Article Text

Abstract
Objective Helicobacter pylori (Hp) is a major risk factor for gastric cancer (GC). Hp promotes DNA damage and proteasomal degradation of p53, the guardian of genome stability. Hp reduces the expression of the transcription factor USF1 shown to stabilise p53 in response to genotoxic stress. We investigated whether Hp-mediated USF1 deregulation impacts p53-response and consequently genetic instability. We also explored in vivo the role of USF1 in gastric carcinogenesis.
Design Human gastric epithelial cell lines were infected with Hp7.13, exposed or not to a DNA-damaging agent camptothecin (CPT), to mimic a genetic instability context. We quantified the expression of USF1, p53 and their target genes, we determined their subcellular localisation by immunofluorescence and examined USF1/p53 interaction. Usf1 -/- and INS-GAS mice were used to strengthen the findings in vivo and patient data examined for clinical relevance.
Results In vivo we revealed the dominant role of USF1 in protecting gastric cells against Hp-induced carcinogenesis and its impact on p53 levels. In vitro, Hp delocalises USF1 into foci close to cell membranes. Hp prevents USF1/p53 nuclear built up and relocates these complexes in the cytoplasm, thereby impairing their transcriptional function. Hp also inhibits CPT-induced USF1/p53 nuclear complexes, exacerbating CPT-dependent DNA damaging effects.
Conclusion Our data reveal that the depletion of USF1 and its de-localisation in the vicinity of cell membranes are essential events associated to the genotoxic activity of Hp infection, thus promoting gastric carcinogenesis. These findings are also of clinical relevance, supporting USF1 expression as a potential marker of GC susceptibility.
- helicobacter pylori infection
- oncogenes
- DNA damage
- genetic instability
- gastric cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Helicobacter pylori (Hp) is a major risk factor for gastric cancer (GC).
Hp promotes p53 proteasomal degradation and inhibits USF1 expression.
In response to DNA damaging agents, USF1 binds to p53 and inhibits its degradation.
What are the new findings?
Low USF1 and p53 levels are associated with low overall survival in human GC patients.
Loss of USF1 accelerates gastric carcinogenesis.
Only Hp and not genotoxic chemicals, leads to USF1 accumulation as structure-like foci at the periphery of the cells.
Hp inhibits USF1/p53 nuclear interaction and impairs DNA repair function.
How might it impact on clinical practice in the foreseeable future?
Depletion of USF1 in gastric tumorous tissue can be an indicator of a poor prognosis and may become a new biomarker to identify subgroup of patients with higher risk of GC.
Identification of drugs able to inhibit cytoplasmic accumulation of USF1 or its nuclear depletion can allow future development of targeted therapies to improve GC treatment.
Introduction
Helicobacter pylori (Hp) is responsible for about 90% of gastric cancer (GC) cases worldwide,1–3 which represents the highest frequency of infectious agents-associated cancer (5.5%).4 Importantly, the detection of preneoplasia5 and Hp eradication during early stages of the precancerous cascade can prevent GC development.6 7 GC is an inflammation-driven disease resulting from the complex interplay between bacterial, host and environmental factors.8 Hp-induced chronic inflammation contributes to neoplastic transformation, via dysregulation of signalling pathways, cell proliferation and genetic instability.9 We previously reported that Hp induces mutations in chronically infected mice.10–12 Hp also causes DNA double strand breaks13 14 and impairs DNA repair pathways, favouring overall mutation load.12 15 16 Importantly, Hp promotes the accumulation of mutations in the tumour suppressor gene TP53,17 which have been reported in 50% of gastric tumours.17 18 In response to genotoxic stress, p53 activates signalling pathways leading to temporary cell cycle arrest allowing DNA damage and cellular repair.19 Its inactivation promotes genome instability, a hallmark of cancer.20 The inhibition of p53 has thus emerged as a strategy of bacterial pathogens to modulate host cellular functions,21 as for Hp which promotes p53 proteasomal degradation.22–24 Together, this results in accumulation of oncogenic changes in infected cells.
Finally, Hp induces aberrant DNA methylation that down-deregulates the expression of genes related to signal transduction pathways and tumour suppression.25–27 We reported that Hp induces DNA hypermethylation in the promoter region of the upstream stimulating transcription factors genes, USF1 and USF2, inhibiting their expression in infected mice concomitantly to the development of gastric preneoplasia.28 USF1 and USF2 are b-HLH-LZ transcription factors ubiquitously expressed. They regulate stress and immune responses, cell cycle control, inflammation and genome stability related genes.29 They may thus act as tumour suppressors.30 31 We previously showed that under ultra-violet (UV) stress, USF1 up-regulates CSA and HR23A genes expression, two actors of the transcription-coupled and global genome nucleotide excision repair pathway (TC-NER and GG-NER), respectively.32 USF1 also binds p53 in response to UV-induced DNA damage, preventing the E3-ubiquitin ligase HDM2-p53 interaction. This results in p53 stabilisation and transient cell cycle arrest.33 How USF1 modulates p53 levels in response to Hp and the consequences on the infection-associated genotoxicity have never been addressed.
In the present study, we investigate the role of both USF1 and p53 transcription factors in gastric carcinogenesis and asked whether USF1 deregulation during Hp infection could impact the p53-response and increase genetic instabilities. Using a mouse model, we showed that the absence of USF1 has strong implications in the oncogenic properties of Hp, triggering the severity of gastric lesions. In line with these data, low expression levels of both USF1 and TP53 and consequently deregulation of their target genes, are observed in a significant number of GC patients, associated with a worse prognosis. Our findings show that USF1 is a key player in the complex regulatory network linking Hp infection to gastric carcinogenesis and pave the way to a better understanding of the mechanisms at the origin of pathogen-induced cancer.
Results
Low USF1 and p53 levels are associated with a worse prognosis in GC patients
Using The Cancer Genome Atlas (TCGA) data sets, GC patients (STAD) are distinguished according to their overall survival (top 25% low vs top 25% high, n=188), based on SLC7A2 expression, the most discriminant gene (online supplementary figure S1A). As observed in the expression heatmap (figure 1A), USF1 and TP53 gene expression levels are correlated with the GC patients overall survival. Low mRNA levels of both USF1 and TP53 are associated with poor 3-year survival (figure 1B and online supplementary figure S1B). Moreover, for every patient, the mRNA expression levels of USF1 and p53 are correlated (online supplementary figure S1C) and consequently impact their transcriptional function leading to a significant downregulation of pathways, notably p53-signalling, DNA repair (BER, NER) and cell cycle regulation, in patients with low versus high survival (online supplementary figure S1D). In order to identify the p53 and USF1-target genes significantly enriched in the two groups (low vs high survival), we performed Gene Set Enrichment Analysis (http://software.broadinstitute.org/cancer/software/gsea) in GC, TCGA dataset (STAD) (online supplementary figure S1E,F). The median expression of the top-genes enriched in the low and high survival groups (online supplementary figure S2), showed a specific survival rate-dependent expression on both USF1-target and p53-target genes (figure 1C). Interestingly, the analysis of another data set from Hippo and colleagues34 (GSE2685), confirmed the decrease of most of USF1 and p53-regulated genes expression in GC patients, in tumorous versus normal tissues (figure 1D). In parallel, we analysed USF1 gene expression in gastric biopsies from GC patients. In 50% of these patients, USF1 expression was lower in the tumorous versus adjacent non-tumoural tissue (fold-change <1; 17/34 patients; p<0.0001) (figure 1E), and 88% (15/17) of patients with low USF1 expression were Hp-positive (online supplementary figure S1G). These data suggest that Hp-associated decrease of USF1 gene expression may define a subgroup of more aggressive gastric tumours. The consequences of Hp infection on both USF1 and p53 target-genes expression were also analysed in gastric cells using expression data from Koeppel and colleagues16 (GSE55699) and Hong and colleagues (E-GEOD-74577). A significant decrease of USF1 and TP53 mRNA levels and target genes, mainly correlated with low survival was observed (figure 1F). These features are also confirmed in Hp-infected mice using both expression data from Galamb and colleagues35 (GSE5081) (online supplementary figure S3A) and our previous study36 (E-MEXP-1135) (online supplementary figure S3B).
Supplemental material

Correlation of p53 and USF1 loss with gastric carcinogenesis and Hp infection. (A) Expression heatmap depicting mRNA expression of genes distinguishing most significantly GC patients according to their overall survival. Expression data for (low survival: 665 days or high survival: 1095 days) were obtained from TCGA (STAD, n=188) (see online supplementary figure S1A). (B) Survival curve for GC patients according to USF1 and TP53 mRNA levels (low: green, medium: blue or high: red). (C) Expression heatmap depicting median mRNA expression of p53 target genes (Fisher_direct_p53_targets_meta_analysis, GSEA) and putative USF1 target genes (genes having at least one occurrence of transcription factor binding site V$USF_01 (v7.4 TRANSFAC) in the regions spanning up to 4 Kb around their transcription starting sites, gene set enrichment analysis (GSEA),) significantly enriched in both low and high survival GC patients (top 50 genes, see online supplementary figure S1E,F). (D) Expression heatmap depicting p53 and USF1-target genes expression (p53-targets: orange and pink; USF1-targets: blue and green; common: black), previously correlated with low (pink and green) or high survival (orange and blue) using data from Hippo and colleagues34 comparing non-cancerous and cancerous tissues. (E) Relative USF1 gene expression in gastric biopsies from GC patients (n=34) measured by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) (tumorous vs adjacent tissue); bar: median value. Mann-Whitney test (low (<1) vs high (>1) expression; ****p<0.0001). (F) Log-fold enrichment of p53-target and USF1-target genes expression in Hp-infected gastric cells. Data from GSE55699 (Koeppel and colleagues)16 and E-GEOD-74577 (Hong and colleagues). GC, gastric cancer; Hp, Helicobacter pylori; TCA, The Cancer Genome Atlas.
Absence of USF1 exacerbates the severity of Hp-induced gastric lesions
To determine the consequences of the absence of USF1 in Hp-associated gastric pathogenesis, we infected Usf1-KO mice (Usf1 -/-)37 and the parental mice (Usf1 +/+) with HpSS1 strain which colonises the mouse stomach.38 At each time-point (9/12 months), the infection status was monitored (online supplementary figure S3C) and histological analysis performed (figure 2A,B). Nine-months postinfection (pi), both Usf1 -/- and Usf1 +/+ mice developed gastric lesions, consisting in infiltration of inflammatory cells, mainly mononucleated cells, in the mucosa and submucosa (figure 2A). A semiquantitative analysis showed an exacerbation of metaplasia and dysplasia in Usf1 -/- mice compared with Usf1 +/+ (figure 2B). After 9 months, only Hp-infected Usf1 -/- mice showed metaplasia and dysplasia that were absent in Usf1+/+ infected mice. An important loss of parietal cells favouring hypochlorhydria and atypia was also observed in Usf1-/- infected mice. In Hp-infected Usf1+/+ mice, these atypia and dysplasia appeared only after 12 months. At 12 months pi, the gastric inflammation was significantly more severe in Hp-infected Usf1 -/- mice, with score-grading of 2.5 for intestinal metaplasia and parietal cell loss, compared with 1 in Usf1+/+ mice. In addition, immunofluorescence (IF) analysis of gastric tissue sections shows that in the absence of USF1 (Usf1 -/- mice), Hp infection strongly promotes p53 loss (figure 2C). This leads to a down regulation of its target genes (GADD45, CDKN1A, PCNA, RAB31) (online supplementary figure S3D), in agreement with previous mice data35 36 (online supplementary figure S3A,B). Together, these results underscored for the first time a role for USF1 in gastric carcinogenesis.

Loss of USF1 exacerbates gastric tumourigenesis associated to Hp infection. Usf1-/- and Usf1+/+ mice were oro-gastrically infected with HpSS1 for 9 and 12 months as described in online supplementary methods. (A) Representative gastric histological changes on H&E stained tissue section, in Usf1 -/- (d, e, f, j, k,l) and Usf1 +/+ (a, b, c, g, h, i) mice, Hp-infected (b, c, e, f, h, i, j, k, l) and non-infected (a, d, g, j), after 9 months (a–f) and 12 months (g–l). As early as 9 months, cysts and atypia are observed in Usf1-/- -infected mice (arrows). Dysplasia is only detected in Usf1-/- infected mice at 9 months pi (arrows). (B) Semiquantitative evaluation of gastric lesions in Hp-infected Usf1-/- and Usf1+/+ mice (see online supplementary information). Mann-Whitney test, infected versus non-infected (*p<0.05). (C) p53 if (green) and nuclei (Hoechst, blue) on gastric tissue sections from Hp-infected Usf1-/- and Usf1+/+ mice at 12 months pi, showing a depletion of p53 in Hp-infected Usf1-/- mice. Scale bar 100 µm.
Hp impairs DNA repair functions by downregulating USF1 and p53
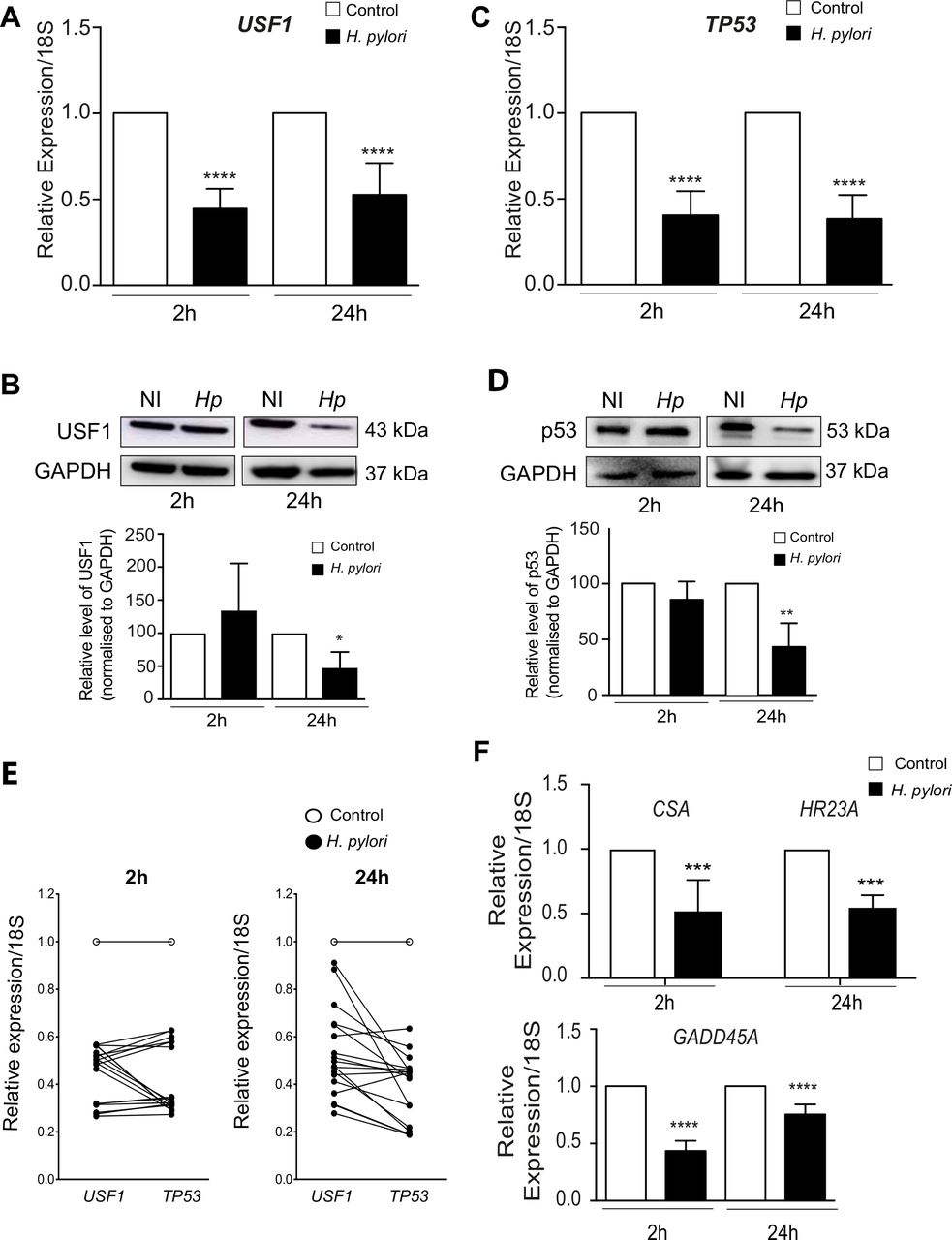
Since USF1 and p53 cooperate to maintain genetic stability, we investigated whether Hp impacts USF1/p53 functioning. We first showed that at 2 and 24 hours after infection of MKN45 gastric epithelial cells, the expression of USF1 and TP53 genes was significantly diminished by the oncogenic strain Hp7.1339 (figure 3A,C,E), with a significant and concomitant decrease of their protein levels at 24 hours pi (figure 3B and D). This resulted in the diminution of the expression of USF1 and p53 DNA repair target genes: respectively CSA, HR23A and GADD45A (figure 3F). Similar results were obtained with cells treated with Hp7.13 total extracts (50 and 100 µg/mL) (online supplementary figure S4A-E), concomitantly with an increase in DNA damage hallmark (phosphorylated-histone H2AX, γH2AX) (online supplementary figure S4F). The infection of MKN45 cells with HpSS1 also, inhibited USF1 and p53 levels (online supplementary figure S5). These data, suggest that Hp-mediated decrease of USF1 and p53 impacts the DNA repair ability of infected cells and consequently affect their genetic stability.
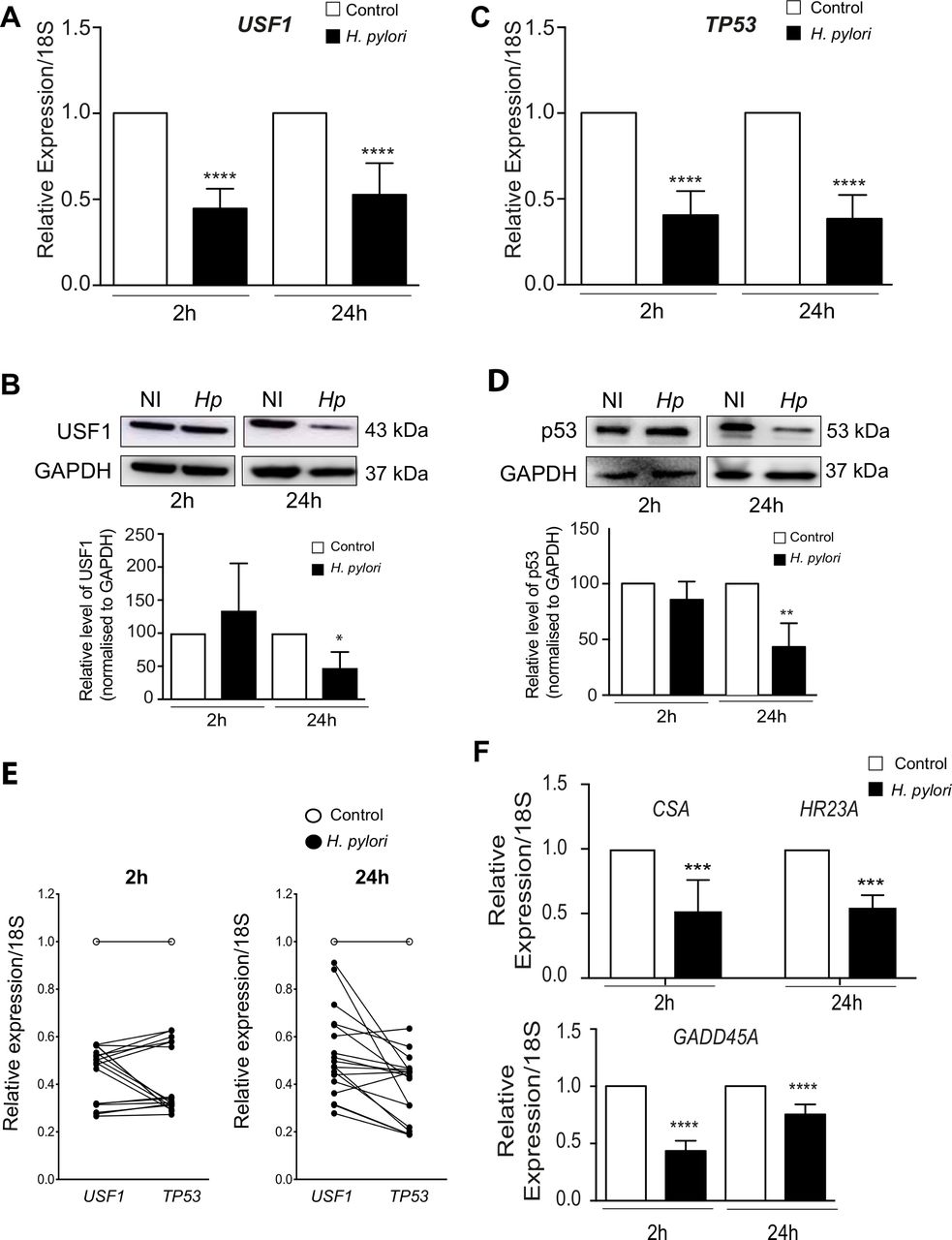
Hp impairs host DNA repair function by downregulating USF1 and p53. MKN45 cells were infected with Hp7.13 (MOI 100:1) for 2 and 24 hours. Control cells were not infected. (A) USF1 (B) Western blot analysis of USF1 (C) TP53 (D) p53 and GADPH (E) paired USF1-TP53 (F) CSA, HR23A and GADD45A mRNA level quantified by quantitative reverse transcriptase-polymerase chain reaction (RT-qPCR). Results are relative to the 18SrRNA. Mean±SD, n=3. (B) WB analysis of (B) USF1 (E) p53 and GAPDH (loading control) in protein extracts from infected and non-infected cells. The histogram below corresponds to immunoblot quantification. Error bars: SD, n=3. Student’s t-test, infected versus non-infected (*p<0.05; **p<0.01; ****p<0.0001). Hp, helicobacter pylori.
Hp leads to USF1 foci accumulation in the cytoplasm and membrane-surrounding regions of gastric cells
Hp has previously been reported to promote the cytoplasmic p53 proteasomal degradation.22 23 USF1 was shown to interact with p53 leading to p53 nuclear stabilisation in response to genotoxic stress.33 Using IF, we observed that, at 2-hour pi, p53 nuclear staining was significantly lower in Hp-infected cells than in non-infected, as also USF1 nuclear staining (figure 4A,B). More importantly, we detected cytoplasmic USF1 foci-like structures, mainly in the membrane-surrounding area of Hp-infected cells at 2 and 24 hours (figure 4A,C; yellow arrows). Quantification of these foci revealed a marked increase with infection time (figure 4C), being present in 80% of Hp-infected cells at 24 hours pi. Importantly, a cytoplasmic accumulation of USF1 was also observed in Hp-infected INS-GAS mice after 6/12 months in the presence of gastric intraepithelial neoplasia (online supplementary figure S6A,B), with a p53 decrease as reported at 12 months pi (online supplementary figure S6C). Together these results indicate that Hp relocates USF1 outside of the nucleus, and promotes USF1 cytoplasmic/membrane accumulation, concomitantly to p53 degradation.
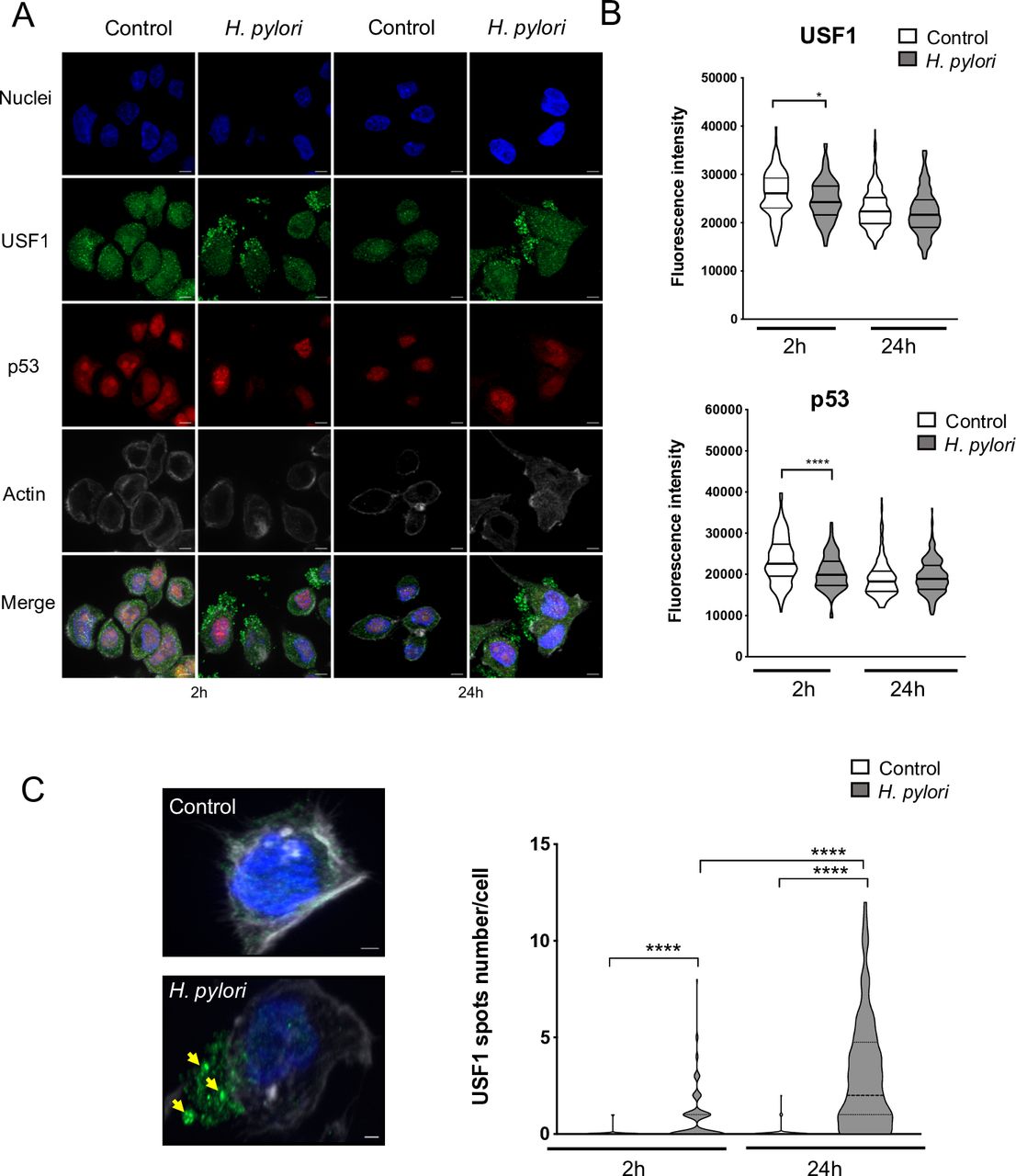
Hp leads to USF1 foci in the vicinity of cell membranes. (A) Immunofluorescence analysis of USF1 and p53 levels and localisation in MKN45 cells infected as in figure 3. p53 immunostaining (red), USF1 (green) and nuclei (Hoechst, blue). Phalloidin actin staining (grey) indicates the cells shape. Scale bar 5 µm. (B) Quantification of USF1 and p53 nuclear if intensity (n=150–220 cells/condition). Mann-Whitney test, infected versus non-infected (*p<0.05; ****p<0.0001). (C) Maximum intensity projection of representative Hp-infected and non-infected cells at 24 hours. USF1 staining (green) shows foci (yellow arrows) in the cytoplasm and vicinity of cell membranes in infected-cells (left part). Quantification of USF1 spots number per cell according to defined spot criteria as indicated in material and methods (right panel) (n=150–220 cells/condition). Experiments in triplicate with 5–7 microscopic fields analysed. Mann-Whitney test, infected versus non-infected (*p<0.05; ****p<0.0001). Hp, Helicobacter pylori.
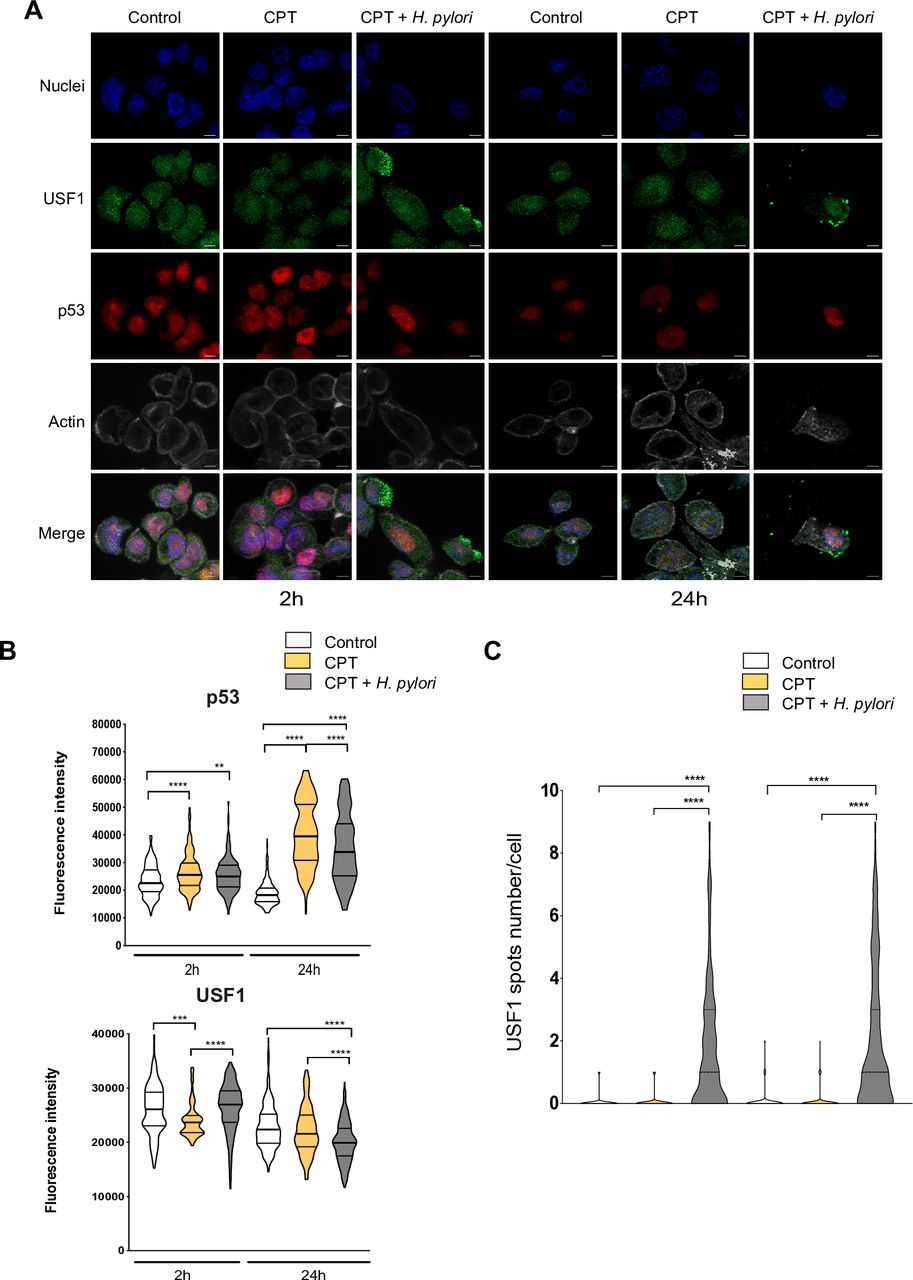
To strengthen this point, we used the well-known DNA-damaging compound, camptothecin (CPT), amplifying the USF1 and p53 genotoxic stress response, as previously reported.33 Briefly, cells were exposed to CPT (50 nM) and infected by Hp or not for 2 and 24 hours. As anticipated, CPT alone induced an immediate DNA-damage response, showed by γH2AX staining (online supplementary figure S7), promoting a strong p53 nuclear accumulation 24 hours post-CPT treatment (figure 5A). This p53 increase was significantly reduced in infected cells, as shown by IF quantification (figure 5B). In parallel, while the impact on USF1 expression was mild (figure 5A,B), an important cytoplasmic/membrane accumulation of USF1 foci was observed in CPT-treated cells only in the presence of Hp, as confirmed by spots quantification (figure 5A,C). Comparable results were obtained when Hp-infection was combined with different genotoxic stress compounds (MMS, H2O2) (online supplementary figure S8 and S9). Together this strongly supports the important and specific role of Hp on USF1 and p53 biological function.
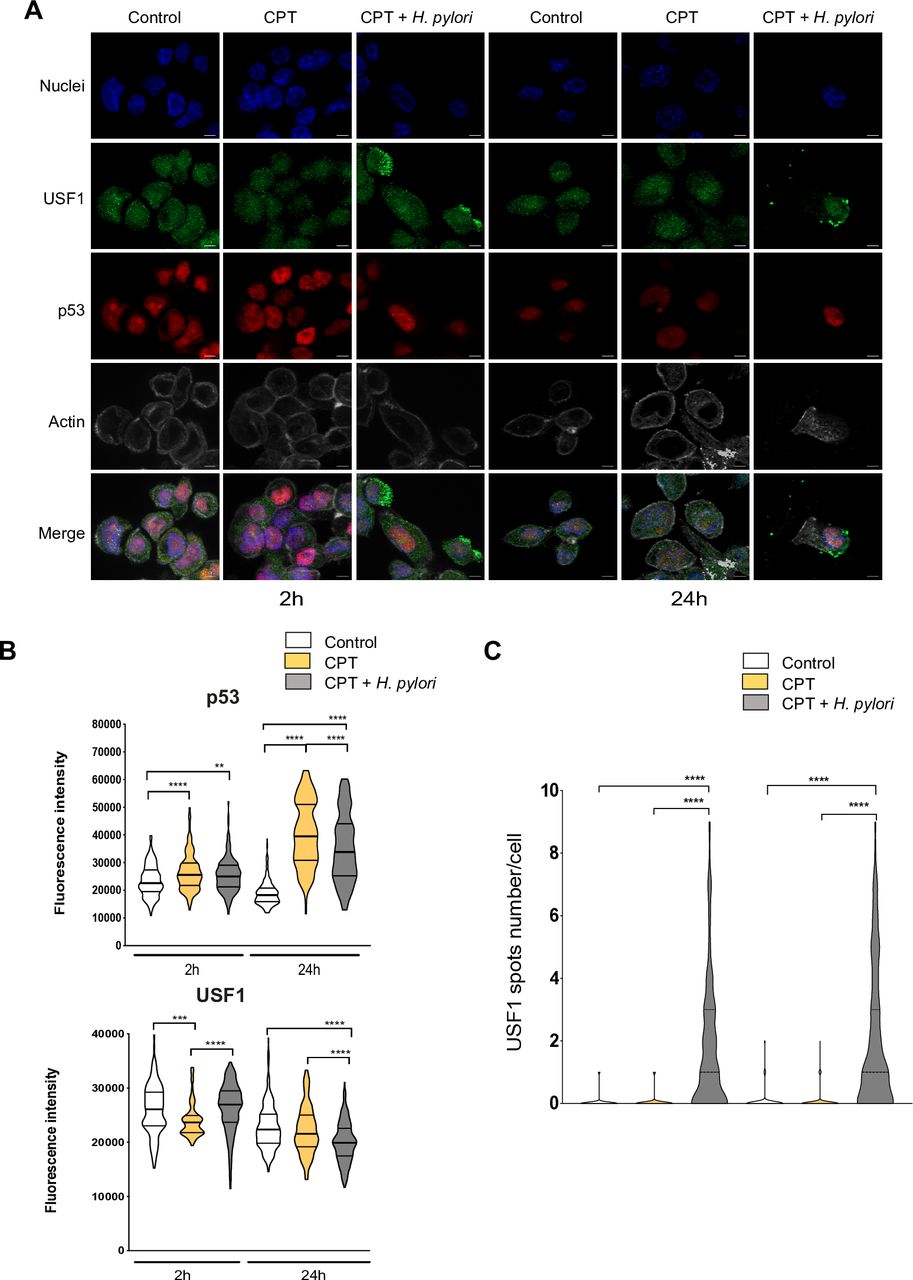
USF1 foci are specifically induced by Hp infection. (A) Immunofluorescence analysis of USF1 and p53 levels and localisation, in MKN45 cells treated or not with CPT (50 nm) and infected with Hp 7.13 for 2 and 24 hours. p53 (red), USF1 (green), nuclei (Hoechst, blue) and phalloidin actin staining (grey). The delocalisation and accumulation of USF1 are specifically observed in the cytoplasm and membrane surrounding area of Hp-infected/CPT-treated cells. Scale bar 5 µm. (B) Quantification of USF1 and p53 nuclear if intensity (n=150–220 cells/condition). (C) Quantification of USF1 spots number/cell as in figure 4. USF1 foci are only observed in the presence of Hp (n=150–220 cells/condition). Mann-Whitney test: treated or treated/infected versus control (**p<0.01; ***p<0.001; ****p<0.0001). Experiments in triplicate with 5–7 fields analysed. CPT, camptothecin; Hp, Helicobacter pylori.
Hp impairs the formation of USF1/p53 complexes
To investigate the mechanism by which Hp impairs USF1 and p53 function, we followed the formation of USF1/p53 complexes in response to CPT-induced genotoxic stress and infection using proximity ligation assay (PLA).40 According to its genotoxic activity, CPT alone induces nuclear USF1/p53 complexes, with a marked increase after 24 hours. In CPT-treated/Hp-infected cells, the formation of these complexes is abrogated. Thus, CPT exacerbated Hp-mediated effects with a stronger inhibition of the formation of USF1/p53 complexes, compared with Hp infection alone (figure 6A,B). Together, this shows that minute nuclear amounts of USF1 in infected cells are associated with the absence of USF1/p53 nuclear complexes, impairing p53 stabilisation in agreement with its Hp-mediated degradation.22 23

Hp inhibits the USF1/p53 complexes in response to a chemical genotoxic stress. (A) Duolink PLA analysis of USF1/p53 complexes (pink foci), in Hp-infected cells either CPT-treated (50 nM) or not as described in methods. nuclei (Hoechst, blue). Experiments in duplicate (5–7 fields analysed). Scale bar: 10 µm for each time-point: right panels zoom: fields delimited in red, scale bar 5 µm. (B) Quantification of USF1/p53 nuclear interaction (5–7 fields analysed). Student’s t-test, CPT-treated and/or infected versus control (*p<0.05; **p<0.01; ***p<0.001). CPT, camptothecin; Hp, Helicobacter pylori; PLA, proximity ligation assay.
Hp infection sensitises gastric cells to genotoxic stress
We next investigated whether Hp infection could sensitise cells to DNA-damage. To address this important clinical question, cells were first infected with Hp7.13 for 24 hours, washed several times prior to their treatment with CPT (50 nM) for 24 hours (figure 7A). Here also, Hp-infected cells displayed accumulation of USF1 foci mainly at their periphery with low p53 staining, while CPT-treatment alone, promotes USF1 and p53 nuclear increase (figure 7B). Sensitising cells with Hp 24 hours prior to CPT-treatment still leads to an important accumulation of USF1 foci in the cytoplasm and surrounding-membrane cell area (figure 7B). Importantly, we noticed the presence of p53-positive micronuclei-like structures (figure 7B, yellow arrows), a signature of elevated genotoxic stress41 known to accumulate p53,42 as under our conditions. Same results were observed with MMS and H2O2-treated cells (1 mM) (online supplementary figure S10). Thus, Hp-induced USF1 cytoplasmic/peripheral accumulation is maintained postinfection, rendering the cells more susceptible to DNA-damaging agents.
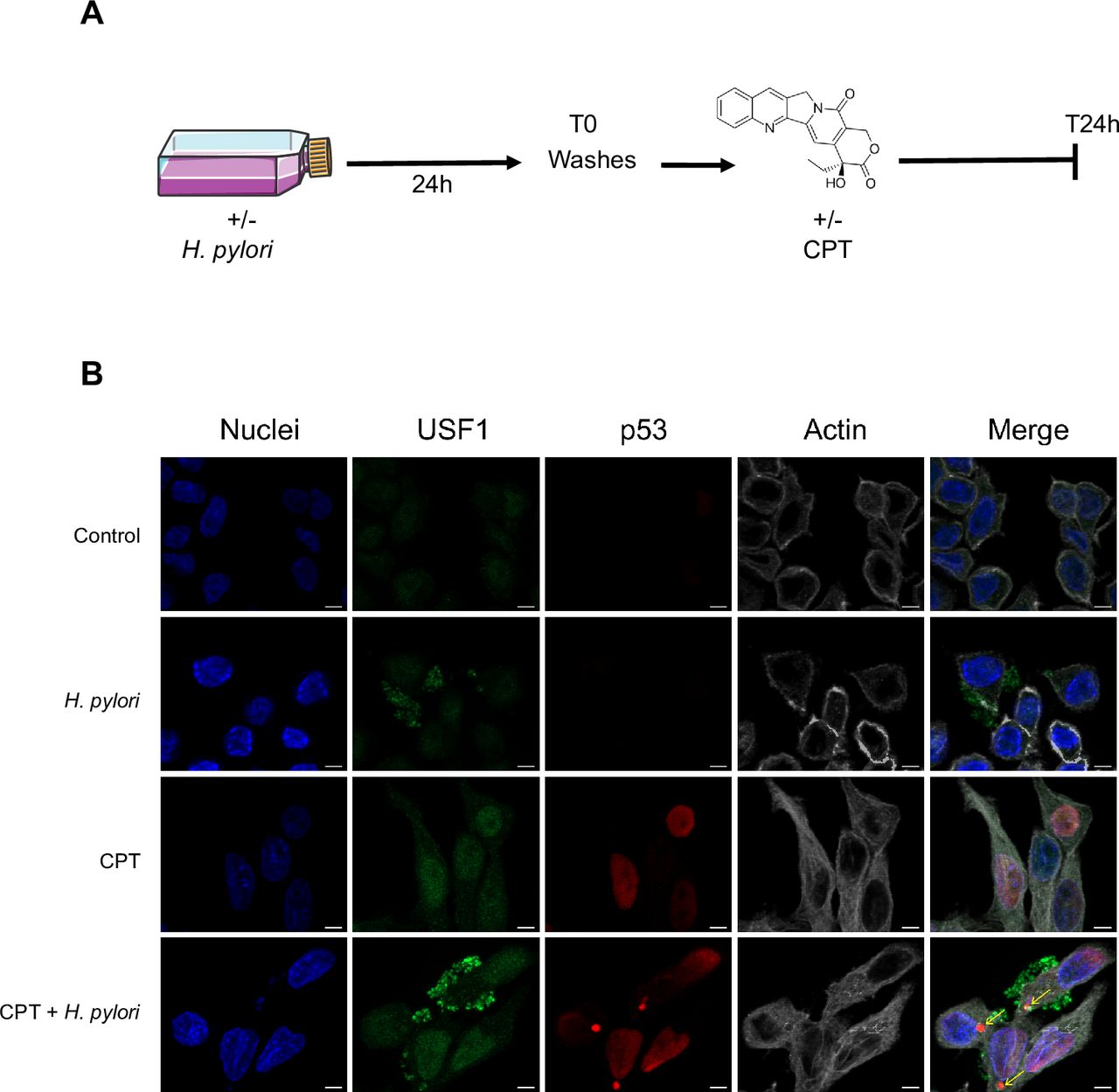
Hp infection makes more susceptible gastric cells to a genotoxic stress. (A) Experimental schedule. MKN45 cells were first infected with Hp 7.13 or not, as in figure 3. After 24 hours, cells were washed three times and either treated or not with CPT (50 nM) for 24 hours. (B) Immunofluorescence analysis of USF1 (green) and p53 (red). nuclei (Hoechst, blue) and phalloidin actin staining (grey). Experiments in duplicate (5–7 fields analysed per condition). Scale bar 5 µm. CPT, camptothecin; Hp, Helicobacter pylori.
Discussion
The impairment of p53 function plays a key role in the promotion of carcinogenesis. We previously showed that UV-induced p53 stabilisation and subsequent transient cell-cycle arrest requires USF1.33 Up to now, no direct in vivo evidence linking USF1 to cancer was provided, although molecular data were in favour of such a role.30 43 44 Studies associated USF1 polymorphisms with increased risk of cancer.45–47 Here, we demonstrate for the first time that loss of USF1 promotes Hp-induced carcinogenesis. First, the in vivo absence of USF1 in Usf1-/- mice, leads to p53 depletion and accelerates the development and triggers the severity of Hp-induced gastric lesions. More importantly, these mice recapitulate the sequential gastric preneoplastic cascade described in human pathology.3 Usf1-/- mice constitute thus an interesting model to study Hp-induced gastric carcinogenesis. Second, in human GC samples, USF1 and TP53 gene expression is associated with patient prognosis (TCGA analysis), as low transcriptional levels correlate with poor 3 years survival. Moreover, USF1 and TP53 expression levels directly impact their target genes such as those related to DNA repair, cell cycle regulation and p53 signalling pathways. Furthermore, low USF1 gene expression in GC patients is mainly associated with Hp status. Thus, Hp-positive gastric tumours with low USF1 and TP53 levels may identify a subgroup of patients with poor prognosis. Together these data demonstrate that USF1 has tumour suppressive functions and that its low level should be considered as a potential marker of cancer susceptibility.
We also show that Hp infection delocalises the nuclear factor USF1 at the periphery of cells into foci that resemble aggregates. This occurs concomitantly with a diminution of its nuclear amount, as schematised in figure 8A. This phenotype is only observed in Hp-infected cells and not after exposure to DNA damaging agents. The unexpected cellular localisation of USF1 may impair its transcriptional regulatory function, reducing the expression of its NER target genes CSA and HR23A in infected cells. It also controls its biological function, impairing nuclear USF1/p53 complex formation. Indeed, Hp-mediated USF1 depletion diminishes the stabilisation of p53 that is known to contribute to genetic instability and oncogenic properties of the infection.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
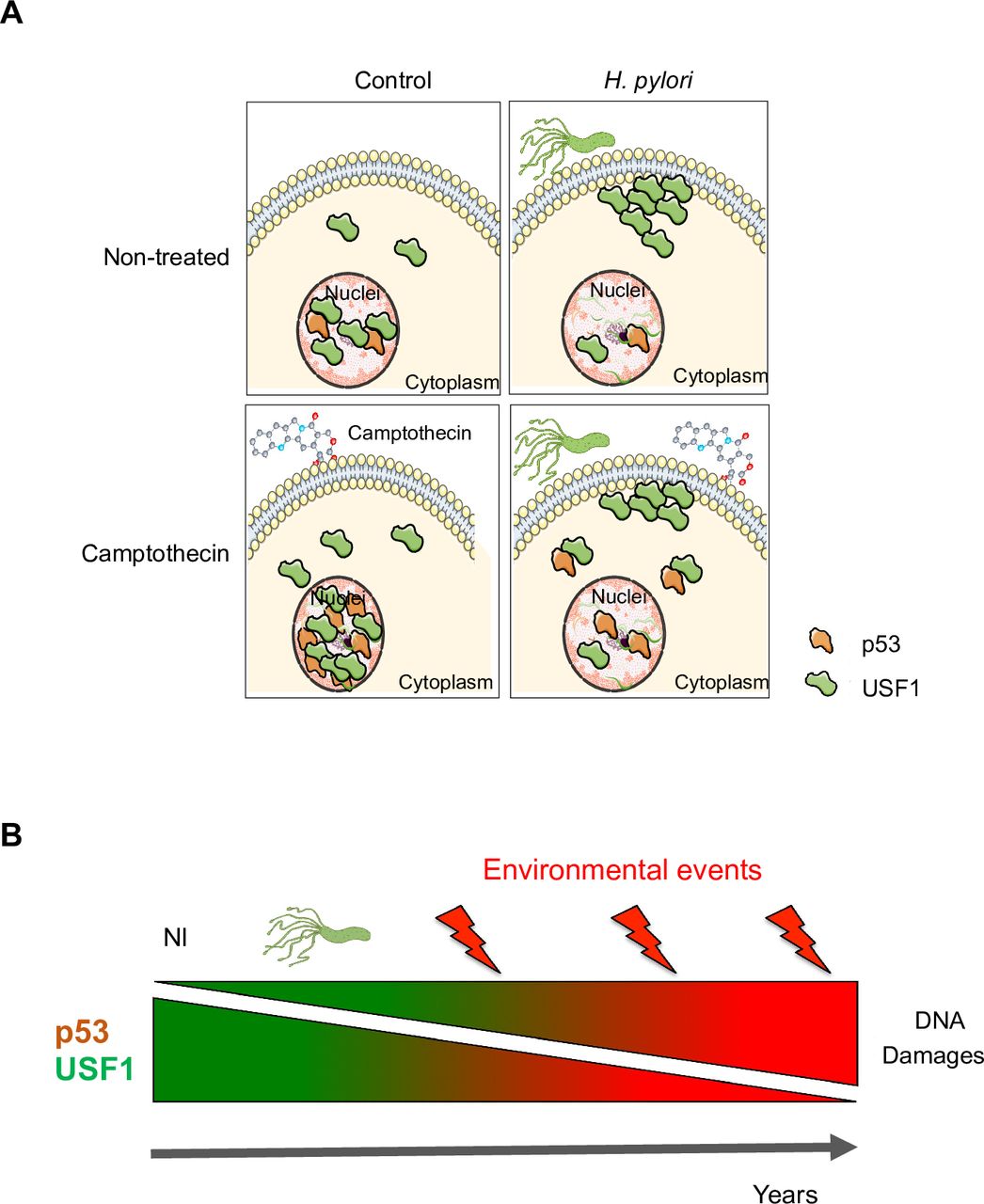
Schematic representation of the data. (A) Gastric epithelial cells infected with Hp show lower nuclear level of USF1 and p53 and the formation of USF1 foci mainly at the periphery of cells close to membranes. Hp infection inhibits USF1 and CPT-induced USF1/p53 complexes in the nuclei. These data support that, in response to a genotoxic stress, the nuclear localisation of USF1 is important to maintain p53 in the nucleus to carry out its function. (B) Exacerbation of gastric carcinogenesis due to synergistic effects of Hp and environmental DNA damaging factors in chronically infected individuals. According to our data, the progressive nuclear decrease of USF1 and p53 in Hp-positive subjects should lead to further accumulation of DNA damage all lifelong. This supports that Hp increases the sensitivity to DNA damaging effects of genotoxic environmental factors, thus promoting the risk of GC. CPT, camptothecin; GC, gastric cancer; Hp, Helicobacter pylori.
USF1 as part of the b-HLH-LZ transcription factor family is well known for its nuclear function.29 The Hp-mediated delocalisation of USF1 outside the nucleus was unexpected. The underlying mechanism and the cellular structure involved remain to be clarified. It may well be that Hp-infection induces USF1 post-translational modifications modulating its nuclear-cytoplasmic trafficking, that results in its cytoplasmic/membrane accumulation. This could represent an Hp strategy to prevent USF1 transcriptional function, impairing its tumour suppressive activity and DNA repair functions. Alternatively, USF1 foci could correspond to protein aggregation due to infection-induced misfolding, as recently reported the formation of aggresomes by Twist1, another b-HLH-LZ transcription factor.48
In response to a genotoxic stress, USF1 and p53 interact promoting p53 stabilisation and blocking its interaction with the E3-ubiquitin ligase HDM2, thereby abrogating subsequent p53 degradation.33 We show that nuclear depletion of USF1 parallels the p53 decrease in Hp-infected cells. Under this condition, we speculate that the nuclear level of USF1 is too low to ensure p53 stabilisation, limiting the formation of USF1/p53 complexes. Importantly, infection of cells by Hp prior to CPT (MMS or H2O2)-treatment maintains the cytoplasmic/membrane delocalisation of USF1, indicating that once initiated this process is sustained even in the absence of a new Hp challenge. Hp-induced accumulation of USF1 outside the nucleus could thus constitute a ‘point of no return,’ after which USF1 is no more available to undertake its nuclear functions. This suggests that Hp-infection may weaken DNA repair ability of cells exposed to genotoxic stress. As illustrated in figure 8B, Hp can persist all lifelong, promoting DNA damage, which thus results from the combined effects of the infection and exposure to genotoxic environmental factors, increasing the risk of GC.
In conclusion, this study demonstrated that USF1 is a new central regulator of DNA damage and repair in response to Hp infection. The absence of USF1 results in the promotion of gastric carcinogenesis as demonstrated in vivo with the Usf1-/- mice, which constitute a new powerful tool to deepen our understanding of the molecular cascade from preneoplasia to GC development. Our findings are also of clinical relevance and pave the way to propose the USF1 level as a potential biomarker for GC.
Methods
Cells culture and bacteria growth conditions, mice infection and histology, analysis of genes expression, proteins and imaging procedures and data banks used in in silico study are reported in online supplementary information.
Bacteria and cells
Human gastric epithelial cells, MKN45 (received from C. Reis’s laboratory, Porto, Portugal), were used in this study and infected with Hp strains 7.1339 and SS1.38
Analysis of protein complexes by PLA
The USF1/p53 complexes were visualised by Duolink PLA,40 as reported in online supplementary information. Imaging analysis was carried out using an inverted widefield microscope Axio Observer Z1 equipped with Apotome grid (Carl Zeiss, Germany).
Human gastric biopsies
All patients were adults, informed and signed a consent letter.
Gastric biopsies (tumorous and adjacent tissue) were from GC patients who attended the Instituto Mexicano del Seguro Social, Medical Center SXXI in Mexico (n=28) and the Florence University Hospital (n=6). For each patient, diagnosis was based on endoscopic examination and histopathological analysis.
Hp infection in mice
Mice experiments were carried out according to the European Directives (2010/63/UE).
Usf1 -/-37 and Usf1 +/+ mice (C57BL/6 j 129SV) were from S. Vaulont (Institut Cochin, Paris, France) and INS-GAS mice49 50 from TC Wang (Columbia University College, New York, USA).
Statistical analysis
Statistical analysis was performed using the Student’s t-test or Mann-Whitney test, after being assessed for normality of samples distribution. Results were considered significant if p<0.05. Kaplan-Meier survival analysis assumption was performed on the TCGA data set (https://cancergenome.nih.gov).
Acknowledgments
We thank Sophie Vaulont (Institut Cochin, Paris, France) for Usf1-/- mice, Timothy C Wang (Columbia University, NY, USA) for providing us couples of INS-GAS mice and Joana Gomes and Celso Reis (I3S-IPATIMUP, Porto, Portugal) for MKN45 cells. We also thank Laurence Bernard-Touami and the Animal Housing ARCHE (UMS Biosit, https://biosit.univ-rennes1.fr, University of Rennes, France), and David Hardy and Magalie Tichit (Unit of Experimental Neuropathology, Institut Pasteur, Paris, France) for her technical help on the histology part and the UtechS Photonic BioImaging (Imagopole), C2RT, Institut Pasteur, supported by the French National Research Agency (France BioImaging; ANR-10–INSB–04; Investments for the Future), both as a part of the FranceBioImaging infrastructure.
References
Footnotes
M-DG and ET are joint senior authors.
Contributors Study concept and design: LC, SC, M-DG, ET; experiments and acquisition of data: LC, SC, VM, KLL, JF, JZ, GJ, NM, DDSB, LF, ET; analysis of data and statistics: LC, SC, AD, M-DG, ET; patient data and samples collection: JT, MC, MMD; drafting of the manuscript: SC, M-DG, ET; critical revision of the manuscript: JT, MMD, HDR; reading and approval of the manuscript: all authors; study supervision and funding: MD-G, ET.
Funding This work was supported by funding from the Odyssey Reinsurance Company to ET, the West committee of the Ligue Nationale Contre le Cancer (LNCC) and the UMS Biosit to MDG. JT received financial support from Fondo de Investigacion en Salud, IMSS, Mexico (FIS/IMSS/PROT/PRIO/13/027). The LNCC provided a doctoral fellowship to LC and the Odyssey Reinsurance company financed JF postdoctoral fellowship. KLL is a doctoral fellow from the University Paris Diderot, Ecole Doctorale Bio Sorbonne Paris Cité (BioSPC).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Local Ethical Committees from the National Council for Research on Health, Instituto Mexicano del Seguro Social, Mexico and Florence University Hospital, Italy. The project was approved by the Comité d’Ethique en Expérimentation Animale (CETEA), Institut Pasteur (Ref 00317.02) and the Federative Structure of Research, Rennes (Ref APAFIS#905–2015060515515795 v4).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.