D871G, D871E or A876P (positions relative to H77 reference strain, GenBank accession number AF009606, table 1). We inoculated Huh7.5 cells with plasma or serum from HCV-infected mice at the sequenced timepoint. The virus spread to >80% of cells in 8–11 days, and the entire ORF was sequenced. We observed high correlation of obtained sequences between in vivo and ex vivo samples (table 1).
Sequence analysis of coding changes in J6/JFH1 and derived viruses from mouse samples and in vitro passages of mouse samples.
In vivo derived NS2 mutations conferred increased infectivity titres of J6/JFH1 in vitro by increasing assembly and release
J6/JFH1 constructs generated with the identified R867L, G868D, D871G, D871E or A876P substitutions, along with the parental control J6/JFH1, were used to generate genomic HCV RNA, which was transfected into Huh7.5 cells. All recombinants infected >80% of the cells 72 h post transfection. Analysing HCV infectivity titres in supernatants revealed that the NS2 mutations increased titres by 5–10-fold (figure 2A). Increased titres were confirmed in a separate transfection (not shown). As A876P displayed the most dramatic effect on infectivity and appeared in vivo in the absence of envelope mutations (table 1), we selected this mutant for further analysis. In a similar transfection experiment, we found that A876P conferred a 10-fold increase in infectivity titres for HVR1-deleted J6/JFH1 (figure 2B). Using transfection supernatants, which had been FFU titrated in triplicate, we performed controlled multiplicity of infection experiments and monitored infectivity titres in resulting culture supernatants (figure 2C, D). For J6/JFH1, we confirmed that A876P and G871G/E increased viral fitness, whereas R867L and G868D had minor, if any, effect compared to the original virus (figure 2C). The HVR1-deleted J6/JFH1 with A876P had faster kinetics and a >10-fold increase in infectivity titres (figure 2D). Direct sequencing of the HVR1 region and the N-terminal part of NS2 of viral RNA at peaks of infection for the different recombinants confirmed their viral identity.

In vitro viability of HCV recombinants J6/JFH1 and J6/JFH1ΔHVR1 harbouring in vivo derived mutations in NS2. (A and B) Huh7.5 cells were transfected with HCV RNA of the indicated J6/JFH1-based recombinants and infectivity titres were determined days 1–4 post transfection. (C and D) Huh7.5 cells were infected with HCV recombinants at a multiplicity of infection (MOI) of 0.003 from transfection supernatants of the indicated J6/JFH1-based recombinants. Supernatants were taken every other day for infectivity titrations; cells were split 1–3. Values represent the mean of triplicate titre determinations and error bars represent SD. FFU, focus forming units; GND, J6/JFH1 with GND polymerase substitutions (negative control).
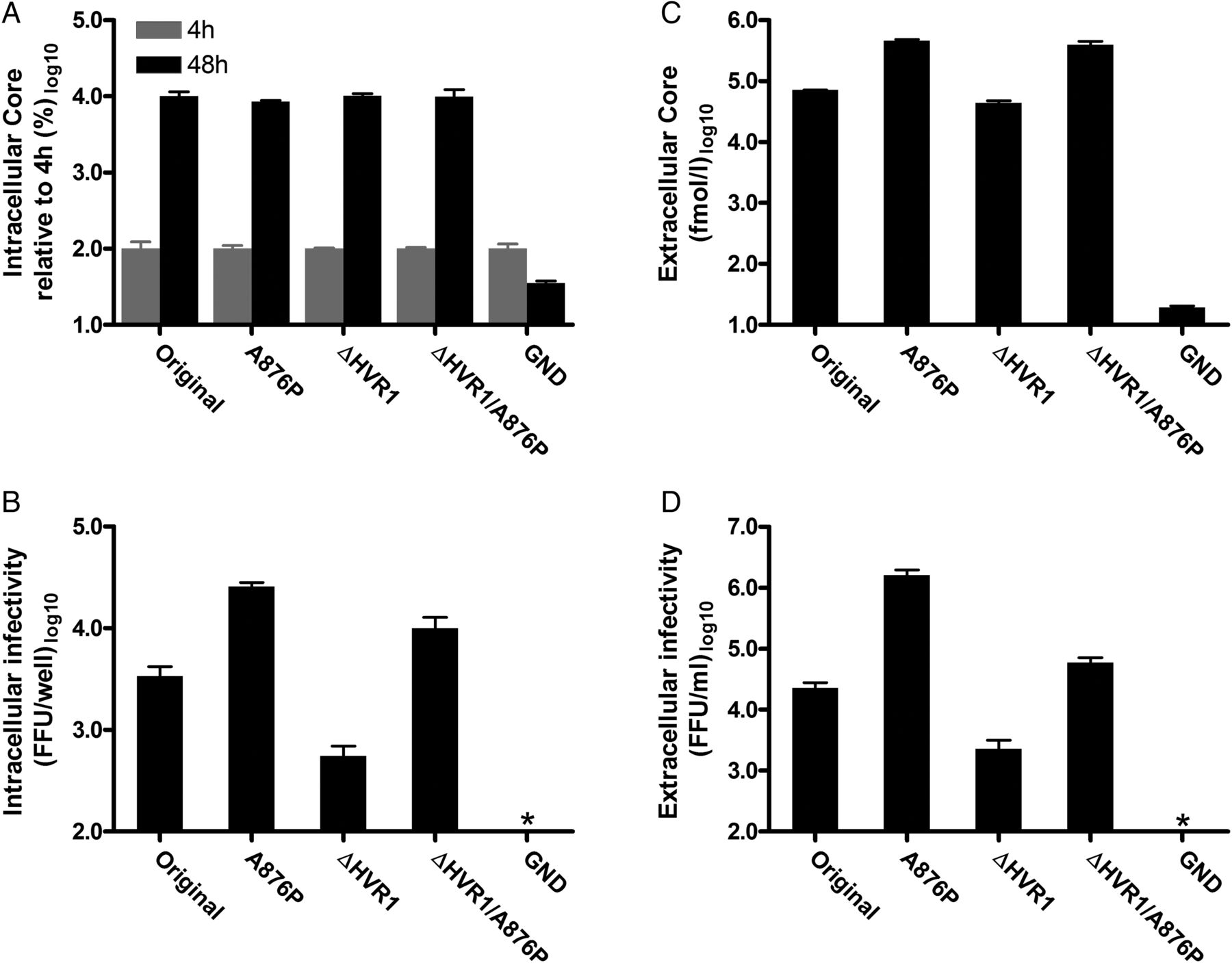
To evaluate which part of the viral life cycle was improved by A876P, we performed transfection of HCV-entry-impaired Huh7-derived S29 cells.26 Intracellular HCV Core values were similar for J6/JFH1 and J6/JFH1ΔHVR1 with and without A876P, suggesting that viral replication was unaltered (figure 3A). However, intracellular infectivity was greatly increased for viruses with A876P (figure 3B), indicating an increase in virus assembly. We observed increased extracellular Core and infectivity titres (figure 3C, D), as well as increased specific infectivity, for viruses with A876P, thus confirming improved assembly as well as either a direct or indirect effect on virus release.
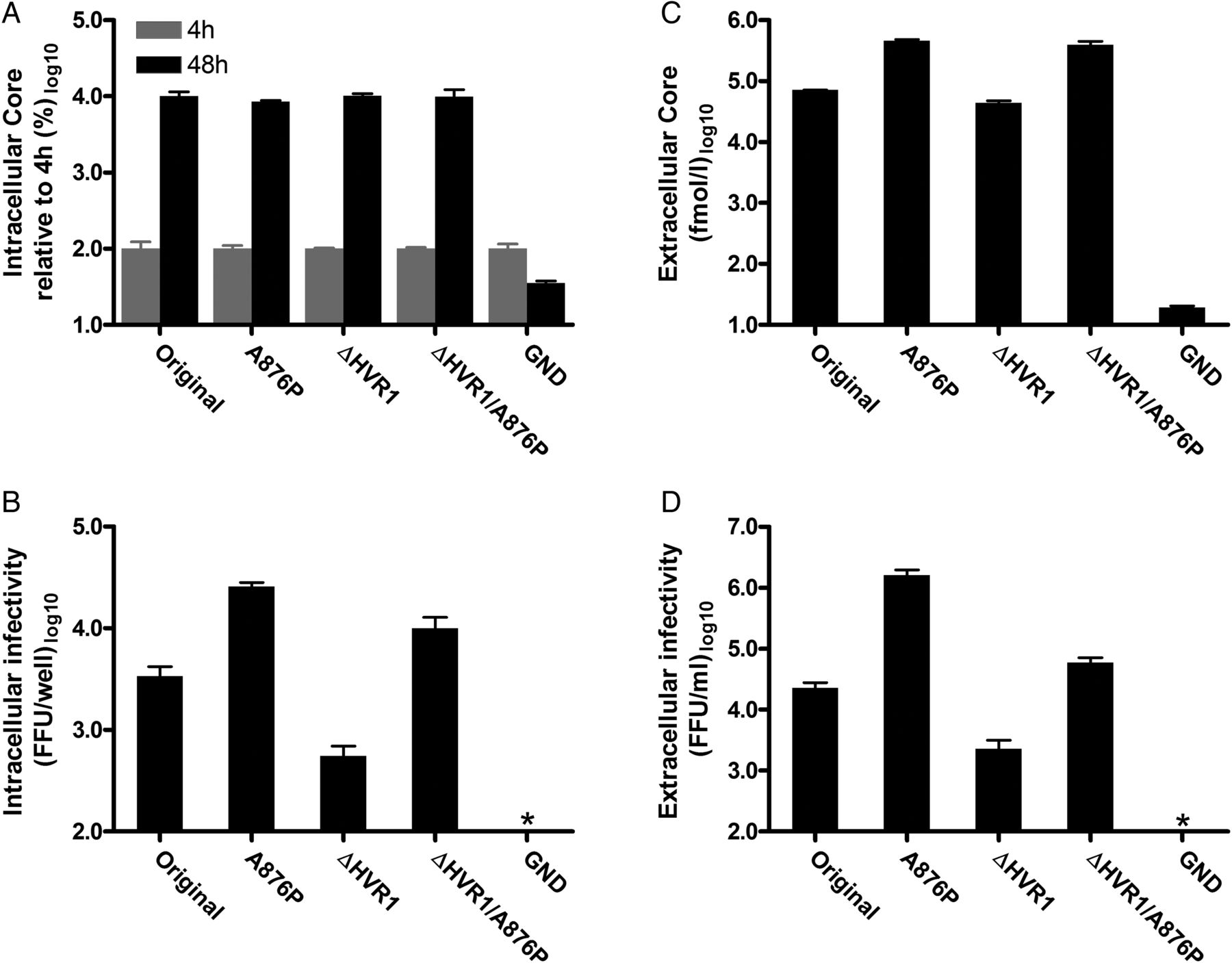
NS2 mutation A876P increased infectivity of HCV recombinants J6/JFH1 and J6/JFH1ΔHVR1 by increasing virus assembly and possibly release. Entry-impaired S29 cells were transfected with HCV RNA of the indicated J6/JFH1-based recombinants. (A) Intracellular Core values were determined in duplicate for 4 and 48 h post-transfection samples. Core values were normalised to the 4 h values as a marker of transfection efficiency. (B) Infectious intracellular HCV was extracted by freeze/thawing and subsequent clearing by centrifugation. Infectivity titres were determined in triplicate. (C) Extracellular Core values in duplicate and (D) extracellular infectivity titres in triplicate were measured directly on the 48 h cell culture supernatants. All bars in the different panels signify mean values with SDs. *Value below assay cut-off. FFU, focus forming units.
Inoculations of human liver chimeric mice with culture-derived J6/JFH1A876P and J6/JFH1ΔHVR1/A876P led to high-titre infections
We generated virus stocks of J6/JFH1A876P and J6/JFH1ΔHVR1/A876P by infecting naive Huh7.5 cells and verified the virus ORF by direct sequencing. Subsequently, three and four mice were inoculated with 104 FFU of culture-derived J6/JFH1A876P (K1207, K1219RL and K1216L) or J6/JFH1ΔHVR1/A876P (K1207R, K1207L, B413L and B413RL), respectively. All animals became infected with HCV genome titres >105.6 IU/mL during the first four weeks (figure 1C, D).
We inoculated Huh7.5 cells with mouse plasma or serum-derived HCV using animal samples from weeks 2 to 3 of infection. The viruses spread to >80% of cells in 6–8 days. Based on our observation that in vivo derived J6/JFH1 (mJ6/JFH1) could be cultured ex vivo without additional changes or reversions in the HCV genome (see above), we sequenced the entire ORF of viruses recovered from cell culture passages of either week 2 or 3 of the in vivo samples. In all ex vivo samples, the presence of A876P was confirmed and no additional NS2 mutations were observed (table 1). In addition, the number of dominant or 50/50 coding mutations in the genomes of passaged viruses was 4.7-fold lower compared with mice inoculated with J6/JFH1 viruses without the A876P mutation (2.8 vs 0.6 average coding changes per genome).
Altered density of culture-derived J6/JFH1ΔHVR1 was not observed for in vivo-derived viruses
We previously reported that the density of infectious HVR1-deleted viruses of several HCV genotypes, including the 2a recombinant J6/JFH1ΔHVR1, changed from a 1.01 to 1.1 g/mL range for the original recombinant to a single peak at ∼1.1 g/mL.25 We performed buoyant density gradient centrifugations of J6/JFH1A876P and J6/JFH1ΔHVR1/A876P derived either from cell culture or from pooled mouse samples. HCV RNA distributions were similar in all cases (figure 4A, B). Comparing the density of culture-derived infectious particles, we confirmed previous findings as J6/JFH1A876P had a heterogeneous density from 1.02 to 1.1 g/mL, whereas J6/JFH1ΔHVR1/A876P displayed a single high-titre infectivity peak at ∼1.1 g/mL (figure 4C). However, in vivo derived mJ6/JFH1A876P and mJ6/JFH1ΔHVR1/A876P displayed comparable density profiles ranging from 1.02 to ∼1.1 g/mL (figure 4D). We repeated this experiment using ex vivo samples pooled from all sampled timepoints of the mouse infections for in vivo derived J6/JFH1A876P with and without HVR1, and confirmed their heterogenous densities.

Equilibrium buoyant density centrifugation of in vitro and in vivo derived J6/JFH1 HCV recombinants. The indicated virus from Huh7.5 cultures (A and C) or from mouse samples (B and D) were layered onto an iodixanol gradient. Following ultracentrifugation, a total of 11 fractions of 1 mL were collected from the bottom and density was calculated. HCV RNA titres (A and B) were determined singly for all fractions and infectivity titres (C and D) were determined in triplicates. Error bars represent SD. Pool of mJ6/JFH1A876P consisted of mouse samples: K1219RL, week 1; K1216L, week 2; K1207, week 2. Pool of mJ6/JFH1ΔHVR1/A876P consisted of mouse samples: B413L, weeks 1 and 2; B413RL, week 1; K1207R, weeks 1 and 4; K1207L, week 2. FFU, focus forming units.
The HVR1 motif protected HCV from nAbs in H06 IgG both in vitro and in vivo
We and others have previously demonstrated an increased neutralisation sensitivity of HVR1-deleted viruses against chronic-phase sera from HCV-infected patients.25 ,35 ,36 Using H06 IgG purified from genotype 1a chronic-phase plasma (patient H), we performed a dose–response in vitro neutralisation assay based on FFU reduction for in vitro generated viruses J6/JFH1A876P and J6/JFH1ΔHVR1/A876P . J6/JFH1ΔHVR1/A876P was highly neutralisation sensitive with an IC50 value of 4.3×10−4 mg/mL, whereas J6/JFH1A876P could only be partially neutralised with an estimated IC50 value of 1.1 mg/mL (figure 5A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypervariable region 1 (HVR1) protects J6/JFH1 from neutralisation both in vitro and in vivo. (A) HCV recombinant J6/JFH1A876P or J6/JFH1A876P/ΔHVR1 were incubated in four replicates for each dilution step of a dilution series of IgG purified from chronic-phase patient serum H06 along with eight replicates of virus only. The virus and virus/antibody mixes were used to infect Huh7.5 cells and the number of focus forming units/well was visualised after 48 h infection by HCV-specific NS5A immunostaining and counted. Data were normalised to virus only and error bars represent SD. GraphPad Prism (V.4.03) was used to fit dose–response curves to the data points. (B and C) Human liver chimeric mice were loaded with either irrelevant or H06 IgG prior to inoculation with in vivo derived mJ6/JFH1A876P (B) or mJ6/JFH1A876P/ΔHVR1 (C). HCV RNA titres were measured on mouse plasma samples at weeks 1–4. HCV RNA cut-off was 750 IU/mL. Lines represent the geometric mean of HCV RNA measurements.
J6/JFH1A876P and J6/JFH1ΔHVR1/A876P were fully viable in vivo (figure 1C, D and table 1) and displayed physiochemical properties similar to that reported for in vivo-derived HCV (figure 4), and we, therefore, used these viruses to evaluate HVR1-mediated antibody protection in vivo. The mouse pools used in these experiments were the same as the ones described above to verify the heterogenous densities of mouse derived J6/JFH1A876P virus with and without HVR1. We ascertained the HCV envelope sequence of the pools by clonal analysis (9 or 10 clones for each pool) and subsequent validation of positions with >20% sequence variation by direct sequencing. We did not find changes in the J6/JFH1A876P pool. However, we did observe a dominant substitution N430D/n (A1629G/a) for J6/JFH1ΔHVR1/A876P, which would abolish the N-linked glycosylation of N430. We generated an HVR1-deleted J6/JFH1 recombinant with this substitution that we passaged, sequence-confirmed in the envelope genes and tested in the in vitro neutralisation assay using H06 IgG, and found no effect on neutralisation susceptibility (not shown). However, this was not surprising as N430 is not a part of the HCV glycan shield.37
Human liver chimeric mice were loaded either with irrelevant IgG or with H06 IgG, and 3 days later they were challenged with either mJ6/JFH1A876P or mJ6/JFH1ΔHVR1/A876P derived from pooled mouse samples (see above). All animals had plasma levels of human albumin >2 mg/mL at the time of loading with human IgG (indicating high-level liver humanisation). The infections were evaluated by measuring HCV RNA titres for 4 weeks post virus inoculation. For mJ6/JFH1A876P, the H06 IgG conferred only partial protection since all animals became infected, albeit with ∼100-fold decreased HCV RNA titres (figure 5B), in agreement with the partial neutralisation observed in vitro (figure 5A). For mJ6/JFH1ΔHVR1/A876P, we observed complete protection with no detectable viraemia in all three animals at all timepoints (figure 5C), indicating that the protection from neutralisation provided by HVR1 in vitro also occurred in vivo. We determined the H06 human IgG plasma level at the time of virus inoculation to be between 1.6 and 2.3 mg/mL. All animals without detectable HCV RNA inoculated with mJ6/JFH1ΔHVR1/A876P had human serum albumin levels >5 mg/mL at weeks 1 and 3 post challenge. Thus, failure of infection was not due to graft failure, but due to efficient heterologous neutralisation in vivo. Interestingly, one of the H06 IgG inoculated animals infected with mJ6/JFH1A876P displayed a rise in HCV RNA titres at week 3. Upon sequencing HCV RNA extracted from this timepoint, we identified the dominant substitution D476G (A1768G) in hypervariable region 2 (HVR2) of E2. We generated a sequence confirmed first passage stock of this virus and performed dose–response neutralisation experiments comparing sensitivity against H06 IgG in vitro. We observed a 6.6-fold decrease in sensitivity (table 2). In addition, when comparing sensitivity against monoclonal nAbs, AR3A, AR4A and AR5A, we observed 2.0–5.8-fold decreases in sensitivity (table 2). Thus, it appeared that D476G was an in vivo escape mutation.
The mutation D476G occurred during H06 IgG treatment in vivo and conferred antibody resistance against H06 IgG and three monoclonal antibodies, AR3–5, in vitro.
Discussion
We identified mutations in NS2 that appeared critical for in vivo infectivity of J6/JFH1-based HCV recombinants and showed that the in vivo derived mutations increased virus titres in cell culture. The most efficient of the identified NS2 mutations, A876P, increased assembly and possibly release, which makes the virus more suited for cell culture applications requiring high yield of particles, such as vaccines based on inactivated culture-derived HCV. Furthermore, since the NS2-adapted virus appeared to be genetically stable both in vitro and in vivo, this provides a valuable tool for bridging HCV studies in cell culture and animal models. Finally, using these viruses we found an increased heterologous neutralisation susceptibility of HVR1-deleted J6/JFH1A876P in vivo, thus supporting the feasibility of developing antigens for generating cross-genotype protective vaccines.
Sequence analysis of HCV from five human liver chimeric mice (figure 1A, B) suggested that culture-derived J6/JFH1 and J6/JFH1ΔHVR1 were not genetically stable in vivo (table 1). We observed consistent clustering of mutations at conserved residues within positions 867–876 of NS2 (R867L, G868D, D871G, D871E or A876P). Analysing J6/JFH1 recombinants harbouring these mutations revealed that they appeared to increase viral fitness (figure 2); A876P had the highest effect, yielding at least 10-fold higher titres. Single-cycle infections of CD81-deficient S29 cells showed that A876P increased assembly and possibly release (figure 3), which fits well with a previous report concerning the role of this region within NS2.38 A876P also increased in vivo genomic stability of both J6/JFH1 and J6/JFH1ΔHVR1 (table 1). These findings indicate virus bottlenecking of the original virus, which could result in lower HCV RNA titres in infected animals or even failure of infection. Furthermore, the increased release of particles in vitro caused by A876P suggests that this virus is an improved candidate for overcoming the inherent difficulties of making large quantities of HCV for inactivated whole-virus vaccines.39 ,40
Having confirmed the full viability, both in vitro and in vivo, of J6/JFH1 viruses harbouring the NS2 mutation, A876P, we analysed the physiochemical properties of in vivo derived mJ6/JFH1A876P and mJ6/JFH1ΔHVR1/A876P. In two independent experiments, we found that the high, homogenous density of culture-derived HVR1-deleted viruses was not observed for in vivo derived viruses (figure 4), as the density of viruses with and without HVR1 fully resembled what has been described for plasma-derived particles.14 ,41 Our finding differed from a recent study reporting that in vivo generated Jc1 without HVR1, another J6/JFH1 variant, maintained a homogenous high-density profile.42 However, in our study the unaltered density of HVR1-deleted viruses meant that we could exclude that differences in neutralisation sensitivity of HVR1-deleted particles was caused by altered physiochemical properties.
H06 IgG (from a genotype 1a-infected patient) had a partial effect against the 2a virus mJ6/JFH1A876P in vivo as all animals became infected with lower viral titres (figure 5B). These results were in agreement with in vitro results (figure 5A) and corroborate our previous in vivo findings that HCV is able to infect in the presence of heterologous chronic-phase patient-derived IgG, even though these contain high titres of cross-genotype reactive polyclonal nAbs.22 However, the partial effect was significant (100-fold titre decreases) and in conjunction with recent findings that HCV protection in vitro from polyclonal IgG can be overcome with monoclonal nAbs targeting critical epitopes,36 our data support the possibility that complete protection in vivo can be achieved, even for hard-to-neutralise HCV strains, such as J6,17 ,23 particularly if higher antibody titres not attainable here were generated. Furthermore, H06 IgG protected the animals from virus challenge (104 IU) of HVR1-deleted mJ6/JFH1 in vivo with no detectable viremia (figure 5C), showing that complete protection is possible with heterologous antibody and indicating that HVR1 plays a major role in protection of HCV in vivo from nAbs. A previous study showed protection of human liver chimeric mice against J6/JFH1, but only with a low challenge dose of 103 IUs and only when the virus was pre-incubated with vaccine-induced antibody prior to virus challenge.39 The apparent exposition of cross-genotype reactive epitopes of an HVR1-deleted virus in vivo suggests that this would be a superior antigen for inducing high titres of these potentially protective antibodies. Interestingly, we identified a putative escape mutation, D476G, in HVR2 of E2 for an animal infected with J6 retaining HVR1. When tested in vitro, this mutation conferred 6.6-fold resistance against H06 IgG as well as 2.0–5.8-fold resistance against monoclonal nAbs AR3A, AR4A and AR5A. While mutations that alter HCV neutralisation sensitivity have previously been described in HVR2,43 this is the first report of an induced escape mutation in vivo. It is, therefore, tempting to speculate whether the described variability in HVR2 could be protecting HCV by modulating nAb sensitivity of the virus. This finding could inspire further studies on the role of HVR2.
Since an IgG preparation from a genotype 1a-infected patient had a greater effect against the HVR1-deleted virus of genotype 2a than it did against the parental genotype 2a virus retaining HVR1, the epitope protection elicited by HVR1 must involve cross-genotype conserved epitopes. We thus confirmed our previous in vitro findings that HVR1 protects cross-genotype conserved neutralisation epitopes,25 although it should be recognised that the in vivo data are based on a single HCV isolate. The mechanism of HVR1-mediated antibody protection remains unclear. However, HVR1 could be obstructing accessibility of neutralisation epitopes as suggested35 or could contain neutralisation inhibitory epitopes.44 ,45 The latter cannot by itself explain increased neutralisation sensitivity of HVR1-deleted viruses as monoclonal nAbs also exhibit greater effectiveness against the HVR1-deleted Jc1 virus.35
The progress in developing an effective HCV vaccine continues and should be viewed in the context of developments in related fields. Thus, recent clinical trials using a tetravalent live attenuated vaccine against dengue virus demonstrated efficient, although serotype-dependent, protection.46 While genetic heterogeneity of HCV is considerable, our in vivo data suggest that a single cross-genotype reactive vaccine might be attainable, possibly negating the need for multivalent approaches and simplifying the development of a vaccine. Furthermore, targeting a cross-serotype conserved epitope for dengue virus was recently accomplished by using unique nAbs identified in patients,47 thus providing promise for the development of an improved vaccine antigen. This was recently accomplished for respiratory syncytial virus by engineering of an optimal, stabilised, vaccine antigen that was shown to elicit protective antibody responses against the virus in animal models.48 Our findings aid both in identifying a possibly superior vaccine antigen, which exposes conserved neutralisation epitopes in the absence of HVR1, while also identifying mutations in NS2 that increase in vitro particle release of relevance for generating sufficient quantities of virus for whole-virus vaccination trials. Thus, our study is highly relevant for efforts to devise an optimal HCV vaccine antigen.
References
Footnotes
Acknowledgements We are grateful to Lotte Mikkelsen and Maria-Louise Deistler (Copenhagen University Hospital, Hvidovre) for technical assistance; to Jesper Bonde, Jens Ole Nielsen, Bjarne Ørskov Lindhardt, and Ove Andersen (Copenhagen University Hospital, Hvidovre) for their support of the project, to Henrik Krarup (Aalborg University Hospital, Aalborg) for performing the Abbott HCV Core antigen analyses, and to Suzanne U. Emerson and Robert H. Purcell (NIH, US), Mansun Law (Scripps Research Institute, US) and Charles Rice (Rockefeller University, US) for providing reagents.
Contributors PM and JB contributed equally. Study concept and design: JP, PM and JB. Acquisition of data: JP, LV, RVM, CB and AF. Generated essential reagents: RW and HA. Analysis and interpretation of data: JP, PM and JB. Manuscript preparation: JP and JB. Revision of manuscript: JP, RVM, HA, PM and JB. Statistical analysis: JP. Funding: JP, PM and JB. Study supervision: PM and JB.
Funding This study was supported by Ph.D. stipends from Faculty of Health and Medical Sciences, University of Copenhagen (JP, RVM), an individual DFF-postdoctoral grant from the Danish Council for Independent Research, Medical Sciences (JP), and research grants from the Lundbeck Foundation (JP, JB), the Novo Nordisk Foundation (JB), the Danish Council for Independent Research, Medical Sciences (JB), and an advanced top researcher grant from the Danish Council for Independent Research (JB). Additional support was provided to PM by the Ghent University (Concerted Action Grant 01G01712), The Research Foundation—Flanders (Project G.0521.12N) and the Belgian state (IUAP P7/47-HEPRO-2).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.