Article Text

Abstract
Objective Liver fibrosis is associated with significant collagen-I deposition largely produced by activated hepatic stellate cells (HSCs); yet, the link between hepatocyte damage and the HSC profibrogenic response remains unclear. Here we show significant induction of osteopontin (OPN) and high-mobility group box-1 (HMGB1) in liver fibrosis. Since OPN was identified as upstream of HMGB1, we hypothesised that OPN could participate in the pathogenesis of liver fibrosis by increasing HMGB1 to upregulate collagen-I expression.
Design and results Patients with long-term hepatitis C virus (HCV) progressing in disease stage displayed enhanced hepatic OPN and HMGB1 immunostaining, which correlated with fibrosis stage, whereas it remained similar in non-progressors. Hepatocyte cytoplasmic OPN and HMGB1 expression was significant while loss of nuclear HMGB1 occurred in patients with HCV-induced fibrosis compared with healthy explants. Well-established liver fibrosis along with marked induction of HMGB1 occurred in CCl4-injected OpnHep transgenic yet it was less in wild type and almost absent in Opn−/− mice. Hmgb1 ablation in hepatocytes (Hmgb1ΔHep) protected mice from CCl4-induced liver fibrosis. Coculture with hepatocytes that secrete OPN plus HMGB1 and challenge with recombinant OPN (rOPN) or HMGB1 (rHMGB1) enhanced collagen-I expression in HSCs, which was blunted by neutralising antibodies (Abs) and by Opn or Hmgb1 ablation. rOPN induced acetylation of HMGB1 in HSCs due to increased NADPH oxidase activity and the associated decrease in histone deacetylases 1/2 leading to upregulation of collagen-I. Last, rHMGB1 signalled via receptor for advanced glycation end-products and activated the PI3K–pAkt1/2/3 pathway to upregulate collagen-I.
Conclusions During liver fibrosis, the increase in OPN induces HMGB1, which acts as a downstream alarmin driving collagen-I synthesis in HSCs.
- BASIC SCIENCES
- HEPATIC STELLATE CELL
- HEPATIC FIBROSIS
- HEPATOCYTE
- SIGNALING
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Osteopontin (OPN) and high-mobility group box-1 (HMGB1) are expressed in human and mouse liver.
We previously demonstrated the mechanisms driving the increase in OPN in liver fibrosis; yet, whether OPN could increase HMGB1 has not been established.
If besides the profibrogenic mechanisms previously identified by us, OPN targets HMGB1 to activate extracellular matrix deposition by hepatic stellate cells (HSCs) remained unknown.
What are the new findings?
OPN and HMGB1 expression correlate with fibrosis stage in humans and mice.
Using in vivo and in vitro loss or gain of function approaches, we demonstrate that OPN is upstream of HMGB1.
Extracellular OPN promotes the acetylation of intracellular HMGB1 in HSCs due to increased NADPH oxidase activity and the associated decrease in histone deacetylases 1/2 leading to upregulation of collagen-I.
Extracellular HMGB1 signals HSCs via the receptor for advanced glycation end-products activating the PI3K–pAkt1/2/3 pathway to increase collagen-I deposition.
How might it impact on clinical practice in the foreseeable future?
Identification of key mediators along with better understanding of the signalling pathways they trigger to promote fibrosis is critical to prevent disease progression and design new therapies. This study reinforces the role of HMGB1, a hepatic sterile damage-associated molecular pattern, in the progression of liver fibrosis. The role of OPN and HMGB1 on collagen-I production by HSCs reveals novel signalling mechanisms that could be targeted for therapeutic benefit.
Introduction
Fibrogenesis encompasses qualitative and quantitative changes in the extracellular matrix (ECM) deposits with a significant buildup of collagen-I fibers, largely produced by activated hepatic stellate cells (HSCs), which extensively distort the normal hepatic architecture. Failure to degrade the progressive increase in scar tissue is a major reason why fibrosis evolves into cirrhosis and hepatocellular carcinoma. To date, extensive research has focused on identifying the key factors involved in the pathogenesis of liver fibrosis; yet, the precise link between injured hepatocytes, HSCs and the fibrogenic response still remains poorly defined.
We have previously shown that osteopontin (OPN), a matrix-bound protein sensitive to oxidant stress and highly induced upon liver damage, plays a central role in the pathogenesis of liver fibrosis by contributing to ECM deposition.1–3 Mechanistic studies revealed first that OPN upregulates collagen-I production by HSCs via integrin αvβ3 engagement and activation of the PI3K–pAkt1/2/3–NFκB signalling pathway;3 and second, that OPN drives ductular reaction contributing to periportal scarring by increasing transforming growth factor (TGF) β production in biliary epithelial cells;1 yet, additional mediators downstream of OPN, perhaps with profibrogenic potential, could participate increasing pathological collagen-I deposition by HSCs.
High-mobility group box-1 (HMGB1) is a nuclear non-histone chromosomal protein that binds the DNA minor groove and is involved in DNA replication, repair and energy homeostasis.4 Initially, it was believed that HMGB1 acted primarily as an architectural protein. However, upon cellular injury and post-translational modifications (PTMs), HMGB1 undergoes translocation from the nucleus to the cytoplasm and is secreted via the lysosomal pathway in most cells.5 HMGB1 signals via the receptor for advanced glycation end-products (RAGE), toll-like receptors (TLRs)-2/4/9, Mac-1, syndecan-1, phosphacan protein-tyrosine phosphatase-ζ/β and CD24.6
Recent work from our laboratory has demonstrated that HMGB1 has noxious effects in the hepatic environment in the setting of alcoholic liver disease.7 When released from injured or necrotic cells due to loss of membrane integrity8 or when secreted by hepatocytes in response to ethanol,7 HMGB1 can trigger harmful responses. Thus, HMGB1 is now considered a member of the family of damage-associated molecular patterns (DAMPs) that communicate injury to neighbouring cells.
Although this alarmin increases in plasma and liver from alcoholic and fibrotic patients,7 ,9 it is unknown whether it plays a direct role in liver fibrosis. Our preliminary in vivo observations suggested that OPN is upstream of HMGB1 in hepatocytes and HSCs. To date, whether OPN by increasing HMGB1 in a paracrine and/or in an autocrine fashion could regulate collagen-I deposition in HSCs has not been demonstrated. Thus, we hypothesised that OPN by upregulating HMGB1 could participate in the pathogenesis of liver fibrosis driving scarring. Using in vivo and in vitro loss-of-function or gain-of-function approaches we focused on dissecting how the OPN and HMGB1 axis in hepatocytes and HSCs regulates the HSC profibrogenic behaviour. Overall, the data show that intracellular OPN increases HMGB1 expression and extracellular OPN induces acetylation of HMGB1 in HSCs due to increased NADPH oxidase (NOX) activity and the associated decrease in histone deacetylases (HDACs) 1/2 leading to upregulation of collagen-I. Thus, OPN has autocrine and paracrine effects in HSCs. Moreover, extracellular HMGB1 upregulates collagen-I expression in HSCs paracrinely due to RAGE activation of the PI3K–pAkt1/2/3 signalling pathway; thus, contributing to the pathogenesis of liver fibrosis.
Materials and methods
Mice
C57BL/6J wild-type (WT) and Opn−/− (B6.Cg-Spp1tm1Blh/J) mice were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). Opn+/− mice were intercrossed and littermates were used in all experiments. The Opn transgenic mice overexpressing OPN in hepatocytes (OpnHep Tg) under the serum amyloid-P component promoter were donated by Dr Mochida (Saitama Medical University, Saitama, Japan).10 These mice were crossbred for 10 generations with the same strain and stock number of C57BL/6J WT listed above. The Hmgb1fl/fl mice were donated by Dr Billiar (University of Pittsburgh, Pittsburgh, Pennsylvania, USA). In these mice, the Hmgb1loxP allele was created by inserting loxP sites within introns 1 and 2 flanking exon 2 of Hmgb1.11 The Hmgb1fl/fl mice were bred with Alb.Cre mice (the Jackson Laboratory) to generate hepatocyte-specific Hmgb1fl/flAlb.Cre mice (abbreviated as Hmgb1ΔHep). All animals received humane care according to the criteria outlined in the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the National Academy of Sciences and published by the National Institutes of Health.
Statistical analysis
Data were analysed by a two-factor analysis of variance. All in vitro experiments were performed in triplicate at least four times. A representative blot is shown in all figures. Eight mice per group were used in all the in vivo experiments, which were repeated twice.
Results
OPN and HMGB1 colocalise and their expression correlates with fibrosis progression in patients with chronic HCV-induced fibrosis
Since we hypothesised that OPN and HMGB1 could upregulate collagen-I deposition in human liver fibrosis, we determined whether there was correlation between the induction of both proteins and scarring. To this end, we analysed the expression of OPN and HMGB1 in paraffin-embedded archived human liver biopsies from deidentified controls and from patients with clinically proven hepatitis C virus (HCV). The latter were paired biopsy specimens, some of which showed progression of HCV-induced fibrosis (progressors) and others did not (non-progressors). Liver biopsies from patients with HCV showed coinduction of both OPN and HMGB1 expression compared with healthy explants (figure 1A, B). While their expression remained akin over time in the non-progressors (figure 1A), both proteins increased with fibrosis stage in the progressors (figure 1B). Computer-assisted morphometry assessment demonstrated correlation between OPN and HMGB1 expression and fibrosis stage in patients with chronic HCV infection (figure 1C). Immunofluorescence analysis proved co-localisation of both proteins in patients with chronic HCV infection (figure 1D). Thus, these results suggest that OPN and HMGB1 colocalise and their expression correlates with fibrosis progression in patients with chronic HCV-induced fibrosis.

Osteopontin (OPN) and high-mobility group box-1 (HMGB1) colocalise and their expression correlates with fibrosis progression in patients with chronic HCV-induced fibrosis. H&E staining, OPN and HMGB1 immunohistochemistry (IHC) in paraffin-embedded archived human liver biopsies from a deidentified control and from a patient with clinically proven hepatitis C virus (HCV)-induced fibrosis that did not progress in disease stage (stage 2) over 3 years show similar expression of OPN (yellow arrows, insets) and HMGB1 (green arrows, insets) (A). H&E staining, OPN and HMGB1 IHC from a patient with clinically proven HCV-induced fibrosis that progressed from stage 2 to 3 in 3 years show increased expression of OPN (yellow arrows, insets) and HMGB1 (green arrows, insets) (B). Total OPN and HMGB1 morphometry analysis according to fibrosis stage. Results are expressed as fold-change of the healthy liver explants, which are assigned a value of 1; n=10/group, ***p<0.001 for stages 1, 2, 3 or 4 vs 0 (C). Immunofluorescence shows colocalisation of OPN (red) and HMGB1 (green) in a patient with chronic HCV-induced fibrosis at stage 3 (D). DAPI, 4′,6-diamidino-2-phenylindole; IOD, integrated optical density.
OPN and HMGB1 colocalise and their expression correlates in CCl4-induced liver injury in mice
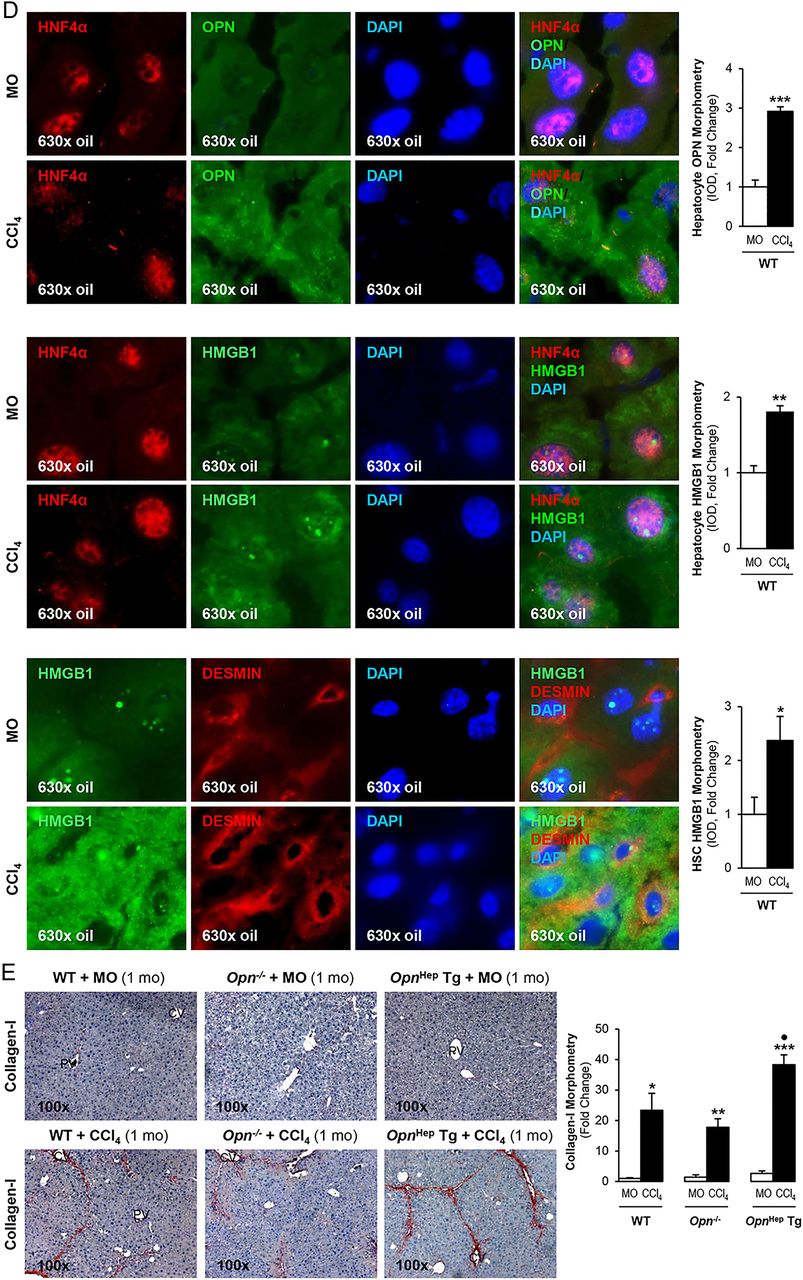
To determine whether OPN is upstream of HMGB1 and dissect if OPN induces HMGB1 therefore contributing to the fibrogenic response to liver injury, we used the CCl4 model of liver fibrosis along with genetic manipulation of Opn using WT, Opn−/− and OpnHep Tg mice as previously.3 Immunohistochemistry (IHC) analysis revealed that CCl4-injected OpnHep Tg mice showed a marked increase in hepatic OPN (figure 2A) and HMGB1 (figure 2B, top) expression compared with mineral oil (MO)-injected WT mice; however, HMGB1 immunostaining was significantly reduced in Opn−/− mice, which was also quantified by morphometry analysis and western blot (figure 2B, middle). Moreover, serum HMGB1 doubled in CCl4-injected OpnHep Tg compared with WT mice (not shown). Since HMGB1 nucleocytoplasmic shuttling is critical for driving downstream events,7 HMGB1 localisation was quantified by computer-assisted morphometry analysis. There was a significant decrease in the ratio of nuclear-to-total HMGB1 along with an increase in the ratio of cytoplasmic-to-total HMGB1 expression in CCl4-injected OpnHep Tg compared with WT and it was lower in Opn−/− mice (figure 2B, bottom). Immunofluorescence analysis demonstrated colocalisation of OPN and HMGB1 along with induced expression in CCl4-injected WT mice (figure 2C). OPN and HMGB1 expression significantly increased in hepatocytes as shown by colocalisation with HNF4α (nuclear staining)12 (figure 2D, top and middle). Similarly, HMGB1 expression was enhanced in HSCs, although to a lesser extent than in hepatocytes, as shown by colocalisation with desmin (figure 2D, bottom). Furthermore, collagen-I deposition was greater in chronic CCl4-injected OpnHep Tg compared with WT but it was much lesser in Opn−/− mice as shown by IHC and morphometry analysis (figure 2E). These in vivo results suggest the possibility that OPN could drive HMGB1 release.
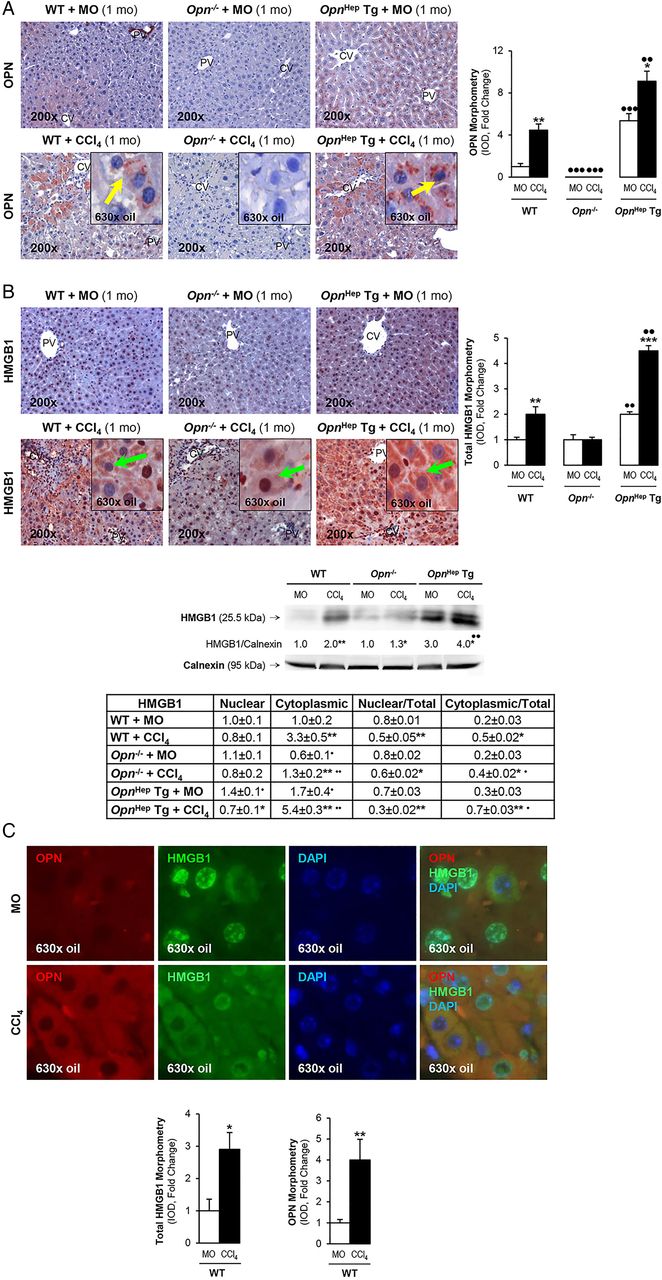
Osteopontin (OPN) and high-mobility group box-1 (HMGB1) colocalise and their expression correlates in carbon tetrachloride (CCl4)-induced liver injury in mice. Wild-type (WT), Opn−/− and OpnHep Tg mice were injected with mineral oil (MO) or CCl4 for 1 month. OPN (A) and HMGB1 (B, top) immunohistochemistry (IHC) and morphometry analysis in livers from CCl4-injected mice show increased OPN (yellow arrows, insets) along with HMGB1 (green arrows, insets) expression, which is greater in CCl4-injected OpnHep Tg than in WT and less in Opn−/− mice. Western blot analysis for HMGB1 in livers from MO-injected WT and CCl4-injected WT, Opn−/− and OpnHep Tg mice. The results from the western blot analysis are corrected by calnexin (loading control) (B, middle). Quantification of nuclear, cytoplasmic, nuclear-to-total and cytoplasmic-to-total HMGB1 expression (B, bottom). Immunofluorescence shows colocalisation of OPN and HMGB1 as well as induction in CCl4-injected WT mice (C, top), which was quantified by morphometry (C, bottom). Immunofluorescence demonstrates colocalisation of OPN and HMGB1 with HNF4α (hepatocyte marker, nuclear) along with induction by CCl4 treatment (D, top and middle). There is also colocalisation of HMGB1 with desmin (hepatic stellate cell (HSC) marker, cytoplasmic) along with induction by CCl4 treatment (D, bottom). Collagen-I IHC and morphometry assessment in livers from MO-injected or CCl4-injected WT, Opn−/− and OpnHep Tg mice (E). In all panels, the results are expressed as fold-change of the WT mice injected MO, which are assigned a value of 1 and are mean values±SEM; n=8/group. *p<0.05, **p<0.01 and ***p<0.001 for CCl4-injected mice versus MO-injected mice; ●p<0.05, ●●p<0.01 and ●●●p<0.001 for OpnHep Tg or Opn−/− versus WT mice. CV, central vein; DAPI, 4′,6-diamidino-2-phenylindole; IOD, integrated optical density; PV, portal vein.
Continued.
Hmgb1 ablation in hepatocytes partially prevents CCl4-induced liver fibrosis in mice
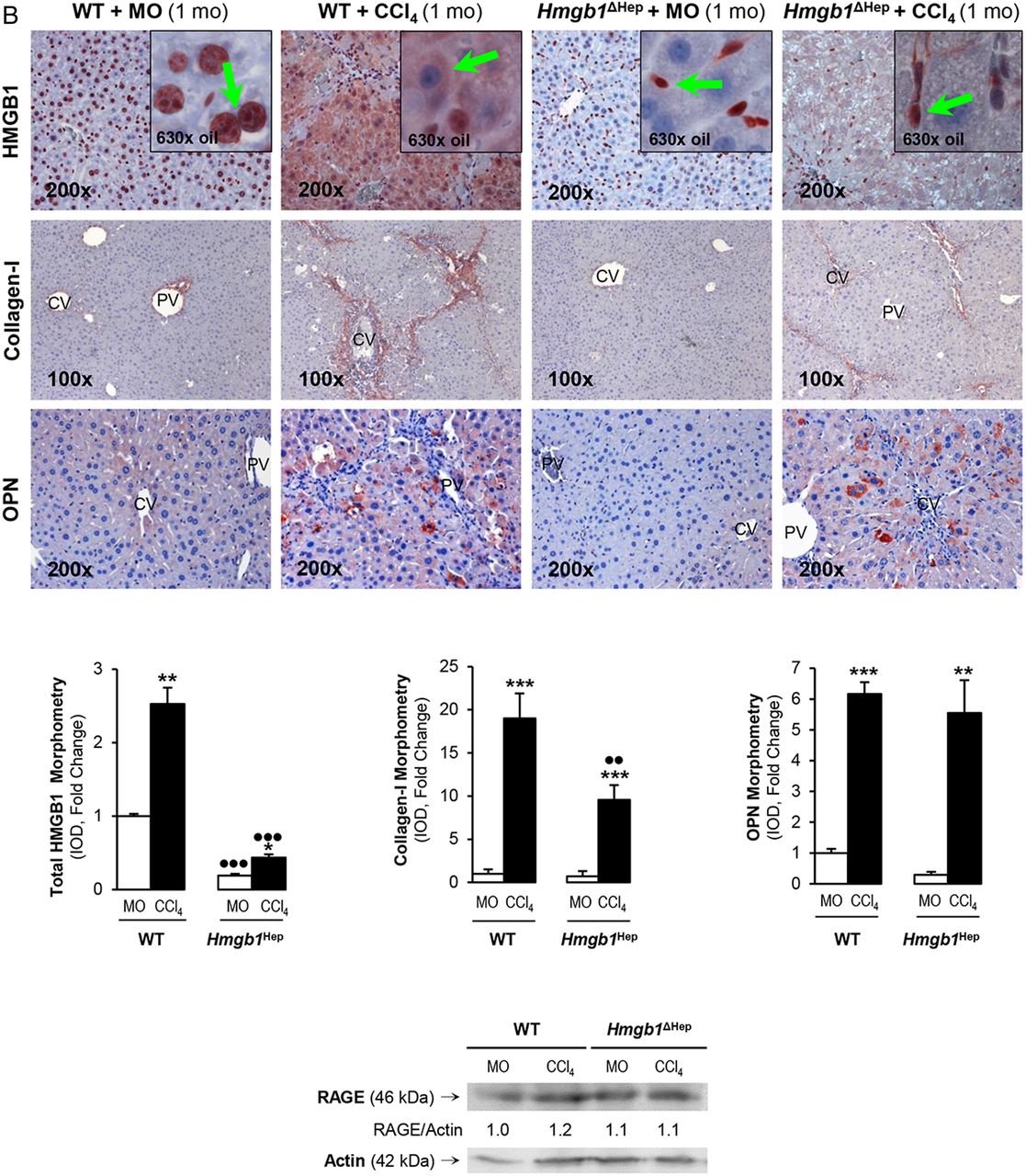
Since the human and mouse data suggested a possible role for HMGB1 of hepatocyte origin in liver fibrosis, to determine the effect of blocking hepatocyte-derived HMGB1, Hmgb1ΔHep and control littermates were chronically injected MO or CCl4. H&E staining, the pathology scores and serum alanine aminotransferase plus aspartate aminotransferase activities demonstrated less necrosis, inflammation, hepatocyte ballooning degeneration and fibrosis in CCl4-injected Hmgb1ΔHep compared with control littermates (figure 3A). Similar results were observed in a second model of liver fibrosis induced by common bile duct ligation (BDL) (see online supplementary figure S1). Hmgb1 deletion in hepatocytes was confirmed by IHC in livers from Hmgb1ΔHep and control littermates (figure 3B, top). IHC revealed less collagen-I expression in CCl4-injected Hmgb1ΔHep compared with control littermates (figure 3B, top). Hmgb1 ablation did not affect OPN expression confirming that OPN is upstream of HMGB1 (figure 3B, top). The intensity of the positive staining from these proteins was quantified by morphometry analysis (figure 3B, middle). Similarly, Hmgb1 ablation did not alter RAGE expression (figure 3B, bottom) or any other known HMGB1 receptor mRNA (not shown). Thus, Hmgb1 ablation in hepatocytes partially prevents CCl4-induced liver fibrosis in mice.
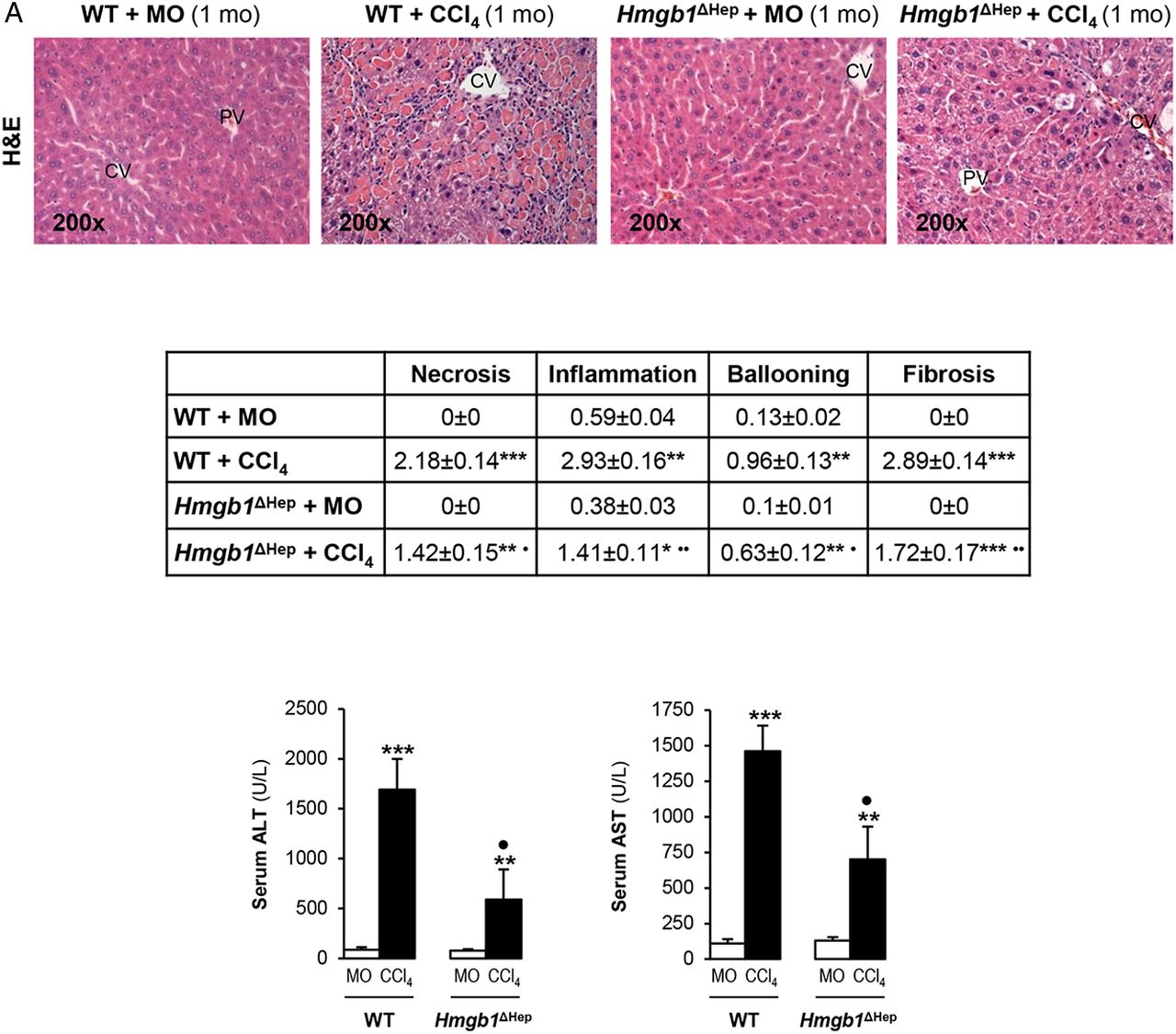
Hmgb1 ablation in hepatocytes partially prevents carbon tetrachloride (CCl4)-induced liver fibrosis in mice. Hmgb1ΔHep and control littermates were injected mineral oil (MO) or CCl4 for 1 month. H&E staining (A, top), the pathology scores (A, middle) and serum alanine aminotransferase (ALT) plus aspartate aminotransferase (AST) activities (A, bottom) show lower necrosis, inflammation, hepatocyte ballooning degeneration and fibrosis in Hmgb1ΔHep compared with control littermates. High-mobility group box-1 (HMGB1); green arrows, insets and collagen-I immunohistochemistry (IHC) and morphometry analysis show reduced HMGB1 and collagen-I deposition in livers from CCl4-injected Hmgb1ΔHep compared with control littermates. IHC and western blot analysis demonstrate similar expression of osteopontin (OPN) and receptor for advanced glycation end-products (RAGE) in these mice (B). The results are expressed as fold-change of the MO-injected control littermates, which are assigned a value of 1 and are mean values±SEM; n=8/group. *p<0.05, **p<0.01 and ***p<0.001 for CCl4-injected versus MO-injected mice; ●p<0.05, ●●p<0.01 and ●●●p<0.001 for Hmgb1ΔHep versus control littermates. CV, central vein; IOD, integrated optical density; PV, portal vein; WT, wild type.

Continued.
OPN is also upstream of HMGB1 in HSCs and they both regulate collagen-I expression in an autocrine fashion in vitro
We previously demonstrated that HSCs express OPN1 and this study revealed that HSCs also produce HMGB1. We next asked whether HSC-derived OPN could exert an autocrine effect on HMGB1 expression in HSCs and eventually on collagen-I synthesis. Freshly isolated mouse WT and Opn−/− HSCs were evaluated for OPN, HMGB1 and collagen-I expression. Opn−/− showed a 90% reduction in intracellular HMGB1 as well as in intracellular and extracellular collagen-I compared with WT HSCs (figure 4A, left). Conversely, WT HSCs infected with an adenovirus to overexpress OPN showed an increase in HMGB1 (figure 4A, right) and collagen-I3 expression compared with HSCs infected with control LacZ adenovirus. To determine if HMGB1 could condition OPN levels, WT and Hmgb1−/− mouse embryonic skin fibroblasts (MEFs) were analysed for OPN, HMGB1 and collagen-I expression. Hmgb1−/− showed similar intracellular OPN but a 90% reduction in intracellular plus extracellular collagen-I expression compared with WT MEFs (figure 4B). These data suggest that OPN is also upstream of HMGB1 in HSCs and they both regulate collagen-I expression in vitro in an autocrine fashion.
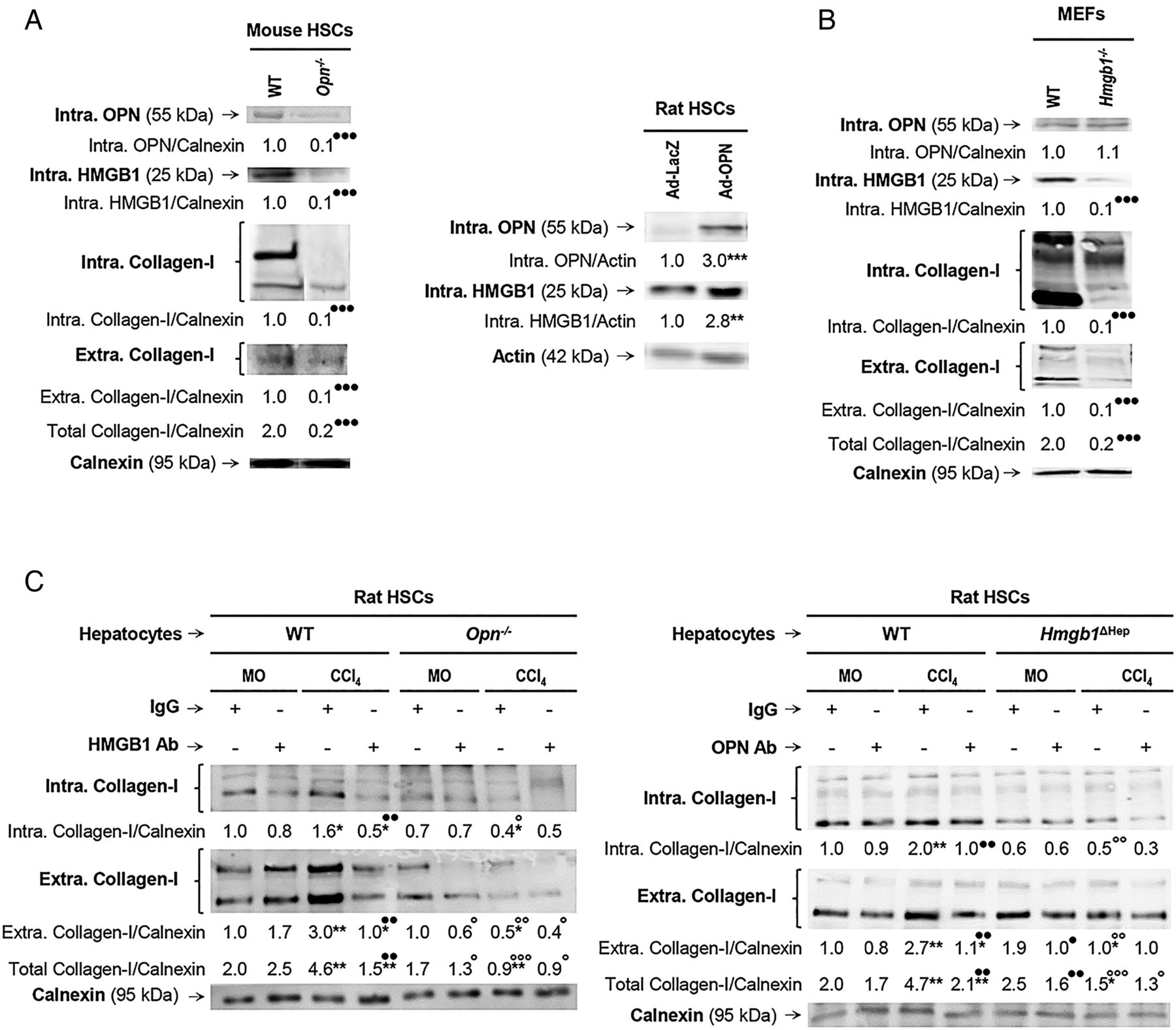
Osteopontin (OPN) is also upstream of high-mobility group box-1 (HMGB1) in hepatic stellate cells (HSCs) and they both regulate collagen-I expression in an autocrine fashion in vitro. Primary HSCs from wild-type (WT) and Opn−/− mice were cultured for 5 days. Western blot analysis of intracellular OPN, HMGB1 and collagen-I plus extracellular collagen-I expression (A, left). Rat HSCs were infected with Ad-LacZ or Ad-OPN for 48 h. Western blot analysis of intracellular OPN and HMGB1 in rat HSCs infected with Ad-LacZ or Ad-OPN (A, right). Mouse embryonic skin fibroblasts (MEFs) from WT and Hmgb1−/− mice were cultured for 1 day. Western blot analysis of intracellular OPN, HMGB1 and collagen-I plus extracellular collagen-I expression (B). Hepatocytes are a major source of OPN and HMGB1 signalling to HSCs to increase collagen-I production. Primary rat HSCs were cultured alone for 5 days and then cocultured with primary hepatocytes from mineral oil (MO)-treated or CCl4-treated Opn−/−, Hmgb1ΔHep and their matching control littermates for 1 day in the presence or absence of non-immune IgG or a neutralising antibody (Ab) to HMGB1 or OPN, respectively. Western blot analysis of intracellular and extracellular collagen-I is shown (C). In all panels, the results are corrected by the specific loading control and are expressed as fold-change of the control, which are assigned a value of 1 and are mean values±SEM; n=3/group. Experiments were performed in triplicate four times. *p<0.05, **p<0.01 and ***p<0.001 for Ad-OPN or CCl4 versus Ad-LacZ or MO; ●p<0.05, ●●p<0.01 and ●●●p<0.001 for Opn−/−, Hmgb1−/−, HMGB1 Ab or OPN Ab versus WT or IgG; °p<0.05, °°p<0.01 and °°°p<0.001 for the Opn−/− and Hmgb1ΔHep coculture versus the WT and the control littermate cocultures.
Hepatocytes are a major source of OPN and HMGB1 signalling to HSCs to increase collagen-I production
Since the human and mouse IHC suggested that hepatocytes are a major source of both OPN and HMGB1, to further define their paracrine involvement in the upregulation of collagen-I production by HSCs, cocultures of primary hepatocytes from MO-treated or CCl4-treated mice and HSCs were established. First, the cocultures were incubated in the presence of neutralising antibodies (Abs) to OPN or HMGB1; and second, the cocultures were established with hepatocytes from MO-treated or CCl4-treated Opn−/−, Hmgb1ΔHep and their respective control littermates. Western blot analysis demonstrated an increase in intracellular and extracellular collagen-I in HSCs cocultured with WT hepatocytes from CCl4-treated mice (figure 4C, left and right, lane 3 in both blots); hence, HSCs were responsive to hepatocyte-derived factors. These mediators were identified as OPN and HMGB1, since incubation with neutralising Abs to each one of them prevented the collagen-I induction in HSCs (figure 4C, left and right, lane 4 in both blots). Moreover, coculture with primary mouse hepatocytes from CCl4-treated Opn−/− or Hmgb1ΔHep mice downregulated intracellular and extracellular collagen-I in HSCs and blunted the CCl4-mediated collagen-I increase (figure 4C left and right, lane 8 in both blots). Therefore, hepatocyte-derived OPN and HMGB1 target HSCs and drive their profibrogenic behaviour.
rOPN induces HMGB1 expression and translocation in HSC and drives the increase in collagen-I production
Next, we examined if treatment with rOPN, as the upstream signal and as a surrogate of the coculture model or paracrine effects, could also replicate the increase in HMGB1 and collagen-I expression observed in HSCs. Primary rat HSCs cultured for 4 days (quiescent) or for 7 days (activated) and stimulated with rOPN increased HMGB1 and collagen-I expression (figure 5A, left). The effects on HMGB1 were at the protein level since Hmgb1 mRNA remained similar after the rOPN challenge (not shown) and inhibition of protein synthesis with cycloheximide blocked the increase in HMGB1 by rOPN in HSCs (figure 5A, right).

Recombinant osteopontin (rOPN) induces high-mobility group box-1 (HMGB1) expression and translocation in hepatic stellate cells (HSCs) and drives the increase in collagen-I production. Primary rat HSCs cultured for 4 days (quiescent) or 7 days (activated) were treated with rOPN for 6 h. Western blot analysis for intracellular HMGB1 and for intracellular plus extracellular collagen-I (A, left). Western blot analysis for HMGB1 in primary rat HSCs treated with 50 nM rOPN for 6 h in the presence or absence of 100 µM cycloheximide (A, right). Primary rat HSCs cultured for 7 days were treated with rOPN for 6 h. Western blot analysis of nuclear plus cytoplasmic HMGB1 and intracellular collagen-I (B). In (A and B), the results are expressed as fold-change of the corresponding control, which are assigned a value of 1 if signal is present and are mean values±SEM; n=3/group in experiments performed in triplicate four times. *p<0.05, **p<0.01 and ***p<0.001 for rOPN versus control; ●●●p<0.001 for cycloheximide cotreated versus rOPN. Primary mouse HSCs treated with 50 nM rOPN for 6 h. Immunofluorescence analysis for HMGB1 (green) and collagen-I (red) (C). HMGB1 structure and schematic representation of the lysine residues targeted in the HMGB1 constructs (D). Rat HSCs were transfected with a series of constructs driving HMGB1 localisation followed by 0–50 nM rOPN treatment for 6 h. The constructs were (1) pGFP, an empty vector as a negative control; (2) wild-type (WT).Hmgb1.GFP containing nuclear localisation signals 1 (NLS1) and NLS2 to overexpress HMGB1 and (3) Hmgb1.NLS1/2(8K→8A).GFP containing all eight lysines in the two NLS mutated to alanines, which cannot be acetylated and result in nuclear localisation. Immunofluorescence for collagen-I (yellow arrows) and GFP fluorescence (HMGB1 localization: white arrows point at nuclear HMGB1 and white arrowheads point at cytosolic HMGB1) were visualised by confocal microscopy (E) and quantified by morphometry assessment (F). In (F), the results are expressed as fold-change of the control WT.Hmgb1.GFP, which are assigned a value of 1 and are mean values±SEM; n=3/group in experiments performed in triplicate four times. *p<0.05, **p<0.01 and ***p<0.001 for rOPN versus control; ●●p<0.01 and ●●●p<0.001 for Hmgb1(8K→8A).GFP versus WT.Hmgb1.GFP. IOD, integrated optical density.

Continued
Since HMGB1 undergoes nucleocytoplasmic shuttling in response to a variety of stressors and PTMs,7 we next examined whether rOPN could promote HMGB1 translocation from the nucleus to the cytoplasm and eventually condition collagen-I synthesis by HSCs. Western blot analysis of nuclear and cytoplasmic proteins from HSCs stimulated with rOPN proved that the increase in cytoplasmic HMGB1 correlated with collagen-I (figure 5B). These results were also validated by immunofluorescence for HMGB1 and collagen-I in primary mouse HSCs (figure 5C).
To further confirm that following rOPN treatment, HMGB1 translocation drives collagen-I deposition, rat HSCs were transfected with constructs driving HMGB1 localisation to the nucleus or to the cytoplasm and collagen-I expression was evaluated. The constructs were (1) pGFP, an empty vector used as a negative control; (2) WT.Hmgb1.GFP, containing nuclear localisation signals (NLS) 1 and 2 to overexpress HMGB1 and allow response to stimuli that could drive the protein to the cytoplasm and (3) Hmgb1.NLS1/2(8K→8A).GFP, containing all eight lysines in the two NLS mutated to alanines that cannot be acetylated therefore resulting in HMGB1 nuclear localisation13 (see scheme on figure 5D).
In the absence of a stimulus, HSCs transfected with the WT.Hmgb1.GFP or the Hmgb1.NLS1/2(8K→8A).GFP vectors showed green fluorescence only in the nucleus corresponding to HMGB1 nuclear localisation (white arrows) compared with HSCs transfected with the pGFP vector, which showed diffuse green fluorescence corresponding to GFP only. Treatment with rOPN increased collagen-I expression in WT.Hmgb1.GFP-transfected HSCs only (red staining); still, rOPN induced greater collagen-I expression (yellow arrows) in HSCs transfected with WT.Hmgb1.GFP showing cytoplasmic HMGB1 (white arrow heads) (figure 5E, top and middle panels, figure 5F). These results suggest that rOPN induces HMGB1 cytoplasmic localisation leading to an increase in collagen-I expression. Moreover, transfection with the Hmgb1.NLS1/2(8K→8A).GFP vector revealed that forced nuclear localisation of HMGB1 decreases the HSCs response to rOPN as less collagen-I was observed compared with the WT.Hmgb1.GFP-transfected HSCs (figure 5E, middle and bottom panels, figure 5F). Overall, these experiments reveal that rOPN induces HMGB1 and collagen-I expression in quiescent and activated HSCs and promotes HMGB1 translocation from the nucleus to the cytoplasm thus driving collagen-I production by HSCs; still, the mechanism for the mobilisation of HMGB1 in HSCs under rOPN treatment remained undefined.
rOPN activates NOX and inhibits HDACs1/2 promoting HMGB1 acetylation and translocation along with collagen-I upregulation in HSCs
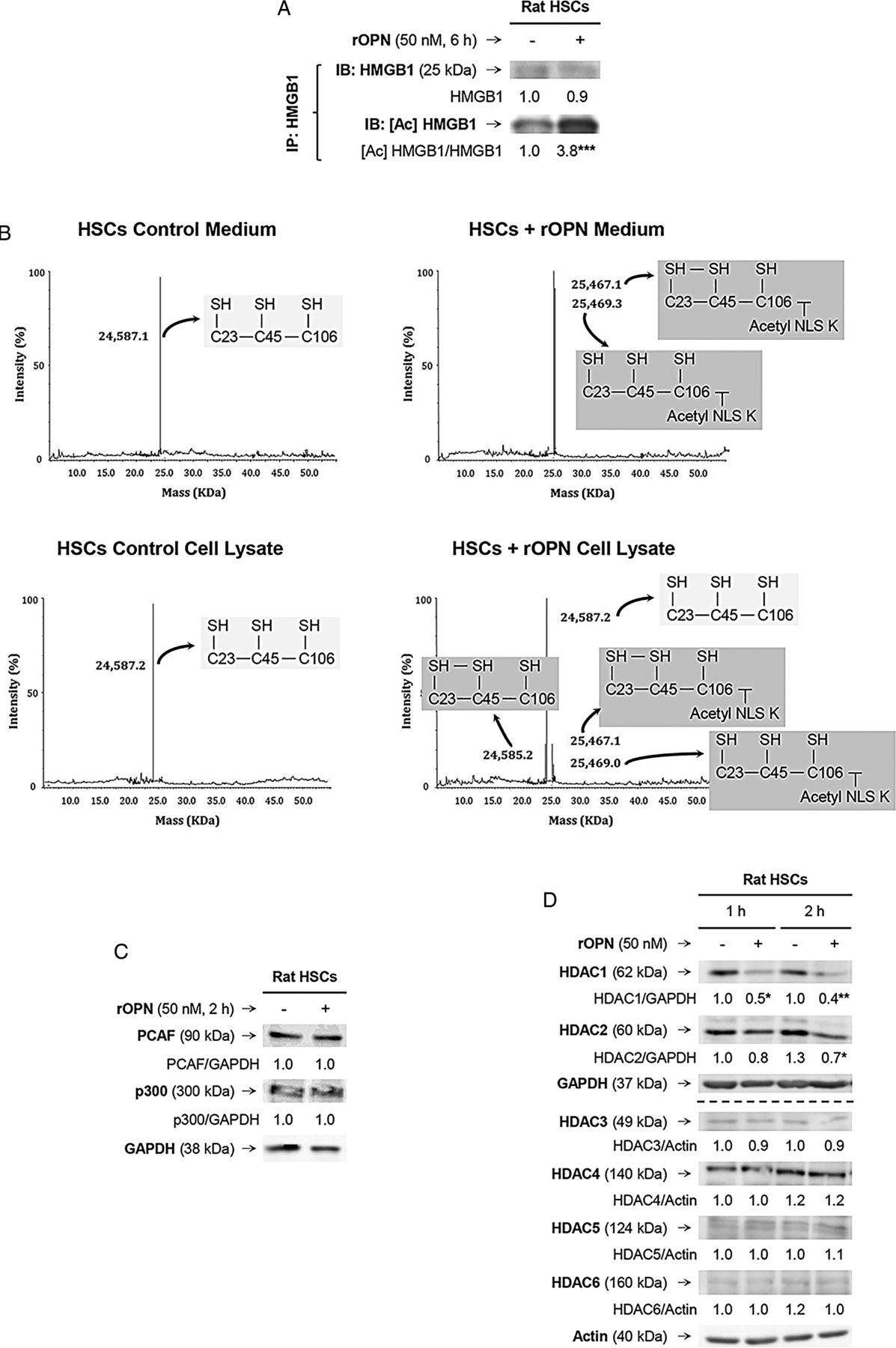
As HMGB1 nucleocytoplasmic shuttling occurs in response to stressors and/or PTMs,7 we examined if rOPN could trigger a specific PTM in HSCs that would increase HMGB1. Immunoprecipitation followed by immunoblotting revealed significant acetylation of HMGB1 in the presence of rOPN (figure 6A). Using electrospray ionisation–liquid chromatography–mass spectrometry,7 we analysed the HMGB1 residues modified under rOPN treatment in HSCs and found significant acetylation in a cluster of eight lysines (28–30, 180 and 182–185), minimal oxidation of cysteines 23 and 45 and no phosphorylation of serine 35 (figure 6B and not shown); however, how these specific PTMs occurred remained unknown.

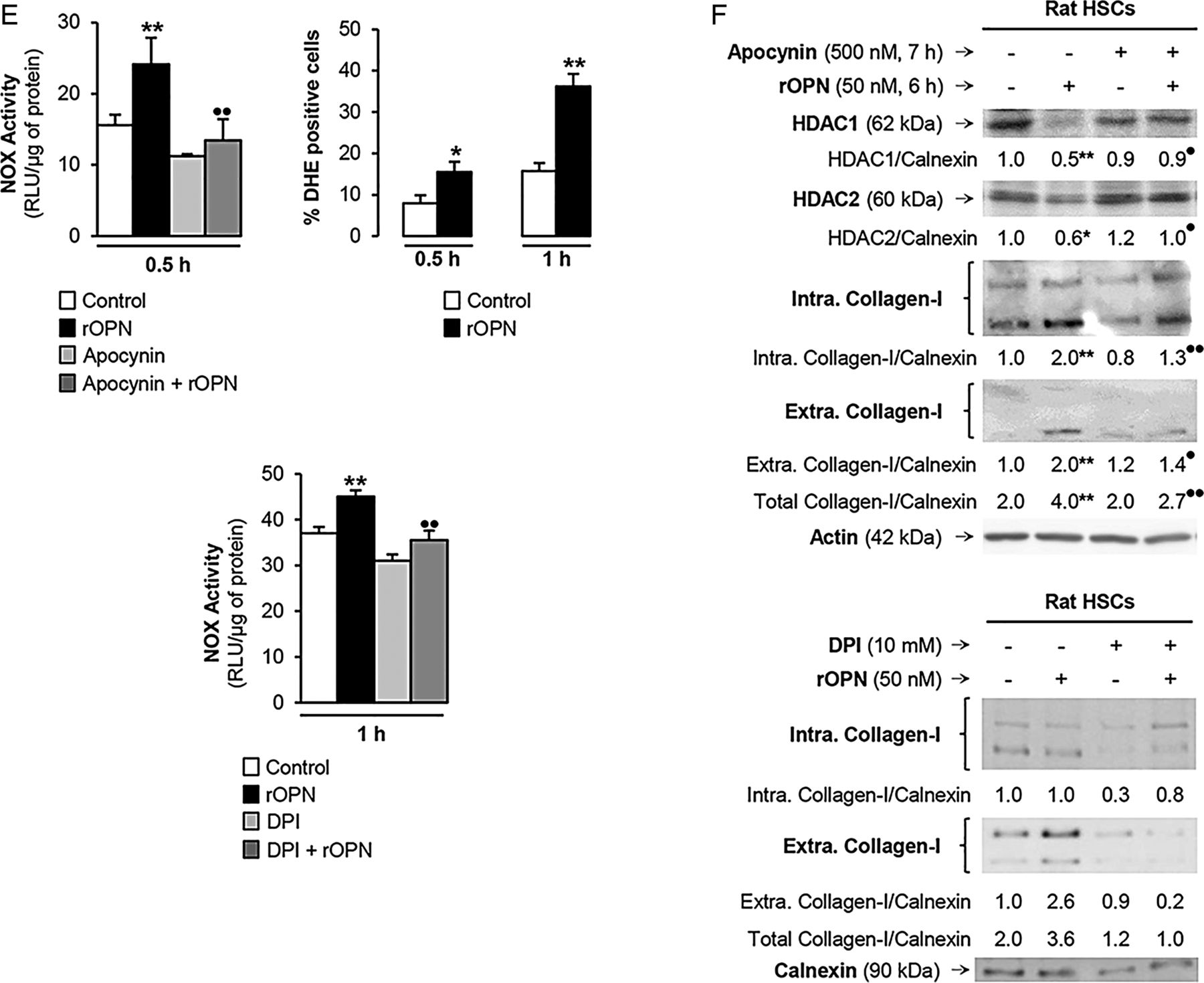
Recombinant osteopontin (rOPN) activates NADPH oxidase (NOX) and inhibits histone deacetylases (HDACs) 1/2 promoting high-mobility group box-1 (HMGB1) acetylation and translocation along with collagen-I upregulation in hepatic stellate cells (HSCs). Rat HSCs were treated with rOPN for 6 h. Immunoprecipitation of intracellular HMGB1 and immunoblotting for acetylated lysines (A). Identification of the HMGB1 isoforms in HSCs lysates and in the cell culture medium. Spectra of whole protein electrospray ionisation–liquid chromatography–mass spectrometry of the HMGB1 isoforms. A schematic representation of each isoform is on each spectra (grey boxes); n=3/group (B). Rat HSCs were treated with rOPN for 2 h. Western blot analysis for PCAF and p300 (C). Rat HSCs were treated with rOPN for 1 and 2 h. Western blot analysis for HDACs1-6 (D). NOX activity in rat HSCs treated with rOPN for 6 h alone or pretreated for 0.5 h with apocynin or diphenyleneiodonium (DPI), the two NOX inhibitors. The percentage of dihydroethidium (DHE)-positive cells was measured by flow cytometry as an indirect measurement of O2.− production (E). Rat HSCs were treated with rOPN for 6 h in the presence or absence of apocynin or DPI. Western blot analysis of HDACs1/2 along with intracellular and extracellular collagen-I (F). The results from the western blot analysis are corrected by the specific loading control and are expressed as fold-change of the controls, which are assigned a value of 1 and are mean values±SEM; n=3/group in experiments performed in triplicate four times. *p<0.05, **p<0.01 and ***p<0.001 for rOPN versus control; •p<0.05 and ••p<0.01 for cotreated versus rOPN. HDACs, histone deacetylases.

Continued
Acetylation typically occurs due to enhanced histone acetyl-transferases (HATs) and/or decreased HDACs activity; thus, we measured the activity of HATs and HDACs. The activity of HATs remained similar; however, there was a decrease in the activity of HDACs in rOPN-treated HSCs compared with control HSCs (not shown). Although PCAF (p300/CBP-associated factor) and p300 have been described to acetylate HMGB1;14 yet, western blot analysis revealed similar PCAF and p300 expression in rOPN-stimulated HSCs compared with control HSCs (figure 6C). Next, we analysed if the expression of HDACs in HSCs changed under the rOPN challenge. Western blot analysis demonstrated a decrease in HDACs1/2, whereas HDACs3–6 remained similar in rOPN-stimulated HSCs compared with control HSCs (figure 6D). Thus, rOPN acetylates HMGB1 likely by inhibiting HDACs1/2, which could contribute to HMGB1 cytoplasmic accumulation. Nevertheless, how inhibition of HDACs1/2 occurred remained undefined.
HDACs activity can be inhibited by activation of NOX with the subsequent generation of reactive oxygen species.15 To establish if rOPN could activate NOX and inhibit HDACs1/2 in HSCs, we measured NOX activity and O2.− production and found an increase in NOX activity and O2.− levels. When NOX induction was blocked by apocynin or diphenyleneiodonium (DPI) chloride, the two NOX inhibitors (figure 6E), they prevented the rOPN-mediated decrease in HDACs1/2 and the increase in collagen-I expression in HSCs (figure 6F). Overall, these results provide proof of concept that rOPN activates NOX to induce the production of O2.−, which inhibits HDACs1/2 expression, allows HMGB1 acetylation and upregulates collagen-I synthesis by HSCs.
rHMGB1 signals via the PI3K–pAkt1/2/3 pathway to upregulate collagen-I expression in HSCs
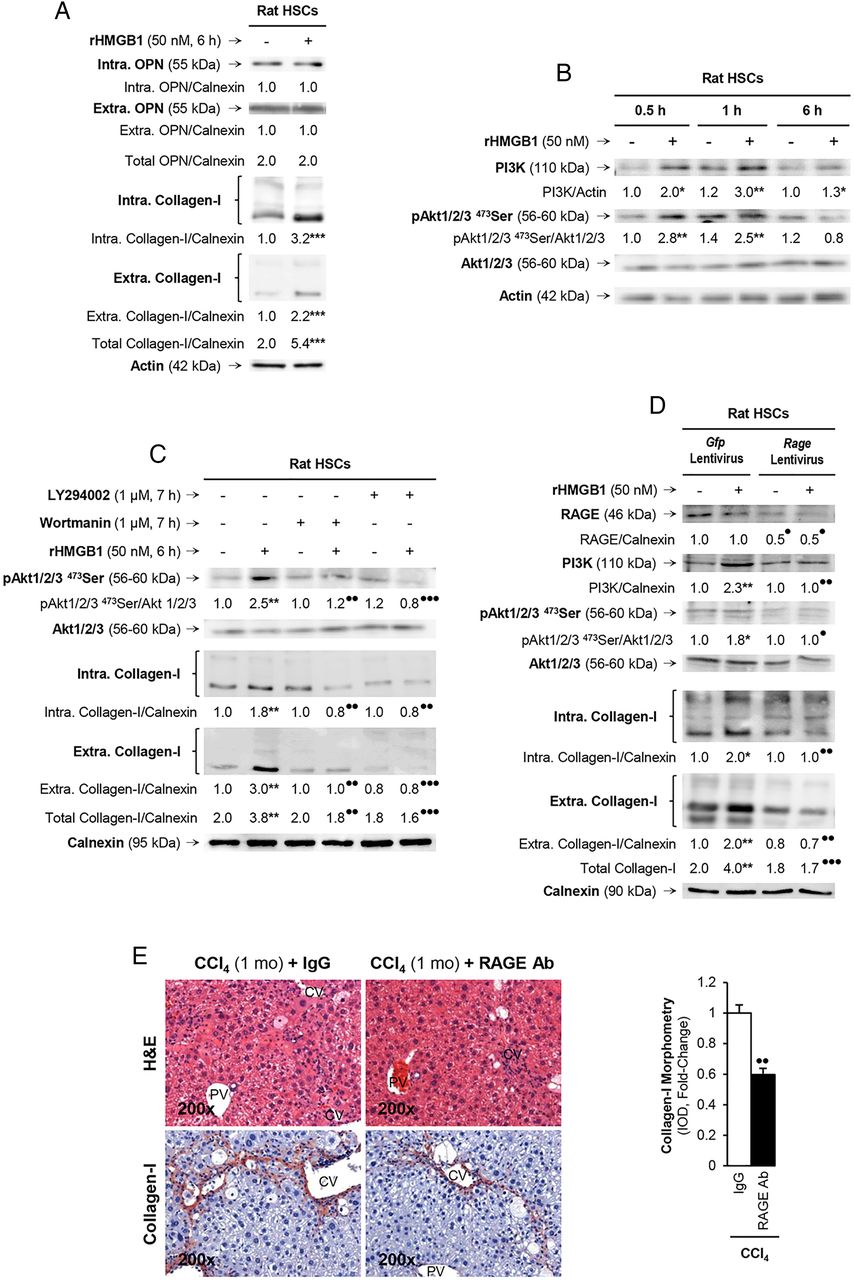
Since the human and mouse data along with the coculture studies suggested that hepatocytes produce and secrete HMGB1, which was identified as downstream of OPN, we next asked if extracellular HMGB1 per se could also signal to HSCs and increase collagen-I synthesis. To address this question, HSCs were challenged with rHMGB1, which did not alter OPN expression, yet increased intracellular and extracellular collagen-I (figure 7A). Since collagen-I production is highly dependent from protein kinase activation, to better understand how rHMGB1 upregulated collagen-I synthesis in HSCs, we analysed the expression of a series of protein kinases to determine their potential activation by rHMGB1. Following evaluation of the expression and phosphorylation state of protein kinases known to activate collagen-I synthesis (ie, ERK1/2, PI3K, Akt, p70RSK, p38, JNK), we identified that rHMGB1 time-dependently increased PI3K and induced the phosphorylation of Akt1/2/3, its downstream target (figure 7B). To confirm that they were indeed involved in the effects of rHMGB1 on collagen-I production by HSCs, cells were preincubated with the PI3K inhibitors wortmannin or LY294002 and then challenged with rHMGB1. Western blot analysis revealed a decrease in collagen-I production under rHMGB1 treatment when cells were pretreated with inhibitor, thus validating the role of PI3K and pAkt1/2/3 in the effects of rHMGB1 on collagen-I expression in HSCs (figure 7C). Therefore, extracellular HMGB1 per se also upregulates collagen-I in HSCs via PI3K–pAkt1/2/3 signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
rHMGB1 signals via receptor for advanced glycation end-products (RAGE) to upregulate collagen-I expression through the PI3K–pAkt1/2/3 pathway in hepatic stellate cells (HSCs). Rat HSCs were treated with 50 nM rHMGB1 for 6 h. Western blot analysis for intracellular plus extracellular OPN and collagen-I (A). HSCs were challenged with rHMGB1 up to 6 h and western blot analysis was performed for PI3K, pAkt1/2/3 and Akt1/2/3 (B). Western blot for pAkt1/2/3, pAkt1/2/3 and intracellular plus extracellular collagen-I in HSCs treated with rHMGB1 in the presence or absence of wortmannin or LY294002 (two PI3K inhibitors) (C). Rage ablation was performed in HSCs by transduction with shRNA lentiviral particles and isolation of stable clones expressing the shRNA via puromycin dihydrochloride selection. Cells were treated with rHMGB1 for 6 h followed by western blot analysis for RAGE, PI3K, pAkt1/2/3, Akt1/2/3 intracellular and extracellular collagen-I (D). The results from the western blot analysis are corrected by the specific loading control and are expressed as fold-change of the controls, which are assigned a value of 1 and are given as mean values±SEM; n=3/group in experiments performed in triplicate four times. *p<0.05, **p<0.01 and ***p<0.001 for rHMGB1 versus control; •p<0.05, ••p<0.01 and •••p<0.001 for cotreated or Rage ablated versus rHMGB1 or Gfp. Wild-type (WT) mice were injected CCl4 for 1 month along with non-immune IgG or RAGE neutralising Ab. H&E staining and collagen-I immunohistochemistry (IHC) and morphometry analysis showing that neutralisation of RAGE protects mice from liver fibrosis (E). The results are expressed as fold-change of the IgG group, which are assigned a value of 1 and are given as mean values±SEM; n=3/group. ••p<0.01 for RAGE Ab versus IgG. CV, central vein; PV, portal vein.
rHMGB1 signals via RAGE to upregulate collagen-I expression through the PI3K–pAkt1/2/3 pathway in HSCs
Last, since HMGB1 binds several receptors, of which RAGE16 and TLRs2/4/917–19 have been described to play a role in the setting of liver fibrosis, we evaluated whether the HMGB1 effects on collagen-I were receptor-mediated. To this end, we ablated Rage or Tlrs2/4/9 using shRNA lentiviral particles or siRNA strategies. Upon successful ablation (figure 7D and not shown), HSCs were treated with rHMGB1 and collagen-I expression was evaluated. RAGE (figure 7D) but not TLRs2/4/9 (see online supplementary figure S2) was critical for the effects of rHMGB1 on collagen-I upregulation in HSCs since western blot analysis showed upregulation of collagen-I following rHMGB1 treatment, without altering RAGE expression but not after Rage ablation (figure 7D). To identify whether the PI3K–pAkt1/2/3 signalling pathway was activated in a RAGE-dependent manner, we analysed the expression of these proteins and found no activation of PI3K and pAkt1/2/3 after rHMGB1 treatment when Rage was ablated (figure 7D). Overall, these results suggest that rHMGB1 signals via RAGE and activates the PI3K–pAkt1/2/3 pathway to upregulate collagen-I in HSCs. Last, to confirm the role of RAGE in the collagen-I upregulation in liver fibrosis in vivo, WT mice were chronically injected CCl4 in the presence of non-immune IgG or a RAGE neutralising Ab. Blockade of RAGE partially prevented liver fibrosis compared with mice injected with an irrelevant isotype-matched control monoclonal antibody (figure 7E).
Discussion
Since the incidence of liver fibrosis is rising worldwide, there is a pressing need to identify novel targets and design new therapies to prevent disease onset and/or progression. To date, most of the research in this field has focused on identifying the events involved in the pathogenesis of liver fibrosis; yet, the precise link between injured hepatocytes, HSCs and scarring remained to be identified. Thus, our goal was to dissect if OPN targets HMGB1 and how the upregulation of both proteins in hepatocytes and in HSCs contributes to the pathogenesis of liver fibrosis by regulating scarring.
This study provides compelling evidence for both co-localisation and correlation of the expression of OPN and HMGB1 with fibrosis progression in patients with clinically proven HCV-induced fibrosis.
To determine if OPN is upstream of HMGB1 and to dissect if it targets it contributing, via yet to be established mechanisms, to the fibrogenic response to liver injury, we used the CCl4 model of liver fibrosis along with genetic manipulation of Opn in mice. In addition to an increase in co-localisation of OPN and HMGB1, rather remarkable in hepatocytes, their expression correlated with the extent of liver fibrosis in mice. Importantly, fibrosis was significantly greater in chronic CCl4-injected OpnHep Tg compared with WT and it was much lesser in Opn−/− mice.3 Moreover, we observed that ageing OpnHep Tg mice showed enhanced expression of hepatic OPN and HMGB1 and developed spontaneous fibrosis3 in the absence of a profibrogenic stimulus (see online supplementary figure S3). Overall, these in vivo results suggested that OPN is upstream of HMGB1 and that the increase in OPN, likely driving HMGB1, plays a major role in the pathogenesis of liver fibrosis in mice; yet, the specific mechanism whereby HMGB1 could act as a profibrogenic paracrine and/or autocrine DAMP remained unknown.
We have previously established two mechanisms whereby OPN contributes to liver fibrosis in vivo. Since the colocalisation studies in human and mice also suggested a possible role for hepatocyte-derived HMGB1 in liver fibrosis, next we evaluated the consequences of blocking HMGB1 in hepatocytes for the development of liver fibrosis. We demonstrated that selective ablation of Hmgb1 in hepatocytes partially prevented CCl4-induced liver fibrosis in mice, which was also validated in the BDL model.
To further define the paracrine involvement of hepatocyte-derived OPN and HMGB1 in the upregulation of collagen-I production by HSCs, cocultures of hepatocytes with HSCs were established. These experiments demonstrated that hepatocytes are a major source of OPN and HMGB1 in addition to a paracrine role in increasing collagen-I production by HSCs. This was further proven by the blocking effect of neutralising Abs to OPN or HMGB1 and by hepatocyte-specific ablation of Opn or Hmgb1 in the cocultures. In both cases, collagen-I synthesis in HSCs was significantly reduced however far more when Hmgb1 was ablated. Hence, hepatocyte-derived OPN and HMGB1 target HSCs and drive their profibrogenic behaviour.
Once the role of hepatocyte-derived OPN and HMGB1 was established, we then asked if intracellular OPN and HMGB1 in HSCs could also play an autocrine role driving scarring. Since we previously demonstrated that HSCs express OPN1 and the present study revealed induction of HMGB1 under OPN treatment, we next ablated both proteins. Analysis of the autocrine effects resulting from (1) regulating Opn expression for HMGB1 production and (2) modulating Hmgb1 expression for collagen-I synthesis suggested that intracellular OPN is also upstream of HMGB1 in HSCs and regulates collagen-I expression.
Next, to dissect the molecular mechanism for the paracrine effects of OPN and HMGB1 for the HSCs profibrogenic behaviour, we treated HSCs with rOPN or rHMGB1. We previously showed that rOPN upregulates collagen-I production in HSCs by binding αvβ3 integrin and activating the PI3K–pAkt1/2/3–NFκB signalling pathway in addition to driving ductular reaction and increasing TGF-β production in biliary epithelial cells.1 ,3 Yet, in view of our data, we also considered that a downstream target of OPN, such as HMGB1, could participate in increasing collagen-I deposition thereby contributing to the pathophysiology of liver fibrosis by regulating scarring.
Extracellular OPN induced HMGB1 and collagen-I expression in quiescent and activated HSCs and also promoted HMGB1 translocation from the nucleus to the cytoplasm driving collagen-I production by HSCs as demonstrated with the transfection experiment using the constructs conditioning HMGB1 subcellular localisation in response to stimuli. The construct preventing HMGB1 acetylation in HSCs under rOPN treatment suggested a key role of a PTM for collagen-I production in this setting. Indeed, analysis of the potential PTMs revealed extensive acetylation of HMGB1 in the cluster of eight lysines (28–30, 180 and 182–185) under OPN treatment in HSCs, which could explain the cytoplasmic increase in HMGB1 since acetylation prevents HMGB1 nuclear re-entry. To understand how this modification occurred, next we measured HATs and HDACs activity along with the expression of each of these proteins. OPN lowered HDACs1/2 activity and acetylated HMGB1 thus contributing to HMGB1 cytoplasmic localisation and increase. Furthermore, OPN activated NOX and stimulated O2.− generation, which ultimately inhibited HDACs1/2 expression allowing HMGB1 acetylation and upregulating collagen-I expression in HSCs. Thus, extracellular OPN can paracrinely promote the autocrine effects of HSC-derived HMGB1 in driving collagen-I deposition.
Finally, the human and mouse data along with the coculture studies suggested that hepatocytes were a major source of HMGB1, which was identified as downstream of OPN. Of note, while hepatocytes secrete a considerable amount of HMGB1,7 HSCs secrete it but to a much lesser degree. Next, we demonstrated that extracellular HMGB1 per se also signals to HSCs via RAGE signalling and activation of the PI3K–pAkt1/2/3 pathway to upregulate collagen-I in HSCs. Therefore, RAGE plays a major role in the HMGB1-mediated effects on collagen-I synthesis in the setting of liver fibrosis.
While the involvement of HMGB1 in other liver diseases has been reported,20–25 the role of HMGB1 in liver fibrosis has not been fully evaluated to date. Hence, we have identified that during the onset of liver fibrosis, the increase in OPN, and as a consequence in HMGB1, drives scarring. As proposed in this study (see online supplementary figure S4), the significant upregulation of this alarmin has critical paracrine and autocrine effects on HSCs and therefore it could be targeted to prevent or slow down the fibrogenic process.
Overall, this study challenged our current view of the mechanisms driving liver disease by reinforcing the role of hepatic OPN and HMGB1, a sterile DAMP, in the onset of liver fibrosis and tested the novel hypothesis that during fibrogenesis the increase in OPN, and as a consequence in HMGB1, acts as a paracrine and autocrine signal to trigger scarring. Development of efficient therapies for liver fibrosis must target the molecular mechanisms driving early fibrosis related to hepatocellular injury to allow rapid intervention. Importantly, HMGB1 has the advantage that, unlike other proteins, provides a wider time frame for clinical intervention due to its longer half-life.26 Thus, it is an attractive target to prevent fibrosis progression. OPN and HMGB1 may also participate in other events taking place in liver fibrosis such as necrosis, inflammation and increased gut permeability. Finally, due to the extent of their production in hepatocytes, it is likely that the overall contribution of hepatocyte-derived OPN and HMGB1 to scarring is far more relevant than that of HSCs. It still remains an open question if their production in other liver cells or in other organs is also relevant or perhaps synergistic for liver fibrosis and if specific PTMs of HMGB1 could also condition the noxious effects of HMGB1 in the liver environment.
Acknowledgments
The authors are very grateful to Drs David T Denhardt (Rutgers University, New Brunswick, New Jersey, USA) for his generous gift of the 2A1 and 2C5 Abs, Marco E. Bianchi (San Raffaele University, Milan, Italy) for providing the Hmgb1 mutants, Timothy R. Billiar (University of Pittsburgh, Pittsburgh, Pennsylvania, USA) for donating the Hmgb1fl/fl and Satoshi Mochida (Saitama Medical University, Saitama, Japan) for providing the OpnHep Tg mice. We are also very thankful to all past and current members from the Nieto Laboratory for their helpful comments and suggestions throughout the course of this project. Confocal microscopy was performed at the Microscopy Shared Resource Facility at the Icahn School of Medicine at Mount Sinai.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
EA and XG contributed equally.
Contributors EA and XG performed in vitro and in vivo experiments and edited the manuscript. T-ML, FM, AL, YL, NK and RU performed some experiments and edited the manuscript. NT provided the human samples and edited the manuscript. DJA performed the analysis of the HMGB1 isoforms and edited the manuscript. NN directed the project, drafted and edited the manuscript and obtained funding.
Funding Short-term Bancaja Fellowship and Postdoctoral Fellowship from the Asociación Española para el Estudio del Hígado, Spain (EA). Postdoctoral fellowships from the Basque Government (Spain) (AL), Keio University School of Medicine (Japan) (NK) and the Government of Navarre, Spain (RU). Wellcome Trust research fellowship (DJA) and support from the Medical Research Council Centre for Drug Safety Science (DJA and NN). UK regenerative medicine platform (UKRMP) (DJA and NN). US Public Health Service Grants R01 DK069286, R56 DK069286 and R56 DK069286-06S1 from the National Institute of Diabetes and Digestive and Kidney Diseases (NN). US Public Health Service Grants P20 AA017067, P20 AA017067-01S1, P20 AA017067-03S1 and U01 AA021887 from the National Institute on Alcohol Abuse and Alcoholism (NN).
Competing interests None declared.
Patient consent Obtained.
Ethics approval IRB.
Provenance and peer review Not commissioned; externally peer reviewed.