Article Text

Abstract
Objective Efforts to manage non-alcoholic fatty liver disease (NAFLD) are limited by the incomplete understanding of the pathogenic mechanisms and the absence of accurate non-invasive biomarkers. The aim of this study was to identify novel NAFLD therapeutic targets andbiomarkers by conducting liver transcriptomic analysis in patients stratified by the presence of the PNPLA3 I148M genetic risk variant.
Design We sequenced the hepatic transcriptome of 125 obese individuals. ‘Severe NAFLD’ was defined as the presence of steatohepatitis, NAFLD activity score ≥4 or fibrosis stage ≥2. The circulating levels of the most upregulated transcript, interleukin-32 (IL32), were measured by ELISA.
Results Carriage of the PNPLA3 I148M variant correlated with the two major components of hepatic transcriptome variability and broadly influenced gene expression. In patients with severe NAFLD, there was an upregulation of inflammatory and lipid metabolism pathways. IL32 was the most robustly upregulated gene in the severe NAFLD group (adjusted p=1×10−6), and its expression correlated with steatosis severity, both in I148M variant carriers and non-carriers. In 77 severely obese, and in a replication cohort of 160 individuals evaluated at the hepatology service, circulating IL32 levels were associated with both NAFLD and severe NAFLD independently of aminotransferases (p<0.01 for both). A linear combination of IL32-ALT-AST showed a better performance than ALT-AST alone in NAFLD diagnosis (area under the curve=0.92 vs 0.81, p=5×10−5).
Conclusion Hepatic IL32 is overexpressed in NAFLD, correlates with hepatic fat and liver damage, and is detectable in the circulation, where it is independently associated with the presence and severity of NAFLD.
- nonalcoholic steatohepatitis
- genetics
- rna expression
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic fatty liver disease (NAFLD) is the leading cause of advanced liver diseases in the Western countries.
Incomplete knowledge of NAFLD pathogenic mechanisms results in a lack of effective strategies for early diagnosis and treatment.
What are the new findings?
The PNPLA3 I48M variant was a major modifier of the liver transcriptome.
Interleukin-32 (IL32) was the most strongly upregulated transcript in severe NAFLD.
IL32 circulating levels correlate with hepatic expression and are increased in patients with NAFLD.
How might it impact on clinical practice in the foreseeable future?
IL32 is a candidate for non-invasive assessment of NAFLD presence/severity and for targeted therapy.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is rapidly becoming a leading cause of advanced liver disease worldwide.1 NAFLD is highly prevalent in the population and is associated with dysmetabolism and qualitative alterations of the diet and microbiota.2 3 However, only a subset of affected individuals progress to advanced liver fibrosis and/or hepatocellular carcinoma. The factors driving liver damage progression are only partially understood, but genetic predisposition plays an important role.4
A common genetic variant, the rs738409 C>G single nucleotide polymorphism encoding for the p.Ile148Met (I148M) aminoacidic substitution in the patatin-like phospholipase domain-containing 3 (PNPLA3), is the major genetic determinant of hepatic fat content and progressive NAFLD.5–7 PNPLA3 is an enzyme expressed in hepatocytes and hepatic stellate cells and it is upregulated by insulin signalling.8 9 The 148M mutant protein predisposes to NAFLD by interfering in the remodelling of triglycerides and phospholipids at the surface of lipid droplets,10–12 resulting in the accumulation of neutral fat due to reduced turnover.13 Furthermore, the I148M variant impairs retinol disposal following activation of hepatic stellate cells, resulting in a proinflammatory and fibrogenic phenotype.14–17 However, to what extent the mechanism of liver disease progression in carriers of the I148M variant differs from those of patients not carrying the mutation is presently unknown. As no effective therapy and accurate non-invasive biomarkers are yet available for progressive NAFLD, this question may have an immediate clinical implication. Indeed, stratification by the I148M variant may identify a distinct subset of patients amenable to specific screening and treatment strategies, enabling a framework of precision medicine.
The aim of this study was therefore to identify new potential therapeutic targets and biomarkers for progressive NAFLD by highlighting and validating the most upregulated hepatic transcripts in patients with fibrosing NAFLD (either NAS≥4, NASH or significant liver fibrosis stage F≥2, hereafter defined as ‘severe NAFLD’), stratified by the presence of the PNPLA3 I148M variant. The study workflow is presented in online supplementary figure S1.
Supplemental material
Patients and methods
Patients
The study was conducted in 125 obese individuals (‘Transcriptomic cohort’) who underwent percutaneous liver biopsy performed during bariatric surgery, and for whom sufficient material for extraction of high-quality RNA was available. Individuals with at-risk alcohol intake (>30/20 g/day in M/F), viral and autoimmune hepatitis or other causes of liver disease were excluded. Interleukin-32 (IL32) plasma levels were retrospectively assessed in 71 obese patients, who underwent percutaneous liver biopsy performed during bariatric surgery (‘Bariatric surgery cohort’). The intersection of the Transcriptomic and the Bariatric surgery cohorts was represented by 16 patients matching both the inclusion criteria. The association between circulating IL32 and liver damage was replicated in an independent cohort of 160 individuals made up of patients with histological NAFLD from the general medicine (n=148), and of healthy blood donors without NAFLD, ruled out by non-invasive assessment (n=12), who presented on a single day and were evaluated at the Fondazione IRCCS Ca’ Granda (Hepatology service cohort). We further examined 44 individuals with dysmetabolism attending a metabolic clinic at the University of Catanzaro (‘Metabolic unit cohort”). Participants were selected using the same criteria described for the Transcriptomic cohort. Since the aim was to validate the association of circulating IL32 with the presence and severity of NAFLD, individuals with autoimmune or inflammatory disorders were also excluded (n=5 in the Bariatric surgery cohort and n=7 in the Hepatology service cohort). For all study cohorts, liver biopsy was performed by needle gauge, and serum/plasma samples were collected at the time of histological or non-invasive assessment of liver damage.
The clinical features of the Transcriptomic, Bariatric surgery and Hepatology service cohorts are presented in table 1, while the clinical features of Transcriptomic cohort stratified by the presence of PNPLA3 I148M and of the Metabolic unit cohort are presented in online supplementary table S1.
Clinical and genetic features of patients included in the study cohorts
Histological and liver damage evaluation
Steatosis was graded based on the percentage of affected hepatocytes as 0: 0%–5%, 1: 6%–33%, 2: 34%–66% and 3: 67%–100%. Disease activity was assessed according to the NAFLD Activity Score (NAS) with systematic evaluation of hepatocellular ballooning and necroinflammation; fibrosis was staged according to the recommendations of the NAFLD clinical research network.18 Liver biopsies scoring was performed by an expert pathologist unaware of patients’ status and genotype.19 20 NASH was defined as the concomitant presence of steatosis, lobular inflammation and ballooning. We arbitrarily defined ‘Severe NAFLD’ as the presence of NASH, and/or NAS≥4, and/or fibrosis stage F2 or higher.
In Metabolic unit cohort, liver steatosis was estimated by assessing the controlled attenuation parameter (CAP). Median CAP value (244 db/m2) was used as cut-off to stratify patients as ‘low CAP’ (CAP<median) or ‘high CAP’ (CAP≥median).
Genotyping
The rs58542926 C>T (E167K, TM6SF2) and rs738409 C>G (I148M, PNPLA3) genetic variants were assessed in duplicate by TaqMan 5'‐nuclease assays (Life Technologies, Carlsbad, California, USA).
Transcriptomic and bioinformatic analysis
The detailed protocol for the transcriptomic and bioinformatic analysis is reported in the Supplementary Methods. Briefly, total RNA was isolated using RNeasy mini-kit (Qiagen, Hulsterweg, Germany). RNA was sequenced in paired-end mode (read length 150nt) using the Illumina HiSeq 4000 (Novogene, Hong Kong, China). Reads were mapped by a custom pipeline,21 encompassing reads quality check (FastQC software, Babraham Bioinformatics, Cambridge, UK), low-quality reads trimming and mapping on GRCh37 reference genome by STAR mapper.22 Reads count (ENSEMBL human transcript reference assembly v75) was performed using RSEM package.23 Counts were normalised using DESeq2 package.24 IL32 transcripts were grouped and reconducted to isoforms described in the literature.25
For principal components analysis (PCA), gene-level expression data were normalised under the null model through DESeq2 standard pipeline, and variance stabilising transformation function was applied.
To identify differentially expressed pathways, pre-ranked gene set enrichment analysis (GSEA) was performed on differentially expressed or significantly correlated genes.26 27 IL32 promoter in HepG2 cells was analysed, exploiting chromatin state segmentation and ChIP-Seq data from the ENCODE project (V.3).28
Cell isolation, culture and gene expression evaluation
The detailed protocol for isolation and culture of primary human cells is presented in the Supplementary Methods. RNA was reverse transcribed using SuperScript VILO cDNA Synthesis Kit, and gene expression measured by quantitative real-time PCR (RT-qPCR) exploiting the SYBR green chemistry (Fast SYBR Green Master Mix) on ABI 7500 fast thermocycler (all from ThermoFisher, Waltham, Massachusetts, USA). All reactions were performed in triplicate. Gene expression levels were normalised using the 2−ΔCt method; β-actin was used as housekeeping gene. For human IL32 expression quantification, 5′-AATCAGGACGTGGACAGGTG-3′ forward and 5′-TCACAAAAGCTCTCCCCAGG-3′ reverse primers were employed.
IL32 and aminotransferase measurement
Circulating levels of IL32 were quantified using Human IL32 DuoSet ELISA kit (R&D Systems, Minneapolis, USA) on fasting plasma (Bariatric surgery) and serum (Hepatology service) samples. The assay is designed to detect IL32α, IL32β and IL32γ with a detection range of 78.5–5000 pg/mL. Samples were measured in duplicate. The intra-assay and inter-assay coefficients of variation were 3.2%±1.2% and 11.3%±4.6%, respectively. Fasting serum level of alanine and aspartate aminotransferases (ALT and AST) were assessed by the IFCC 37°C method at the Fondazione IRCCS Ca’ Granda core laboratory. FIB4 score was calculated applying the formula:  .
.
Statistical analysis
For descriptive statistics, continuous variables were shown as mean and SD. Highly skewed biological variables were reported as median and IQR and were log-transformed before analyses. Categorical variables were tested by χ2 test and are presented as number and proportion. In all transcriptome-wide unsupervised analysis and GSEA, p values were corrected for multiplicity by Benjamini-Hochberg false discovery rate method, and adjusted p values<0.1 were considered statistically significant. Differences between groups were evaluated by two-tailed Student’s t-test or analysis of variance followed by post hoc Tukey’s honest significance test, when appropriate. Diagnostic accuracy was reported according to the Standards for Reporting of Diagnostic Accuracy (STARD) guidelines.29 Differences in diagnostic performance were tested by paired or unpaired Delong test, when appropriate. Multivariate regressions were performed by fitting data into generalised linear models. In particular, logistic regression models were fit to examine binary traits (presence of severe NAFLD or NAFLD), and ordinal regression models were fit for ordinal traits (severity of steatosis). Linear combinations of IL32-ALT-AST were generated by predicting responses of generalised linear models. P values<0.05 (two-tailed) were considered statistically significant. Statistical analyses were carried out using the R software V.3.5.0 (http://www.R-project.org/).
Results
PNPLA3 I148M is a major determinant of hepatic transcripts variability in obese patients
To identify the major determinants of hepatic transcriptome variability, PCA was performed (figure 1A). The main component of transcriptome variability (PC1) was independently associated with histological ballooning (p<0.01), lobular inflammation (p<0.05) and female sex (p<0.01). PC1 was also strongly associated with carriage of the PNPLA3 I148M variant (p<0.001). PC2 was similarly associated with the I148M variant (p<0.001), as well as with female sex (p<0.05) and steatosis. Therefore, carriage of the PNPLA3 I148M variant was strongly linked with the main components of hepatic transcriptome variability. The correlation of PC1/2 with GSEA pathways is presented in the Supplementary Results.

Principal components correlation and differential pathway expression. (A) Correlation matrix of the main five principal components (PCs) of hepatic transcriptome variability with clinical features as assessed by multivariate regression. (B, C, D, E) Pathways enriched in genes differentially (multiplicity adjusted p<0.1, negative binomial generalised linear model) expressed in: PNPLA3 I148M carriers versus non-carriers (B), severe NAFLD versus NAFLD and healthy individuals (C), severe versus non-severe in PNPLA3 I148M non-carriers (D), severe versus non-severe NAFLD among PNPLA3 I148M carriers (E). BMI, body max index; EMT, epithelial-to-mesenchymal transition; IFN, interferon; NAFLD, non-alcoholic fatty liver disease; NES, normalised enrichment score; PC, principal component; PNPLA3, patatin-like phospholipase domain-containing protein 3; TM6SF2, transmembrane 6 superfamily 2 human gene.
Impact of the PNPLA3 I148M variant on gene expression
To dissect the impact of the PNPLA3 I148M variant on the liver transcriptome, we performed differential expression analysis, which was further adjusted for disease stage. A set of 642 genes was differentially expressed in the presence of the PNPLA3 I148M variant (online supplementary dataset A, online supplementary figure S3A). Of these, 161 genes were downregulated, while 481 were upregulated. The most significant differentially expressed genes in I148M variant carriers are shown in table 2 (upper panel). In PNPLA3 I148M variant carriers, but not in non-carriers, GSEA revealed an overexpression of pathways related to inflammation, carcinogenesis and downregulation of metabolic pathways (figure 1B). Collectively, these results are in line with a reduction of lipid metabolism/turnover along with the promotion of liver inflammation and cell proliferation in the presence of the PNPLA3 I148M variant.
Supplemental material
Most differentially expressed genes in the liver of 125 patients with severe obesity (Transcriptomic cohort) according to the presence of PNPLA3 I148M variant and of severe NAFLD
Pathways and specific transcripts associated with severe NAFLD
We next examined genes and pathways differentially expressed in severe versus non-severe NAFLD and normal liver. We found 320 genes differentially expressed in severe NAFLD (online supplementary dataset B, online supplementary figure S3B). Of these, 16 genes were also deregulated by carriage of the PNPLA3 I148M variant (online supplementary figure S3C). As expected, we found a higher expression of genes involved in hepatic fibrogenesis, such as keratin 8 (KRT8), collagen type 1 α1 chain (COL1A1) (table 2, second panel). IL32 was the most robustly upregulated gene in severe NAFLD (adjusted p=1.2×10−6), together with suppressor of cytokine signalling 1 (SOCS1) and aldose reductase (AKR1B10). Consistently, GSEA revealed the overexpression of pathways related to inflammation (eg, tumour necrosis factor α (TNFα) signalling, IFNγ response), cell death (eg, apoptosis, p53 pathway), metabolism (eg, MTORC1 signalling), hypoxia and epithelial-to-mesenchymal transition (EMT) (figure 1C).
Taken together, these results outline inflammation and fibrogenesis as the hallmark of NAFLD progression towards NASH/fibrosis and identify IL32 as a NAFLD-related cytokine and candidate regulator of this biological response.
Pathways and specific transcripts associated with severe NAFLD in carriers of the I148M variant
We next examined the differences in the transcriptome associated with severe NAFLD in patients stratified by the PNPLA3 variant. We found differential expression of 193 and 148 genes (online supplementary datasets C and D, online supplementary figure S3D and S3E) in severe NAFLD in PNPLA3 I148M carriers and non-carriers, respectively. Distance analysis revealed a more marked difference in hepatic gene expression due to severe NAFLD condition as compared with normal liver in I148M carriers as compared with non-carriers (online supplementary figure S4, p=0.002). Only four genes were deregulated in severe NAFLD in both carriers and non-carriers (online supplementary table S2). However, IL32 was overexpressed in both carriers and non-carriers with a similar fold induction (log fold change 1.45 vs 1.32, adjusted p=0.006 vs 0.03, in carriers vs non-carriers, respectively).
Among the most significant genes altered in severe NAFLD in non-carriers (table 2, third panel), we found increased levels of IL8, while GSEA highlighted overexpression of pathways linked to inflammation, epithelial-to-mesenchymal transition, fibrosis and hypoxia (figure 1D).
In I148M carriers (table 2, bottom panel), in severe NAFLD we found upregulation of several genes related to inflammation and fibrogenesis, including KRT8/KRT18 and SOCS1. GSEA revealed the enrichment of pathways related to inflammation (IL6-JAK-STAT3 signalling), KRAS signalling, hypoxia and apoptosis (figure 1E). Overall, this analysis suggested that the PNPLA3 I148M variant may influence the mechanisms associated with liver disease progression in NAFLD, while confirmed IL32 upregulation as a robust marker of severe disease.
IL32 is associated with inflammatory and metabolic response in severe NAFLD
We next evaluated IL32 isoform expression profile. Transcripts coding for IL32β were the most represented (60% of gene products, p<0.001 vs all the others, figure 2A), but there was an appreciable contribution of non-coding transcripts (18% of gene products) and of the IL32ε (15% of gene products). Expression of IL32γ was also detectable (4% of gene products). We could not identify any isoform switch associated with severe NAFLD (figure 2A). However, only IL32β (p<0.001), IL32ε (p<0.001) and non-coding transcripts (p<0.01) were significantly overexpressed in severe versus non-severe NAFLD patients (figure 2B).
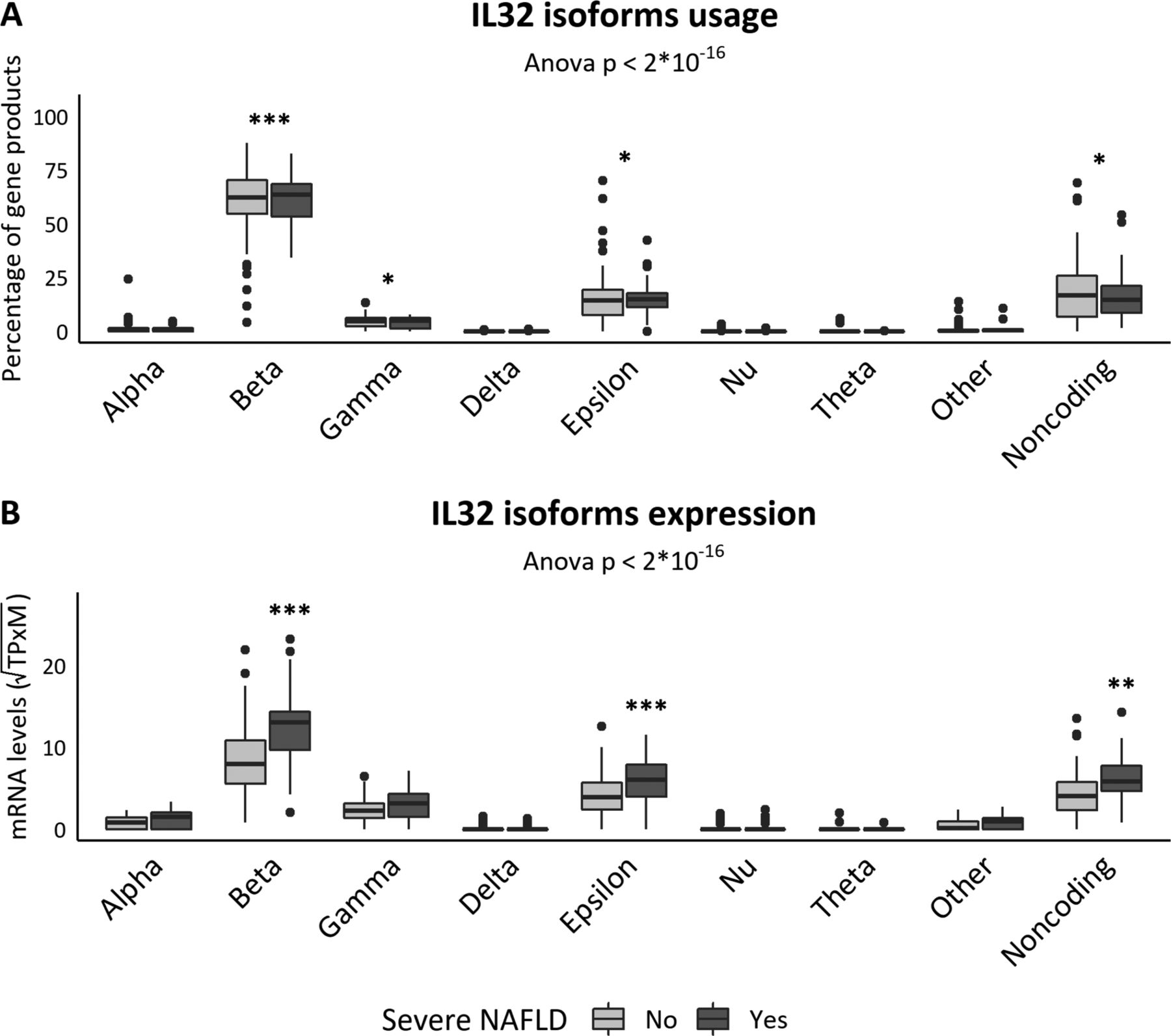
IL32 isoform expression. (A) Percentage of the IL32 gene products. ***p<0.001 versus all the other isoforms, post hoc Tukey’s test. (B) Isoform absolute expression value. ***p<0.001 versus NAFLD and healthy patients, post hoc Tukey’s test. ANOVA, analysis of variance; IL32, interleukin-32; NAFLD, non-alcoholic fatty liver disease; PNPLA3, patatin-like phospholipase domain-containing protein 3; TPM, transcripts per million.
To investigate the transcriptional pattern related to IL32 expression, coregulation analysis was performed. We found a correlation of hepatic IL32 with genes involved in liver damage, inflammation and cell proliferation (online supplementary table S3, upper panel). In particular, there was a direct correlation between IL32 mRNA levels and several chemokines and growth factors (online supplementary table S3, lower panel). Conversely, CCL14 showed a strong inverse relationship with IL32. Consistently, GSEA highlighted an enrichment in pathways related to inflammation, liver damage and metabolism (figure 3A).

IL32 expression correlations. (A) Pathway enriched in IL32 correlated genes (adjusted p<0.1). (B) IL32 expression stratified by steatosis grade; statistical analysis was performed by ordinal logistic regression. (C, D, E) IL32 expression stratified by the presence of lobular inflammation (C), ballooning (D), fibrosis (E). **p<0.01; ***p<0.001, Student’s t-test. EMT, epithelial-to-mesenchymal transition; IFN: interferon; IL32, interleukin-32; NAFLD, non-alcoholic fatty liver disease; NES, normalised enrichment score; PNPLA3, patatin-like phospholipase domain-containing protein 3; ROS, reactive oxygen species.
Hepatic IL32 was overexpressed in patients with NASH as compared with those without NASH (n=12 vs 113, respectively; log2(fold change)=1.2±0.35, adjusted p=0.048; as shown in supplementary dataset E). Concerning the histological features of liver damage, IL32 expression was associated with steatosis grade (figure 3B, p=9.9×10−5), lobular inflammation (figure 3C, p<0.01), ballooning (figure 3D, p<0.001) and fibrosis (figure 3E, p<0.001).
We then investigated IL32 expression in different cell types (figure 4A). We found higher IL32 mRNA levels in primary hepatocytes and HepG2 hepatoma cells (p<0.001 vs hepatic stellate cells, and monocytes/macrophages). However, IL32 was also expressed at high levels in lymphocytes and endothelial cells. ChIP-seq data analysis revealed binding sites for several fatty acid-sensitive transcription factors (peroxisome proliferator-activated receptor gamma coactivator 1-alpha, PPARGC1A and retinoid X receptor alpha, RXRA) in the human IL32 promoter, as confirmed by chromatin immunoprecipitation in HepG2 cells (online supplementary figure S5A), suggesting that IL32 expression may be regulated by fatty acids and retinoids in hepatocytes. In keeping with these findings, HepG2 exposure to free fatty acids resulted in a dose-dependent increase of IL32 expression (online supplementary figure S5B, p=2×10−5, fold increase=1.4±0.1 at 0.5 mM and 1.9±0.3 at 1 mM, adjusted p<0.05 for both).

IL32 expression by cell type and IL32 plasma levels. (A) IL32 expression in different cell types assessed by RT-qPCR. ***p<0.001 versus human hepatocytes (HH), post hoc Tukey’s test. (B) Spearman correlation between IL32 liver expression and plasma levels in 16 patients, who belong to both transcriptomic and Bariatric surgery cohorts. (C, D, E, F, G, H) IL32 plasma levels in patients stratified either by the presence of NAFLD or severe NAFLD as assessed in the Bariatric surgery cohort (C and D), Hepatology service cohort (E and F) and the overall cohort (G and H). *p<0.05; **p<0.01; ***p<0.001, Wilcoxon rank-sum test. ANOVA, analysis of variance; IL32, interleukin-32; HSCs, hepatic stellate cells; HUVEC, human umbilical vein endothelial cell; M1, M1-like human macrophages; M2, M2-like human macrophages; NAFLD, non-alcoholic fatty liver disease.
IL32 is detectable in plasma and correlates with NAFLD
We then examined whether circulating IL32 levels reflected its hepatic expression. There was a strong correlation between hepatic mRNA expression and plasma IL32 levels (figure 4B, R=0.73, p=0.002), although this could be only assessed in a subset of patients. In the Bariatric surgery cohort, circulating levels of IL32 were increased in the NAFLD group compared with patients with normal liver (figure 4C, p<0.01). Moreover, circulating IL32 was higher in patients with severe NAFLD versus those without severe NAFLD (figure 4D, p<0.01). The association of circulating IL32 with both NAFLD and severe NAFLD was independently replicated in the Hepatology service cohort (figure 4E and F, p<0.001 and p<0.01, respectively) and considering both cohorts (figure 4G and H, p<0.001 for both). In keeping, in the Metabolic unit cohort, IL32 correlated with CAP (online supplementary figure S6A, R=0.32, p=0.03) and was increased in the ‘high CAP’ (CAP≥median, 244 db/m2) group compared with the low CAP patients (p<0.05, online supplementary figure S6B).
At ROC analysis, the diagnostic accuracy of IL32 in predicting the presence of either NAFLD or severe NAFLD was similar to that of aminotransferases. At multivariate analysis, the association between circulating IL32 levels and both NAFLD, and severe NAFLD was independent of AST and ALT levels both in the Bariatric surgery and Hepatology service cohorts, and overall (online supplementary table S4).
The association of IL32 with severe NAFLD was independent also of the FIB4 score (n=61, p=0.003, online supplementary table S5). In the Bariatric surgery cohort, we did not detect any significant correlation between the FIB4 score and severe NAFLD (p=0.07, online supplementary table S5).
Thus, we investigated whether IL32 evaluation improved aminotransferases diagnostic performance for both NAFLD and severe NAFLD. In the Bariatric surgery cohort, IL32 evaluation improved the diagnostic accuracy of ALT-AST for NAFLD (AUC=0.85 vs 0.72; p=0.01, figure 5A, online supplementary tables S6 upper panel and S7A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL32 diagnostic accuracy. (A, B, C) NAFLD diagnostic accuracy in the Bariatric surgery cohort (A), the Hepatology service cohort (B) and the overall cohort (C). (D, E, F) Severe NAFLD diagnostic accuracy in the Bariatric surgery cohort (D), the Hepatology service cohort (E) and the overall cohort (F). IL32-ALT-AST score (red curve) was compared with ALT-AST score (blue). Comparison was performed by Delong test. AUC, area under the curve; NAFLD, non-alcoholic fatty liver disease.
The same IL32-ALT-AST and ALT-AST models proposed for NAFLD diagnosis was applied also to the Hepatology service cohort (online supplementary table S6 upper panel and S7B). In these patients, the IL32-ALT-AST model showed an improved accuracy in identifying patients with NAFLD compared with the ALT-AST model (figure 5B, AUC=0.95 vs 0.81, p=7×10−4). Overall, IL32 introduction in the model resulted in a 24% improvement of AUROC (figure 5C, AUC=0.92 vs 0.81, p=5×10−5). In the Metabolic unit cohort, the IL32-ALT-AST model showed an improved accuracy in identifying patients with a high CAP (CAP≥median, 244 db/m2) compared with the ALT-AST model (online supplementary figure S6C, AUC=0.72 vs 0.55, p=0.04).
In the Bariatric surgery cohort, the AUROC of the IL32-ALT-AST score for severe NAFLD diagnosis was slightly higher than that of the ALT-AST model (0.88 vs 0.85; figure 5D and online supplementary tables S6 and S7, middle and lower panels), but the difference was not statistically significant (p=0.06). In the Hepatology service cohort and overall, IL32 introduction did not improve the diagnostic accuracy for severe NAFLD over that of ALT-AST alone (figure 5E, F).
Discussion
In this study, we identified IL32 as an NAFLD-related hepatic cytokine and a potential novel circulating biomarker of this condition.
We first confirmed that carriage of the PNPLA3 I148M variant is one of the major factors contributing to disease heterogeneity, by showing that in obese individuals it was a major determinant of the liver transcriptome variability. Indeed, the I148M variant was associated with higher expression of genes involved in the inflammatory response and carcinogenesis independently of liver disease stage, possibly involved in mediating the predisposition to NASH and liver cancer.7 30 This was associated with downregulation of oxidative metabolism, in keeping with reduced lipid turnover in carriers of the variant.13 These data suggest that, independently of the liver disease stage, PNPLA3 I148M variant carriage modifies the liver biology predisposing to NAFLD.
Next, we set out to determine gene expression pathways associated with severe NAFLD. In line with literature data, our dataset revealed the overexpression of pathways linked with insulin resistance, apoptosis, inflammation and EMT. Despite the fact that these results need to be confirmed in larger samples, the presence of the I148M variant was associated with activation of distinct pathways. Specifically, in variant carriers, there was an upregulation of IL6 and p53 signalling and of KRAS. Conversely, TNFα signalling activation was only appreciable in non-carriers.
The most remarkable finding was that, at the single gene level, IL32 was the most robustly upregulated transcript in severe NAFLD. This was consistent in both carriers and non-carriers of the PNPLA3 variant. Among the IL32 isoforms,25 31 IL32-β showed the highest hepatic expression, and most importantly, correlated with the circulating levels of the protein. IL32 activates TIR-domain-containing adapter-inducing interferon-β protein, through an indirect interaction with protease-activated receptor 2. This process leads to the upregulation of the expression of type I interferons and TNFα in macrophages.32 Concerning liver diseases, IL32 was previously shown to be upregulated during HCV and HBV infection, and is associated with liver disease severity.33 34 Furthermore, an increased expression of IL32 in the adipose tissue and plasma of patients with type 2 diabetes was previously reported.35 However, the role of IL32 in liver disease was difficult to study, as this gene is not expressed in mice, thus limiting the reliability of mechanistic studies in this model.
Among liver cells, IL32 was expressed at high levels in hepatocytes. In the whole parenchyma, IL32 correlated with induction of inflammatory chemokines and chemokine receptors involved in recruitment of T cells and neutrophils,36 but also with activation of the PI3K-AKT-mTOR axis and fibrogenesis. Consistently, hepatic IL32 transcription correlated with steatosis grade and histological severity of liver damage. Circulating IL32 was strongly associated with hepatic mRNA levels and it was higher in patients with NAFLD, and in particular in those with severe disease. The correlation of IL32 with the histological steatosis grade is consistent with the presence of several lipid responsive transcription factor binding sites in the promoter of this gene.37 In keeping, exposure of HepG2 hepatoma cells to fatty acids induced IL32 expression in an in vitro model of NAFLD.
A few previous studies examined the association of gene expression profile with liver damage in patients with NAFLD and controls by unbiased approaches.38–42 Although there were wide discrepancies in the results, possibly related to the different design, study population and inclusion criteria, significantly smaller sample size in most previous studies, and methodological and analytical differences, a few common conclusions could be drawn. First, in keeping with our data, pathways related to inflammation, oxidative stress, fibrogenesis and carcinogenesis were upregulated in severe disease, while intracellular signalling pathways and lipid catabolism were downregulated.38–42 Furthermore, consistent with our results, COL1A1,39 40 42 AKR1B1039 41 and ubiquitin D (UBD)41 42 were among the most upregulated genes in severe disease. While COL1A1 encodes for a major constituent of fibrous tissue during hepatic fibrogenesis (collagen type 1), AKR1B10 is implicated in intracellular metabolism and regulation of intracellular signalling, and it has robustly been implicated in hepatic carcinogenesis.43 UBD encodes for ubiquitin D, which is implicated in the formation of Mallory-Denk bodies during steatohepatitis and is also upregulated in liver cancer.44 Supporting our findings, while this manuscript was under revision, Dali-Youcef et al also reported upregulation of hepatic expression of IL32 and UBD in obese individuals with NASH, providing initial evidence consistent with a role of IL32 in inducing hepatic insulin resistance.45
In the present study, we also generated data suggesting that circulating IL32 may represent a novel candidate non-invasive biomarker of NAFLD. Although the diagnostic accuracy of IL32 alone was not superior to that of aminotransferases, IL32 plasma levels correlated with NAFLD independently of ALT and AST. In addition, IL32 improved the discriminative accuracy of aminotransferases for NAFLD diagnosis in two independent cohorts with different clinical features. The IL32-ALT-AST model showed an improved performance compared with the ALT-AST model in discriminating patients with a high CAP to that with a low CAP also in an independent cohort of patients with metabolic disorders. Further investigations in large cohort of individuals from the general population are required to validate the potential utility of IL32 as biomarker of NAFLD presence and severity.
In conclusion, by exploiting hepatic transcriptome analysis in patients stratified by the presence of PNPLA3 I148M variant, we highlighted key pathways involved in disease progression in specific subsets of obese individuals. Even if further studies are required to clarify mechanistic relationship among liver fat accumulation, IL32 overexpression and inflammatory response, we identified IL32 as a possible new link between hepatic fat accumulation and inflammatory and metabolic complications of NAFLD. Furthermore, the association between circulating IL32 and NAFLD suggests that this cytokine may be considered as a candidate non-invasive biomarker and a therapeutic target for this condition, worthy of evaluation in further studies.
Acknowledgments
We thank Lorenza Lazzari, from the Department of Transfusion Medicine and Hematology – Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, for providing primary human umbilical vein endothelial cells. Part of the results of this study has been presented during the poster session at the AASLD Liver Meeting 2019.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SR and LV are joint senior authors.
Twitter @lucavalenti75
GAB and PD contributed equally.
Contributors GAB, PD, SR, LV: study design and manuscript drafting; GAB, PD, SR, LV: data analysis and interpretation; RR, MM, PP: data generation; SP, MM, SB, TM, GR, ALF, DP: patient recruitment and characterisation; LV, SR: study supervision.
Funding The study was supported by myFIRST AIRC grant no.16888 for the EPIDEMIC-NAFLD project (LV), Ricerca Finalizzata 2016 Ministero della Salute - RF-2016-02364358 (LV), Ricerca Corrente Fondazione IRCCS Ca’ Granda (LV, SF). SR was supported by the Swedish Research Council (Vetenskapsrådet (VR), 2016-01527), the Swedish state under the Agreement between the Swedish government and the county councils (the ALF-agreement) (SU 2018-04276), the Novonordisk Foundation Grant for Excellence in Endocrinology (Excellence Project, 9321-430), Wallenberg Academy Fellows from the Knut and Alice Wallenberg Foundation (KAW 2017.0203) (SR).
Competing interests LV reports having received speaking fees from MSD, Gilead, AlfaSigma and AbbVie; having served as a consultant of Gilead, Pfizer, Astra Zeneca and Novo Nordisk; and received research grants from Gilead.
Patient consent for publication Not required.
Ethics approval Informed consent was obtained from each patient and the study protocol was approved by the Ethical Committee of the Fondazione IRCCS Ca’ Granda and University of Catanzaro and conformed to the ethical guidelines of the 1975 Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Deidentified gene expression data are available on request to the corresponding authors.