Article Text

Abstract
Coeliac disease (CD) is a frequent immune enteropathy induced by gluten in genetically predisposed individuals. Its pathogenesis has been extensively studied and CD has emerged as a model disease to decipher how the interplay between environmental and genetic factors can predispose to autoimmunity and promote lymphomagenesis. The keystone event is the activation of a gluten-specific immune response that is driven by molecular interactions between gluten, the indispensable environmental factor, HLA-DQ2/8, the main predisposing genetic factor and transglutaminase 2, the CD-specific autoantigen. The antigluten response is however not sufficient to induce epithelial damage which requires the activation of cytotoxic CD8+ intraepithelial lymphocytes (IEL). In a plausible scenario, cooperation between cytokines released by gluten-specific CD4+ T cells and interleukin-15 produced in excess in the coeliac gut, licenses the autoimmune-like attack of the gut epithelium, likely via sustained activation of the Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathway in IEL. Demonstration that lymphomas complicating CD arise from IEL that have acquired gain-of-function JAK1 or STAT3 mutations stresses the key role of this pathway and explains how gluten-driven chronic inflammation may promote this rare but most severe complication. If our understanding of CD pathogenesis has considerably progressed, several questions and challenges remain. One unsolved question concerns the considerable variability in disease penetrance, severity and presentation, pointing to the role of additional genetic and environmental factors that remain however uneasy to untangle and hierarchize. A current challenge is to transfer the considerable mechanistic insight gained into CD pathogenesis into benefits for the patients, notably to alleviate the gluten-free diet, a burden for many patients.
- gluten sensitive enteropathy
- autoimmunity
- coeliac disease
- lymphoma
- T lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
Gluten-specific CD4+ T cells play a driver role in coeliac disease (CD) pathogenesis and can persist for decades under gluten-free diet (GFD).
Flow cytometry detection of circulating gliadin-specific CD4+ T cells and induction of interleukin (IL)-2 in serum may provide useful biomarkers to detect a recall response after gluten challenge.
Gluten-specific CD4+ T cells cooperate with IL-15 produced by intestinal epithelial and myeloid cells to promote the activation of cytotoxic CD8+ T intraepithelial lymphocytes and license the autoimmune-like destruction of epithelial cells.
Most patients are cured by GFD; a small fraction of patients may develop refractory CD (RCD). The most severe cases are due to a low-grade intraepithelial lymphoproliferation called type 2 RCD (RCD2).
RCD2 malignant cells are characterised by somatic mutations, notably in the Janus kinase 1-signal transducer and activator of transcription 3 pathway, which give them a selective growth advantage in the cytokine-rich coeliac intestine.
Patients with RCD2 are at high risk of developing enteropathy-associated T cell lymphomas that share the same oncogenic mutations.
Besides HLA-DQ2/8, the essential genetic predisposing factor and gluten, the environmental driver, several genetic and environmental cofactors may contribute to considerable variability in disease presentation. The role of genetic variants controlling the immune response, of early childhood feeding practices, enteroviral infections and intestinal microbiota are under scrutiny.
A strict GFD remains the only treatment in uncomplicated CD but is difficult to maintain.
Several novel therapeutic approaches are under development to replace or, at least, alleviate diet.
Introduction
Coeliac disease (CD) can be defined as a chronic autoimmune-like enteropathy driven by an abnormal immune response to dietary gluten in genetically predisposed individuals.1 It is a common disease in countries consuming gluten where prevalence has increased over the past 50 years and is currently estimated between 0.5% and 1.5%.2 It can manifest at all ages in life by a variable spectrum of intestinal and extraintestinal symptoms that are the consequence of duodenal damage and/or immune activation.1 Diagnosis relies on the detection of serum IgA antibodies against the autoantigen tissue transglutaminase 2 (TG2), and, except in children with very high titres of IgA autoantibodies, on the demonstration of duodenal villous atrophy and increased number of intraepithelial lymphocytes (IEL).1 Most patients are cured by a strict life-long gluten-free diet (GFD). Yet, this diet is difficult to maintain and mucosal healing is delayed or remains incomplete in many patients.1 A very small number of adult patients can become truly refractory to GFD and develop either type 1 or type 2 refractory CD (RCD). While RCD1 may be due to a switch towards gluten-independent autoimmunity, RCD2 is a low-grade intraepithelial lymphoma that can frequently evolve into enteropathy-associated T cell lymphoma (EATL), a very rare but most severe complication of CD.1 3
Over the past 30 years, CD has thus emerged as a model disease to decipher how the interplay between environmental and genetic factors can predispose to autoimmunity and how chronic lymphocyte activation may ultimately lead to lymphomagenesis. The interactions between three actors unique to CD: gluten, the main environmental trigger, HLA-DQ2/8, the main genetic predisposing factor and TG2, the CD-specific autoantigen lead to the activation of gluten-specific CD4+ T cells.4 Their activation is indispensable to initiate CD but not sufficient to induce epithelial damage which depends on the activation of cytotoxic CD8+ IEL.4 Recent work indicates that IEL activation and tissue damage require the cooperation between cytokines released by gluten-specific CD4+ T cells and interleukin 15 (IL-15) produced in excess by epithelial and myeloid cells in active CD.5 6 The onset of RCD2 and EATL is likely driven by the chronic production of the same cytokines that promote the outgrowth of malignant IEL carrying somatic mutations that enhance their signalling pathway.7 8 If our understanding of CD pathogenesis has considerably progressed, several questions and challenges remain ahead. One unsolved question concerns the considerable variability in age at disease onset as well as in disease penetrance, severity and presentation.1 This variability, observed in many immune-mediated diseases, points to the role of additional genetic and environmental factors. Another important challenge ahead is to transfer the considerable mechanistic insight gained in CD pathogenesis into benefits for the patients. Several therapeutic avenues are currently explored in order to complement or to alleviate GFD and to treat RCD.
The driver role of the gluten-specific immune response
All studies converge to the driver role of gluten-specific CD4+ T cell in CD pathogenesis. The first clue was provided in the 1990s by demonstrating the link between CD and major histocompatibility complex (MHC) class II molecules.9 Thus, over 90% patients express HLA-DQ2.5 (DQA1*05-DQB1*02) with most remaining patients expressing HLA-DQ8 (DQA1*03-DQB1*03:02) or HLA-DQ2.2 (DQA1*02:01-DQB1*02).1 4 In 1993, the seminal observation of HLA-DQ2-restricted gluten-specific CD4+ T cells in the duodenal biopsies of patients with CD brought the first demonstration that HLA-DQ2 molecules, in keeping with their role in antigen presentation, could selectively present gluten peptides to CD4+ T cells.10 The next key finding was the identification of TG2 as the target of the CD-specific autoantibodies.11 It was soon recognised that this ubiquitous enzyme could bind and modify gluten peptides in a way that licenses their binding to HLA-DQ2/8 and the activation of CD4+ T cells12 13 (figure 1). Further studies have unravelled how the characteristics of gluten peptides allow their selective interactions with HLA-DQ2/8 and TG2.

Schematic representation of the driver antigluten response. The richness of gluten proteins in proline and glutamine underlies their ‘toxicity’ for patients with coeliac disease. Due to the lack of luminal endo-prolylpeptidases, proline-rich gluten proteins are incompletely digested and release large immunogenic peptides which can be modified by transglutaminase 2 (TG2). This enzyme selectively targets a subset of proline-rich and glutamine-rich sequences in gluten peptides and deamidate neutral glutamine residues (Q) into negatively charged glutamic acid (E). Gluten peptides harbouring negative charges on specific positions can bind with high avidity the peptide pockets of HLA-DQ2 or HLA-DQ8 molecules at the surface of antigen-presenting cells. The latter cells can then efficiently stimulate activation and expansion of gluten-specific CD4+ T cells that produce proinflammatory cytokines such as interleukin (IL)-2, interferon gamma (IFNγ) and IL-21. Created with Biorender.com.
Gluten, the viscoelastic blend obtained by mixing flours from wheat, barley or rye with water, comprises hundreds of proteins of the prolamin family. These proteins (eg, gliadins and glutenins in wheat) contain repeated sequences rich in proline and glutamine that underlie their pathogenicity in CD. Since luminal enzymes lack endoprolylpeptidase activity, digestion of proline-rich proteins is incomplete and releases large immunogenic peptides,14 which can reach the subepithelial tissue. Proline-rich and glutamine-rich sequences in gluten peptides are also privileged targets for TG2, which can deamidate neutral glutamine residues into negatively charged glutamic acid.12 13 The negative charges introduced by this enzymatic modification allow the formation of stable HLA-DQ-gluten complexes at the surface of antigen-presenting cells and thereby the efficient activation of gluten-specific CD4+ T cells.15 Following their priming in gut-associated lymphoid tissues, gluten-specific CD4+ T cells can migrate into the intestinal lamina propria and produce proinflammatory cytokines such as interferon gamma (IFNγ), IL-214 and, likely, IL-216 when exposed to gluten peptides (figures 1 and 2). The repertoire of gluten peptides that can be modified by TG2 and elicit T cell responses has been extensively characterised. Most patients with CD respond to a limited and shared set of peptides defined as immunodominant epitopes.15 A larger number of these epitopes are recognised in the context of HLA-DQ2.5 than of HLA-DQ2.2 and HLA-DQ8, likely accounting for the preferential association of CD with HLA-DQ2.5. The much higher risk of CD in individuals homozygous for HLA-DQ2.5 further suggests a quantitative model, in which disease development depends on the amplitude of the gluten-specific CD4+ T cell response.15 17

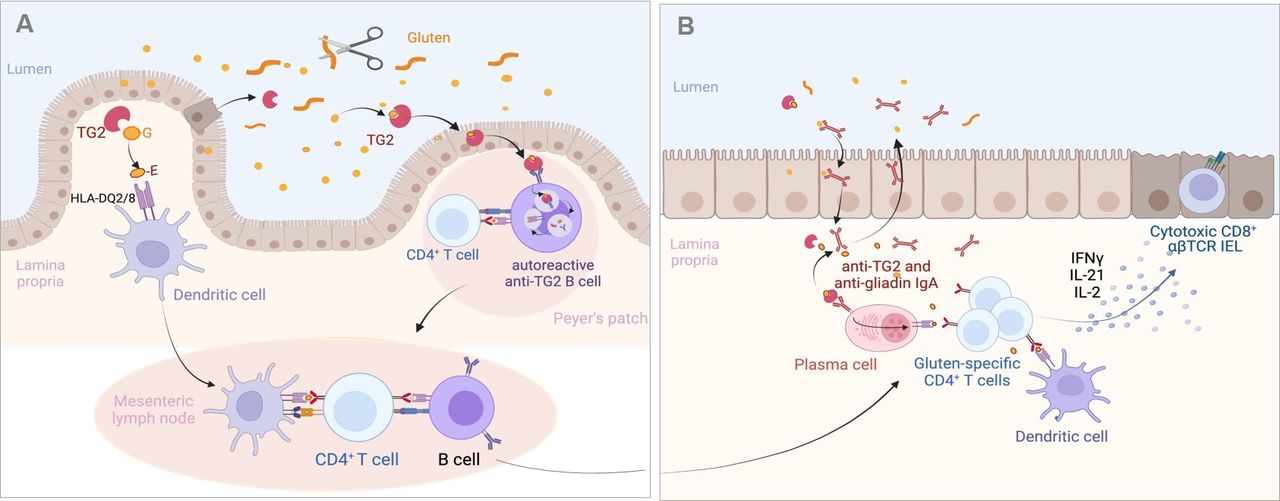
Proposed mechanisms of the antigluten and antitransglutaminase 2 (TG2) responses in the coeliac intestine. (A) Initiation of the adaptive immune response against gluten and TG2. Gluten peptides left undigested are deamidated by TG2 either in the intestinal lumen where TG2 can be released by extruded dead enterocytes or after crossing the epithelium by TG2 liberated in the extracellular matrix. Deamidated peptides bind HLA-DQ2/8 molecules at the surface of dendritic cells in Peyer’s patches (not shown) or in lamina propria. Dendritic cells can then initiate the activation of gluten-specific T cells in the Peyer’s patches (not shown) or after migration in the mesenteric lymph nodes. Gluten-specific CD4+ T cells cooperate with naïve antigluten B cells to induce the antigluten humoral response. Alternatively, TG2-gluten complexes may cross the Peyer’s patch epithelium and bind to the B cell receptor of autoreactive anti-TG2 B cells present in the lymphoid follicle. TG2-gluten complexes bound to the B cell receptor are endocytosed, processed in B cell endolysosomal compartments, where released gluten peptides can be loaded on HLA-DQ molecules. HLA-DQ-gluten complexes are then translocated at the surface of autoreactive B cells, allowing the activation of gluten-specific CD4+ T cells and simultaneously cooperative interactions that license the IgA response against TG2. (B) Activation of the effector antigliadin CD4+ T cell response. Following their activation in gut lymphoid tissues and mesenteric lymph nodes, gluten-specific effector CD4+ T cells, antigluten and anti-TG2 IgA plasma blasts migrate into the lamina propria. On re-encounter with HLA-DQ-gluten complexes displayed by dendritic cells and by anti-TG2 plasma cells, effector CD4+ T cells secrete cytokines and notably interferon gamma (IFNγ), interleukin (IL)-21 and IL-2, which participate in the activation of CD8+ cytotoxic T intraepithelial lymphocytes (IEL). Plasma cells produce IgA antibodies against gluten and TG2 antibodies that are transported into the intestinal lumen. IgA-gluten and IgA-TG2-gluten complexes may be retrotransported across the epithelium and further foster the antigluten response. Created with Biorender.com.
Many efforts have recently aimed at monitoring this response in vivo. One specific method relies on the use of cocktails of fluorescent tetramers made of recombinant HLA-DQ2.5 molecules linked to the main immunodominant peptides. These soluble fluorescent complexes selectively label cognate gluten-specific CD4+ T cells, which can be then quantified by flow cytometry or sorted for further characterisation.18 These approaches have allowed in-depth analysis of the T cell repertoire of gliadin-specific CD4+ T cells19 as well as of their phenotype and dynamics.20 21 Strikingly, tetramer+ cells are undetectable in healthy individuals but they expand in the lamina propria of untreated CD20 and circulate in small numbers in blood. Their number decreases after GFD but some can persist for decades.20 Although it cannot be excluded that occasional intake of small amounts of gluten may foster the persistence of gluten-specific memory T cells, these findings support the need for a life-long GFD in order to prevent relapse. Moreover, a peak of tetramer+ cells is detected in the blood 6 days after oral gluten challenge in most treated patients with CD,20 22 likely reflecting the transient circulation of gluten-specific T cells on re-stimulation in the gut. The possibility to demonstrate a gluten-specific immune response in the blood after oral challenge is of practical interest. It may allow CD diagnosis in patients who have initiated GFD without prior testing for serum anti-TG2 antibodies. It may also be useful to monitor therapeutic trials.23 Since tetramers are expensive and only reveal a fraction of gluten-reactive CD4+ T cells, other methods have been or are being developed. One method consists in the detection of CD4+ T cells producing IFNγ after in vitro re-exposure to gluten.24 A newer promising method derives from the extensive phenotypic characterisation of tetramer+ cells. This analysis has identified a combination of 7–11 membrane markers that can be used for detecting gliadin-specific CD4+ T cells.25 26 Finally, the peak of IL-2 released in the serum 4 hours after oral gluten challenge in treated CD might be a useful biomarker to detect the recall response of gluten-specific CD4+ T cells residing in the gut.27 This peak of IL-2 correlates with the early onset of symptoms such as nausea which can occur rapidly after gluten consumption.27
Despite considerable progresses in our understanding of the anti-gluten CD4+ T cell response, several questions are not fully solved. First, how gluten peptides cross the intestinal epithelium is unclear. It is a significant issue given the proposition to use of larazotide, a drug that presumably blocks the paracellular pathway, to treat CD.28 Besides doubts on the mechanism of action of larazotide,29 it is important to stress that only the transcellular passage of gluten peptides through the endocytic pathway has been formally demonstrated in active CD.30 This passage may be facilitated by the retro-transcytosis of IgA-gliadin complexes formed in the duodenal lumen31 (figure 2B). A second question concerns the site where TG2 encounters gluten and engages the immune system. The answer may help optimising delivery of anti-TG2 blockers, which have shown promising results in a recent clinical trial.32 The deamidating function of TG2 requires Ca2+ concentrations found in the extracellular milieu. The common assumption is that gluten peptides meet catalytically active TG2 in the extracellular matrix beneath the gut epithelium (figure 2A). Alternatively, recent evidence indicates that TG2 is released by enterocytes shed in the intestinal lumen where the enzyme can bind and modify gluten peptides. TG2-gluten complexes may then be endocytosed by the epithelium covering Peyer’s patches where they can prime anti-gluten and anti-TG2 immune responses33 (figure 2B). Finally, a third question concerns the autoreactive immune anti-TG2 response. Recent work suggests that autoreactive anti-TG2 B cells play an instrumental role in the activation of gluten-specific CD4+ T cells, and that, in turn, the latter cells can provide help to the anti-TG2 B cells and license their production of auto-antibodies. Accordingly, Iversen et al have shown that, in vitro, anti-TG2 B cells incubated with TG2-gluten complexes can activate gluten-specific CD4+ T cells.34 Conversely, du Pré et al have demonstrated in an elegant mouse model that anti-TG2 autoreactive B cells were present in Peyer’s patches but remained quiescent due the lack of T cell help. Their activation could however be achieved in the presence of gluten-specific CD4+ T cells and of TG2-gluten complexes.35 A plausible scenario illustrating how TG2-gluten complexes that have crossed the epithelium covering the Peyer’s patches may initiate simultaneously the gluten-specific anti-CD4+ T cell response and the activation of anti-TG2 B cells is shown in figure 2A. Work by Hoydahl et al further suggests that autoreactive anti-TG2 IgA plasma cells, which represent 10%–20% of intestinal plasma cells during active CD, may also present gluten peptides to the CD4+ T cells that have migrated into lamina propria following their priming in Peyer’s patches or mesenteric lymph nodes (figure 2).36 37 Given the high number of anti-TG2 plasma cells, this mechanism may potently amplify the activation of effector gluten-specific CD4+ T cells and their production of cytokines. Comforting an important role of B cells in CD pathogenesis, in vivo elimination of B cells by anti-CD20 antibody attenuated villous atrophy in a CD mouse model of CD.38 Since the autoantibodies do not seem to induce intestinal damage in vivo,39 the most likely hypothesis to date is indeed that autoreactive B cells are instrumental for gluten presentation to T cells. Overall, these data explain why anti-TG2 antibodies are exclusively present in patients exposed to gluten and why these antibodies represent a highly reliable biomarker to monitor the antigluten response.
In conclusion, the adaptive immune response against gluten is now well characterised and drives disease pathogenesis. Yet, gluten-specific CD4+ T cells do not mediate directly tissue damage. Rather, they act by promoting expansion and activation of cytotoxic CD8+ T IEL that execute the autoimmune-like destruction of epithelial cells.
Intraepithelial lymphocytes, interleukin-15 and epithelial damage in active coeliac disease
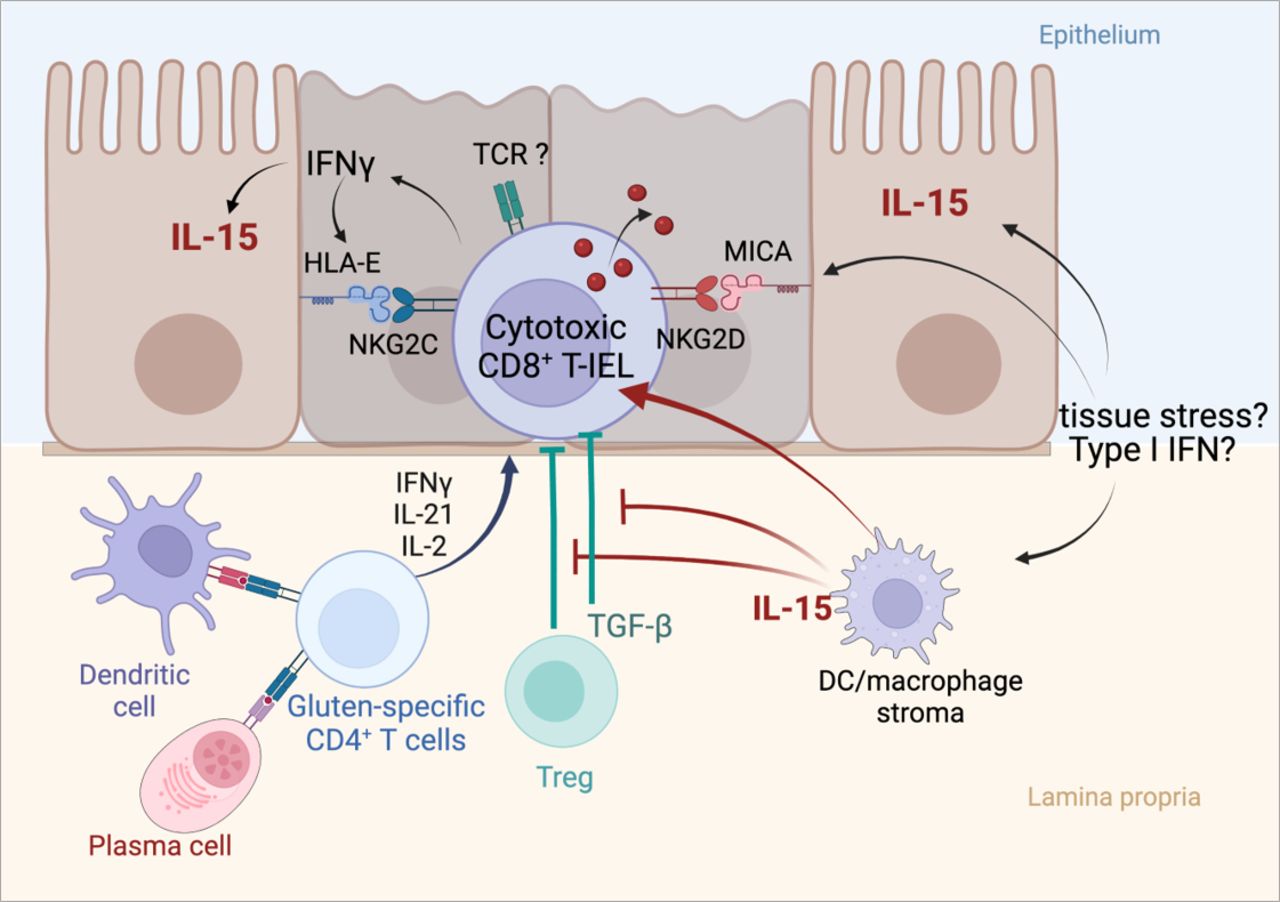
Ferguson and Murray were the first to attract attention on IEL in CD when they reported their massive expansion in untreated CD.40 There is now compelling evidence that CD8+ T IEL with an αβ T cell receptor (TCR), the major subset of IEL in the normal small intestine, play an instrumental role in epithelial damage.4 Thus, in untreated CD, CD8+ T IEL display all the phenotypic and functional characteristics of activated cytotoxic T cells. They notably synthesise large amounts of IFNγ and granzyme B and upregulate NKG2D and CD94/NKG2C, two surface receptors of the natural killer receptor (NKR) family. The latter receptors are thought to facilitate the cytolytic attack of enterocytes on interaction with their ligands, two unconventional MHC class I molecules that are upregulated in the gut epithelium in untreated CD4 41 42 (figure 3). The mechanisms that initiate IEL activation are however not definitively solved. Many data suggest the role of cooperative interactions between cytokines produced by gluten-specific CD4+ T cells and a distinct cytokine, IL-15, that is produced by intestinal epithelial and myeloid cells and is upregulated in untreated CD.43 In vitro, IL-15 can largely recapitulate the state of activation that characterises CD8+ T IEL in untreated CD. Thus, IL-15 induces the expansion of CD8+ T IEL, stimulates their expression of IFNγ, granzyme B and NKG2D and ultimately promotes the cytotoxicity of IEL in response to various signals.41 42 44–46 Moreover, IL-15 impairs immunosuppression of cytotoxic CD8+ T IEL by transforming growth factor beta (TGF-β)47 and by regulatory FOXP3 T cells,48 two important regulatory mechanisms (figure 3). Accordingly, spontaneous enteropathy with expansion of cytotoxic CD8+ T IEL has been observed in transgenic mice engineered to release massive amounts of IL-15 from the gut epithelium.49 However, in mice where IL-15 transgenic expression is weaker, complementary signals derived from CD4+ T cells are necessary to license the cytolytic attack of the gut epithelium. This cooperative role has been demonstrated in an elegant mouse model humanised for HLA-DQ8, which developed gluten-dependent villous atrophy on overexpression of IL-15 in enterocytes and in dendritic cells.5 Importantly, epithelial damage was reduced by depleting either CD8+ or CD4+ T cells and by blocking IFNγ and at a lesser degree IL-21, indicating a contribution of gluten-specific CD4+ T cells via their production cytokines.5 In a distinct model using ovalbumin as model dietary antigen, it was shown that cooperative interactions between ovalbumin-specific CD4+ T cells and IL-15 overexpressed in the intestine were necessary to induce the activation of cytotoxic CD8+ T IEL and epithelial damage on feeding by ovalbumin.6 Strikingly, the cooperative effect of CD4+ T cells could be fully recapitulated in vitro as well as in vivo by IL-2 that was secreted by CD4+ T cells in response to ovalbumin.6

Activation of cytotoxic intraepithelial lymphocytes and induction of tissue damage in active coeliac disease (CD). Cooperation between the cytokines released by activated gluten-specific CD4+ T cells and interleukin (IL)-15 produced by epithelial and myeloid cells drive the activation of cytotoxic CD8+ T intraepithelial lymphocytes (IEL) and license epithelial cell killing. Cytotoxic CD8+ T IEL synthesise interferon gamma (IFNγ) and cytotoxic molecules such as granzyme B and perforin, upregulate the activating natural killer receptor NKG2D and CD94/NKG2C, thereby facilitating the interactions of cytotoxic CD8+ T IEL with enterocytes which, in active CD, display MICA (MHC class I polypeptide-related sequence A), the ligand of NKG2D (induced by tissue stress) and HLA-E, the ligand of NKG2C (induced by IFNγ). DC, dendritic cell; TCR, T cell receptor; TGF-β, transforming growth factor beta; Treg, regulatory T cell. Created with Biorender.com.
Overall, these results provide a plausible scenario for epithelial damage in CD (figure 3). Yet, several questions remain. First, it is still uncertain whether the cytotoxicity of IEL against enterocytes is totally independent of signals via their TCR. Direct recognition of gluten peptides seems unlikely.4 6 Their recognition of endogenous antigens is however not excluded. Second, it is unclear if IL-15 is upregulated in all patients with CD. Unlike most cytokines, IL-15 is poorly secreted and remains sequestered at the surface of producing cells bound to the α chain of its receptor (IL-15 Rα). Its detection in the serum and in tissues is challenging, perhaps explaining discrepancies.50 Third, the mechanism(s) underlying IL-15 upregulation of IL-15 in CD remains elusive. An inducing effect of the p31-43 gluten peptide common to the N-terminus of α-gliadins has been suggested.45 Despite efforts to gain insight into the properties of this peptide,51 52 this role awaits further demonstration. A simpler hypothesis might be the upregulation of IL-15 by IFNs. Thus, both type I IFNs and IFNγ can stimulate the expression of IL-15-IL-15Rα complexes at the surface of epithelial cells where these complexes can activate nearby lymphocytes.53 Type I IFNs are induced by viruses and, as discussed below, enterovirus infections are a suspected trigger of CD. Moreover, IFNγ and IL-15 may create a positive feedback loop amplifying their reciprocal production and the activation of IEL (figure 3). Finally, another question concerns the role of IL-2 in the cooperative interaction of CD4+ T cells with IL-15. Several clues support this role. IL-2 is the main cytokine detected in the serum of patients on GFD after oral challenge.27 IL-2 binds to the same signalling module as IL-15 on CD8+ T cells and induces a largely overlapping activation programme.54 Moreover, the capacity of low doses of IL-2 to stimulate the expansion of cytotoxic CD8+ T cells in vivo in humans was inadvertently demonstrated during a recent clinical trial in type I diabetes.55
In conclusion, current data converge to indicate that, in untreated CD, the autoimmune-like attack of the gut epithelium by CD8+ T IEL is orchestrated by the three cytokines (IFNγ, IL-21 and IL-2) produced by gluten-specific CD4+ T cells and by IL-15 (figure 3). One major pathway collectively activated by these cytokines is the Janus kinase 1-signal transducer and activator of transcription 3 (JAK1-STAT3) pathway. In keeping with a key role of this pathway, CD8+ T IEL activated in CD share many overlapping features with the CD8+ T cells driving autoimmunity in patients and mice carrying constitutive STAT3 gain-of-function (GOF) mutations.56 The importance the JAK1-STAT3 pathway in CD pathogenesis is further illustrated by its central role in the onset of lymphomas complicating CD (see below).
While effector cytotoxic CD8+ T IEL are the main culprit in CD, two other subsets of lymphocytes have recently attracted attention in CD. This is the first case of TCRγδ+IEL, which represents 10%–15% IEL in the normal adult small intestine and which expand in CD either untreated or treated.57 Recent elegant work has shown how chronic inflammation may jeopardise the epithelial niche of resident TCRγδ+IEL and promote their replacement by newly recruited TCRγδ+IEL, which produce IFNγ and may contribute to tissue damage in untreated CD.58 Why their expansion persists durably after GFD is however not known. A second intriguing subset revealed in a very recent article are regulatory CD8+ T cells. These cells can be identified by their surface expression of KIR3DL1 and KIR2DL3, two NKR distinct of those involved in epithelial damage. They were shown to expand in untreated CD and to kill gluten-specific CD4+ T cells at least in vitro.59 Whether and when these cells, which also need IL-15 for their expansion, can effectively restrain the activation of gluten-specific CD4+ T cells in vivo remains to be defined.
Refractory coeliac disease: from chronic inflammation to autoimmunity and lymphomagenesis
Most patients with CD are cured by GFD, which switches off immune activation, allowing mucosal healing and clinical improvement. Mucosal healing may take however 1–2 years in many adults and persistent villus atrophy is frequent, mainly due to inadvertent intake of gluten. Indeed, as little as 50–100 mg/day of gluten may trigger epithelial damage in patients with CD and traces of gluten are present in most processed foods.1 Overall villus atrophy truly unresponsive to GFD, which defines RCD, is rare with a prevalence estimated at 0.3%–0.5% among patients with CD. RCD can develop immediately after first diagnosis or after a period of response to GFD, notably in patients with a history of dietary transgression.1 3
Depending on the characteristics of the IEL, two types of RCDs have been individualised. In RCD1, IEL display a phenotype (mainly CD8+ TcRαβ+) comparable to that observed in uncomplicated CD and they remain polyclonal. To date, no biological or immunological marker allows differentiating RCD1 from active CD and the mechanism underlying resistance remains elusive.3 Chronic intestinal inflammation may perhaps promote the development of autoreactive CD8+ T IEL. Indeed, substantial numbers of autoreactive CD8+ T cells circulate in the normal human blood,60 61 where they remain generally quiescent. Yet, they can be recruited and activated in inflamed tissues and cause tissue damage, as shown in type I diabetes.61 62 In line with this hypothesis, extraintestinal autoimmunity is more frequent in RCD1 than in uncomplicated CD (30% vs 15%–20%)63 (G. Malamut, unpublished observations) and patients are improved by oral budesonide or immunosuppressive drugs.1 3 RCD1 prognosis is good with over 90% survival at 5 years and limited risk of lymphoma.64 65
RCD2 is a much more severe condition often associated with profound malnutrition and hypoalbuminemia. In 60% of cases, patients display ulcerative jejunitis, a characteristic endoscopic feature also reported in patients developing EATL.64 65 This invasive lymphoma is very rare (see below) but remains the most severe complication of CD with an overall survival of <15% at 5 years.66 In fact, RCD2 can be defined as an intraepithelial lymphoma and the frequent first step to EATL, as 40% of patients develop an EATL within 5–10 years after RCD2 diagnosis.3 64 65 In contrast with malignant EATL cells, which are actively dividing medium-sized to large cells, RCD2 IEL are non-dividing small lymphocytes with a normal cytological appearance. Accordingly, they cannot be identified by routine histology and diagnosis of RCD2 relies on a combination of immunohistochemistry, flow cytometry and molecular biology approaches in order to demonstrate the distinctive characteristics of IEL.3 Typically, RCD2 IEL lack surface expression of CD3, of TCR and generally of CD8 but they contain intracellular CD3ε; they express NKR, notably NKG2D and NKP46. In addition, they contain clonal TCRγ rearrangements that be detected in DNA extracted from biopsies.3 67 68 It has often been suggested that RCD2 IEL derived from the malignant transformation of the CD8+ TcRαβ+IEL that expand in CD. We have shown that it is not case and that they generally arise from a small subset of IL-15-dependent innate-like lymphocytes with dual NK and T cell traits that are present and develop in the normal gut epithelium7 (figure 4A). In keeping with their NK features, RCD2 IEL can exert NK-like cytotoxicity against enterocytes in vitro. This killer function is boosted in the presence of IL-15 and depends notably on NKG2D and CD103.43 45 As IL-15 and MICA, the ligand of NKG2D, are upregulated in the gut epithelium in RCD2, it is likely that RCD2 IEL can attack the gut epithelium and provoke the severe epithelial lesions associated with this condition43 45 (figure 4A).
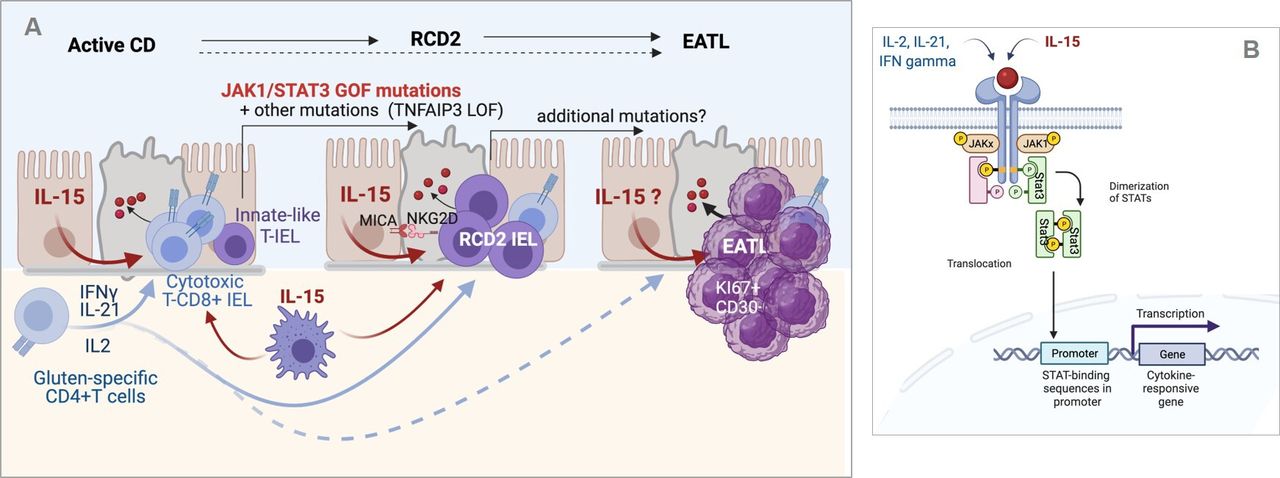
Inflammation-driven lymphomagenesis in coeliac disease (CD). (A) Left panel: in active CD, cytokines produced by gluten-specific CD4+ T cells cooperate with interleukin (IL)-15 to stimulate the expansion and cytotoxic activation of polyclonal CD8+ T intraepithelial lymphocytes (IEL) that drive a cytolytic attack of epithelium. Middle panel: in type 2 refractory CD (RCD2), innate-like T IEL that have acquired somatic mutations and notably gain-of-function (GOF) mutations in Janus kinase 1 (JAK1) or signal transducer and activator of transcription 3 (STAT3) can clonally expand and outcompete CD8+ T IEL in the cytokine-rich CD intestine. Transformation is fostered by the acquisition of additional mutations, notably in TNFAIP/A20, which enhances the nuclear factor kappa B pathway, and in epigenetic regulators. Due to their natural killer (NK)-like functions, RCD2 IEL can induce severe epithelial lesions. Right panel: further accumulation of mutations in the clonally expanded RCD2 IEL ultimately lead to enteropathy-associated T cell lymphoma (EATL). While EATL generally develops from RCD2, it can also develop without a prior step of RCD2. (B) Schematic representation of the JAK1-STAT3 pathway. Its activation both by cytokines produced by CD4+ T cells and IL-15 explains its importance in the activation of normal CD8+ T IEL and RCD2 IEL. Created with Biorender.com.
The malignant nature of RCD2 IEL has been disputed. Several clues support however this assumption. First, RCD2 IEL can progressively replace normal T IEL all along the GI tract; they may also disseminate into the lamina propria and blood and finally within different organs such as the liver, the skin and the lungs.69 Second, when EATL complicate RCD2, malignant cells share the same clonal TCR rearrangement indicating their origin from a common clonal precursor.65 70 Third, RCD2 IEL contain a broad spectrum of somatic mutations and chromosomal abnormalities that are recurrent between patients and largely overlapping with those found in EATL whether or not this lymphoma develops de novo in CD or after RCD28 (figure 4A). Overall, these mutations point to a common mechanism of lymphomagenesis driven by chronic inflammation. Thus, over 80%–90% of RCD2 and EATL display GOF mutations in the JAK1-STAT3 pathway, a major signalling pathway downstream IFNγ, IL-2, IL-6, IL-21 and IL-15 (figure 4B). Mutations in STAT3 are present in 40% of RCD2 and EATL. As in many other cancers, they are predominantly found in the SH2 regulatory domain, where they enhance STAT3 activation. JAK1 mutations are present in 50% of RCD2 and EATL and cluster in p.1097 in the kinase domain, a highly conserved position and the site of interaction of JAK1 with SOSC1, one major negative regulator of the JAK1-STAT3 pathway. Strikingly, loss-of-function (LOF) SOCS1 mutations are observed in some rare cases without STAT3 or JAK1 mutations.8 The JAK1-STAT3 signalling cascade has been extensively characterised and is known to switch on a broad transcriptional programme that induces lymphocyte activation, survival and proliferation.71 JAK1 and STAT3 GOF mutations may thus provide malignant IEL with a selective advantage to outcompete normal lymphocytes in the inflamed gut intestine. Accordingly, RCD2 IEL show enhanced in vitro responses to IL-15 compared with normal IEL.7 8 In a substantial number of cases, this advantage is likely reinforced by LOF mutations in TNFAIP3/A20, a potent negative regulator of the nuclear factor kappa B pathway, which is activated by other inflammatory cytokines, such as tumor necrosis factor α or IL-1.8 72 Importantly, RCD2 IEL carry many additional oncogenic mutations such as recurrent LOF mutations in the DNA methylase TET273 and in the histone-methyl-transferase KMT2D,74 which may foster transformation and ultimately lead to invasive EATL8 (figure 4A).
Overall, recent insight in the oncogenic landscape of RCD2 and EATL authenticates the link between gluten-driven chronic inflammation and the onset of intestinal lymphomas. This hypothesis was first suggested in 1967 by Harris et al who reported 13 intestinal lymphomas in 202 adult patients with CD, who had been followed up between 1941 and 1965 in Birmingham.75 Later follow-up of the ‘Birmingham’ cohort until 1985 led Holmes et al to conclude to a 43-fold increase in the risk of lymphomas in CD. Key observations were the long delay (20 years) between diagnosis of CD and onset of lymphoma and the protective role of the GFD. Thus, no patient developed a lymphoma after 5 years on GFD.76 The protective role of GFD may explain the much lower incidence of lymphomas since GFD is the rule. Thus, the increase in relative risk was only 3 in the last meta-analysis published in 2005.77
Additional factors and environmental triggers in coeliac disease
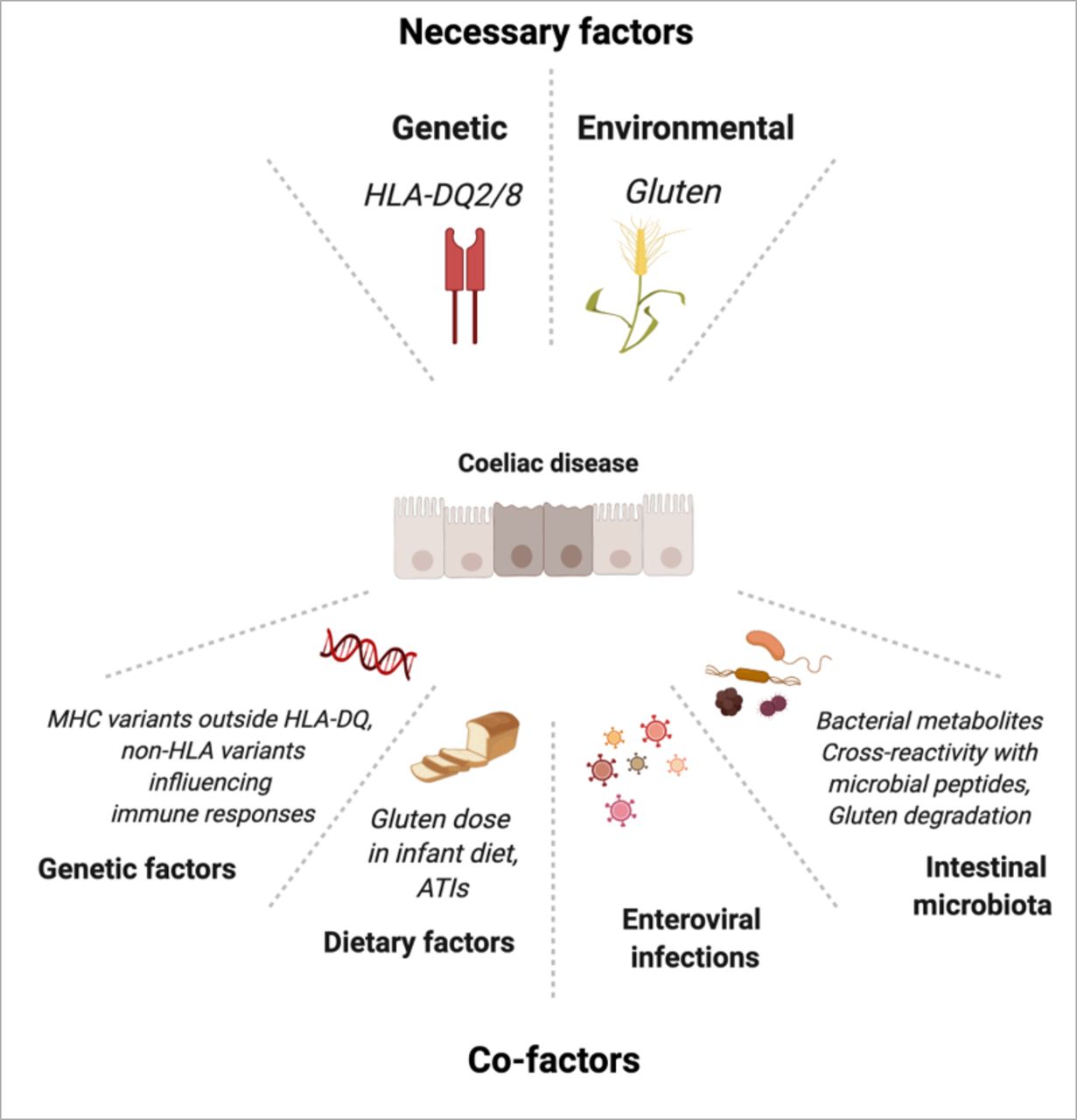
If the pathogenesis of CD is overall well understood, many interrogations remain on the additional genetic and environmental factors that may explain the considerable variability in age at disease onset as well as in disease penetrance and severity (figure 5).

Genetic and environmental factors in coeliac disease. ATI, amylase trypsin inhibitor; MHC, major histocompatibility complex. Created with Biorender.com.
Genetic factors
The concordance of about 75% between monozygotic twins vs 12% in monozygotic twins stresses the importance of genetic factors.78 HLA-DQ2/8, the major risk factor is generally considered to account for 35% of the genetic predisposition.79 A recent very large genome-wide association study (GWAS) suggests however a lesser contribution of 23% when calculations are based on a CD prevalence of 1%.80 Importantly, there is a dose effect as individuals homozygous for HLA-DQ2 have a higher risk to develop CD81 and to progress to RCD. Thus, HLA-DQ2 homozygosity has been reported in 40% of RCD1 and 66% of RCD265 compared with 25%–30% in uncomplicated CD and 5% in controls.81 As discussed above, the higher density of HLA-DQ2 molecules on antigen-presenting cells of homozygous individuals can increase the amplitude of the antigluten CD4+ T cell response15 17 and explain this dose effect. Over the past 15 years, GWAS have attempted to identify other genes that may account for the remaining inheritability. Fine mapping of the MHC region outside HLA-DQA1-DQB1 has revealed five new variants including two in the HLA-B region. Initial estimation that an additional 18% of CD heritability may be explained by these variants,80 has been revised and calculated to be only 2.3%.82 A possible influence of the HLA-B variants in the activation of CD8+ IEL was suggested, but functional data are lacking. Other analyses have identified 57 common variants in 39 non-HLA-regions.83 Supporting the link of CD with autoimmunity, they largely overlap with variants predisposing to other autoimmune diseases.84 Yet, altogether they account for no more than 15% of the heritability. The individual contribution is thus very weak and, since over 95% are located in intronic non-coding regions, their functional role remains largely elusive despite elegant system biology analyses that suggest their influence on the transcription of immune genes.85 86 In line with this hypothesis, it is interesting that some variants associated with CD may impact the expression of genes such as CTLA-4, PTPN2 and SOCS1, in which monoallelic LOF mutations can cause severe autoimmune enteropathies.87 It is also notable that CD has been observed in several patients with IPO8 deficiency, a rare disease impairing signalling downstream TGF-β, supporting the suggested immunoregulatory role of this cytokine in CD.47 88 GWAS remains difficult in RCD due to the small size of the cohorts. One study in patients with RCD2 from Dutch and French origins has pointed to a predisposing genetic locus in 7p14.3 (p=2.37 Å~10−8, OR=2.36), which may control expression of FAM188B.89 The exact contribution of this variant to CD-associated lymphomagenesis remains elusive but, interestingly, FAM188B was recently implicated in the regulation of P53, a central player in tumour surveillance.90 In conclusion, despite considerable efforts, genetic heritability outside HLA-DQ remains difficult to apprehend and it is still uncertain whether and how non-HLA common variants may become used to predict CD onset or severity.
Environmental triggers
Exposure to gluten is necessary but not sufficient in many genetically predisposed patients to trigger CD. Both dietary and microbial factors likely contribute to promote or, on the contrary, to protect against the break of tolerance to gluten.
In children, considerable attention has been paid to feeding practices. Recent meta-analyses conclude that neither breast feeding nor the time of gluten introduction significantly influence CD development in children at risk.91 In contrast, three observational studies showed a significant impact of the cumulative amount of gluten uptake between 1 and 5 years in CD in genetically predisposed children.92–94 Complementary events may however be needed. Thus, in the PREVENTCD cohort, children who became coeliac before 6 years had higher serum concentrations of inflammatory cytokines at 4 months before being exposed to gluten than at-risk children who remained disease-free.94 Moreover, positive interaction between cumulative intake of gluten at 2 years and cumulative exposure to enteroviral infections was observed in another birth cohort.95
Another unsolved question concerns the role of gluten peptides not recognised by CD4+ T cells or of non-gluten wheat components. A proinflammatory role of the peptide 31–43 common to the N-terminus of A gliadins has been suggested. Despite evidence that this peptide may alter intracellular trafficking in epithelial cells,51 52 its role remains unclear. Other studies have put forward the role of amylase trypsin inhibitors (ATIs) that are present in the non-gluten fraction of wheat. Thus, ATIs can activate toll-like receptor 4 in myeloid cells,96 97 induce barrier dysfunction and IL-15-dependent increase in IEL in B6 mice, exacerbate gluten-induced immunopathology and promote dysbiosis in non-obese diabetic HLA-DQ8 humanised mice.98 Whether contamination by ATIs may have increased in commercial gluten preparations and contribute to promote CD remains however unknown.
The role of GI infections as a trigger of CD is a long-lasting hypothesis first put forward by M. Kagnoff, who suggested mimicry between A-gliadins and adenovirus.99 Association studies did not confirm this hypothesis but they have suggested the favouring role of repeated rotavirus infections in children.100 The favouring role of GI infections101 and notably of enteroviral infections95 102 103 has been confirmed in several birth cohorts of children at risk, although none of them could conclude on the respective role of individual viruses. A contribution of enterovirus is supported by studies in humanised HLA-DQ8 mice. In these mice, intestinal infection by the T1L reovirus prevented systemic tolerance induced by oral feeding with gluten. Loss of tolerance was ascribed to the induction of type I IFN, which promoted the activation of an antigluten TH1 response and the appearance of anti-TG2 antibodies.104 Yet, mice did not develop villous atrophy, indicating that the T1L reovirus was not sufficient to trigger disease. Identification of the enterovirus(es) that promote CD onset may open prevention by vaccination. Thus, protection by the rotavirus vaccine has been observed in the TEDDY birth cohort, notably in at risk children exposed to gluten before 6 months.101 Protection was also reported in a general population study after a randomised placebo-controlled trial led in Finland in 2006.105 Such protection was however not confirmed in a very large general practice cohort of children followed between the age of 6 months and 7 years and the years 2010–2015 in the UK.106
Besides possible infectious triggers, a strong focus has recently been made on the microbiota. It seems indeed possible that recent changes in the microbiota might have contributed to the twofold to threefold increase in CD prevalence observed over the past 50 years.2 107 Numerous observational studies have documented differences in the microbiota of patients with CD compared with controls, yet with little consistency except for an increase in proteobacteria and a decrease in species associated with anti-inflammatory properties, a finding reported in many chronic inflammatory intestinal conditions.79 Interestingly however, several studies have provided mechanistic insight into the potential beneficial versus deleterious effects of the microbiota in CD.108 In keeping with findings in IBDs,109 Lamas et al observed that tryptophan metabolites were decreased in the faeces of patients with active CD.110 Since supplementation by Lactobacillus reuteri and a diet enriched in tryptophan could reduce gluten-induced small intestinal immunopathology in the HLA-DQ8 mouse model, the authors suggested that tryptophan metabolism might be a marker of dysbiosis in CD and a therapeutic target.110 Other studies by the group of E. Verdu have shown how microbes may diversely influence gluten degradation in the intestinal lumen and either promote the release of highly immunogenic peptides (eg, Pseudomonas aeruginosa) or, on the contrary, reduce their luminal concentration (Lactobacillus spp).111 Finally, and in line with observations made in other immune conditions, Petersen et al have used a combination of sequence homology search, functional and structural data in order to demonstrate molecular mimicry between two immunodominant DQ2.5-restricted α-gliadin T cell epitopes and a spectrum of microbial peptides derived from commensal and pathogenic bacteria.112 Of note, a large set of bacterial mimics were derived from Pseudomonas fluorescens, against which 86% of patients with CD display positive serology.113 In addition, most of the selected candidate peptides could efficiently activate patient-derived T cell clones. Yet, some of the latter clones only recognised the gliadin epitopes. Moreover, only a minority of patients orally challenged with gluten developed a gliadin-specific T cell response that cross-reacted with the bacterial mimic epitopes. The authors therefore suggested that a pathogen or a commensal bacterium might trigger a T cell response, that cross-reacted with gluten epitopes and was then relayed by gluten-driven expansion of a cross-reactive T cell repertoire with epitope spreading.112 Yet, given the degeneracy of T cell recognition, it is not excluded that T cells activated by gluten may randomly cross-react with bacterial peptides. The fact that CD subsides after GFD further suggests that the presence of bacteria carrying the mimic peptides is not sufficient to sustain inflammation. Along the same line, titres of antibodies against the P. fluorescens-specific I2 sequence were comparable in patients with CD responsive or not to GFD, pleading against the idea that the antibacterial response may sustain tissue damage.114 Altogether, these studies support the hypothesis that intestinal microbiota may play a role in disease onset or severity. Yet, more work is needed to define their exact contribution and whether it may be targeted for improving the burden of the diet or for disease prevention.
Coeliac disease: from pathogenesis to therapy
A strict GFD remains the only treatment of CD. Yet, as discussed above, this diet is difficult to maintain. Therefore, considerable efforts have been undertaken to identify treatments that may replace or, at least, alleviate diet (figure 6). In uncomplicated CD, almost all strategies aim at reducing the antigluten CD4+ T cell response. A first set of approaches intend to prevent its activation. Drugs have been designed to sequester gluten in the intestinal lumen,115 or to improve its intraluminal digestion.116 Unfortunately, they have failed to protect against histological damage when tested in clinical trials.116 A new glutenase with superior gluten-degrading activity seems promising but clinical efficacy remains to be proven.117 A putative inhibitor of the paracellular pathway, larazotide, was proposed to prevent gluten epithelial passage. Yet its mechanism is controversial.28 Moreover, protection against tissue damage was not assessed during clinical trials, limiting conclusions.29 In contrast, very promising results have been recently obtained using a selective TG2 inhibitor (ZED1227) in a 6-week phase II trial. The inhibitor was administered orally 30 min before a daily 3 g gluten challenge. Compared with patients receiving placebo, those treated with ZED1227 showed lesser increase in IEL, lesser reduction in the villus height/crypt depth ratio and no increase in serum anti-TG2 IgA antibodies.32 The study was however of short duration and relied on the controlled administration of relatively low amounts of gluten. Further studies are thus needed to assess safety and efficacy. A second set of approaches inspired by allergen-specific immunotherapy aims at restoring tolerance to gluten.23 A first clinical protocol has consisted in repeated intradermal injections of three synthetic peptides encompassing five HLA-DQ2.5-restricted common gluten epitopes (Nexvax2). Initial results suggesting that Nexvax2 administration might prevent the systemic release of IL-2 induced by oral gluten challenge have not been confirmed.118 A second method of immunotherapy based on the intravenous infusion of biodegradable nanoparticles loaded with a mixture of native and deamidated gliadin peptides was tested in a recent small clinical trial119 after demonstrating efficacy in several mouse models of gluten sensitisation.120 Patients in remission were infused with nanoparticles on days 1 and 8 and orally challenged with gluten (6–12 g/day) between days 15 and 29. At day 29, the increase in circulating gliadin-sensitised T cells observed in the placebo group was not seen in patients treated by gluten-loaded nanoparticles; moreover, histology suggested mild villus flattening only in the placebo group. Yet, the number of IEL increased in both groups, and it cannot be excluded that the gliadin-specific T cells were entrapped in the intestine. In addition, the 2-week gluten challenge is too short to observe a histological relapse. Further studies with more prolonged follow-up will be necessary to assess if the antigluten response is truly attenuated and if so, how long. In addition to therapies designed to inhibit the antigluten CD4+ T cell response, a recent trial has tested whether blocking IL-15 with the humanised anti-IL-15 antibody AMG-714 could protect patients in remission during challenge by 2–4 g/day gluten. Despite improvement of diarrhoea and a significant lesser increase in IEL at the highest dose, this treatment failed to prevent mucosal damage.121

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed rationale-based therapies for coeliac disease (CD). GOF, gain-of-function; IFN, interferon; IL, interleukin; JAK, Janus kinase; TG2, tissue transglutaminase 2. Created with BioRender.com.
In RCD, strict GFD remains indispensable to turn off gluten-induced inflammation and cytokine production by gluten-specific CD4+ T cells. Additional treatments are however necessary to further curb inflammation and, in RCD2 to prevent progression to EATL. In RCD1, a large set of immunosuppressive treatments have shown efficacy.122 Currently, the first-line option is open-capsule budesonide, which induced clinical and histological responses in 90% of patients with RCD1.1 123 Thiopurines can be used in case of dependence or resistance to steroids. The design of more personalised therapies awaits the identification of (a) precise mechanism(s) driving resistance. The importance of cytokines activating the JAK-STAT pathway in CD, suggests that JAK inhibitors may be an interesting option, notably if in situ phosphorylation of STATs is detectable. In RCD2, the objectives are to correct malnutrition and to prevent the onset of overt EATL. The treatment should ideally eradicate RCD2 IEL. Yet, due to their low proliferative rate, these cells are insensitive to most ablative chemotherapies and the first-line treatment remains, as in RCD1, open capsule budesonide. This treatment induced clinical and histological responses in 90% of RCD2.123 It may curb the production of inflammatory cytokines and induce the apoptosis of RCD2 IEL.124 However, this treatment does not prevent EATL, stressing the need to identify other therapeutic options. Purine analogues such as cladribine, pentostatine or fludaribine125 might reduce the number of RCD2 IEL but they induce severe immunosuppression and, in monotherapy, they favour progression to EATL.65 126 They are used in combination with melphalan for conditioning before autologous stem cell transplantation, a therapeutic option for patients <65 years with severe RCD2 resistant to steroids who are at very high risk of EATL.127 Onset of EATL has been observed during the conditioning phase but long-term outcome is favourable in the small number of severely sick patients who have been treated127 (unpublished observations). Progresses in RCD2 pathophysiology have led to consider more targeted approaches. A 3-month treatment with the human anti-IL-15 antibody AMG 714 disappointingly failed to significantly decrease the number of RCD2 IEL.128 Tofacitinib, an oral JAK1/JAK3 inhibitor is currently tested in a phase II trial (NTR 7529, EudraCT 2018-001678-10). Blocking IL-15 or the JAK1/3 pathways raise however several caveats. First, it may impair putative antitumour responses that are strongly stimulated by IL-15.46 Second, the mutational profile of RCD2 IEL is very complex and, as observed in other cancers, targeting one signalling pathway may promote selection of resistant clones.129 An alternative approach may be to selectively deplete RCD2 cells with antibodies specific of the malignant cells. One candidate antibody is NKp46 conjugated to a cytotoxic drug with the possible inconvenient however to deplete also NK cells.68 Finally, it will be important to define whether RCD2 is associated with an antitumour response that may be boosted to prevent progression to EATL.
Conclusion and perspectives
At a time when humans realise the huge cost to pay for the benefits of technical progresses, CD can be viewed as the unanticipated consequence of one first major advancement in human history, the domestication of cereals 8000 years ago in the Fertile Crescent. CD also provides a striking example on how human encounter with a novel environmental factor can overwhelm the multiple immunoregulatory mechanisms evolved to preserve homeostasis of the immune system and trigger chronic inflammation in individuals with certain genetic traits. Along this line, the high frequency of HLA-DQ2/DQ8 haplotypes in populations consuming gluten-containing cereals seems paradoxical. Disease penetrance may be too low and the frequency of mild presentations too high for negative genetic selection. Conversely, some mild degree of chronic intestinal inflammation may perhaps have conferred protection against enteric pathogens. To date, these considerations remain however speculative. Strikingly, insight into CD pathogenesis participates in a better understanding of many other human immune diseases. The mechanism behind the predisposing role of HLA-DQ in CD provides an archetypal example of the role of HLA molecules in such diseases. The key role of cytokines activating the JAK1-STAT3 pathway in driving the autoimmune-like attack of the gut epithelium in promoting lymphomagenesis stresses the importance of a tight control of this pathway to maintain immune homeostasis, notably in the small intestine. This is further illustrated by the recent demonstration of constitutive mutations enhancing this pathway in patients with non-coeliac autoimmune enteropathies, who are also at risk of lymphomas.87 RCD2 and EATL also exemplify how the interplay between acquired somatic mutations and chronic inflammation driven by prolonged exposure to a luminal antigen can foster lymphomagenesis in the GI tract, a scenario already well established in mucosa-associated lymphoid tissue B lymphomas or in alpha chain immunoproliferative diseases.130
If to date the GFD proposed in the mid-1950s remains the only treatment for uncomplicated CD, mechanistic insight into CD pathogenesis has opened many new diagnosis and therapeutic avenues. The discovery that TG2 is the target of coeliac-specific autoantibodies has led to develop highly specific diagnostic tests. The demonstration of its role in the activation of the antigluten response has provided the rationale to develop selective inhibitors, which show promising therapeutic results in a first clinical trial. In-depth characterisation of the gluten-specific CD4+ T cell responses has allowed designing desensitisation approaches and defining tools and methods to monitor related therapeutic trials. These tools are also promising to facilitate diagnosis in patients already on GFD, a frequent issue nowadays. The identification of highly recurrent somatic mutations in the JAK/STAT pathway as putative driver of CD lymphomagenesis and of specific surface markers of the malignant cells highlight potential therapeutic targets. Accordingly, CD is attracting increasing interest from drug companies providing hope that it will be soon possible to implement new strategies to alleviate or complete GFD in order to facilitate patients’ daily life. Yet, it will be necessary to assess precisely the risks and benefits of each of these strategies, notably in uncomplicated CD where GFD, despite its difficulties for the patients, remains a safe and relatively cheap treatment.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Contributors AL wrote the review and prepared the figures; GM wrote the review; NC-B conceived and wrote the review.
Funding Work of the authors is supported by institutional grants from INSERM and Université Paris-Cité and by grants from ANR (Nr18-CE14-0005), Foundation ARC-Recherche Clinique (PGA1 RF20180206809), Inserm-Plan Cancer (SCILD), Foundation Princesse Grace and Association Française Des Intolérants au Gluten (AFDIAG). Institute Imagine is supported by the Investissement d’Avenir grant ANR-10-IAHU-01. AL has been/is supported by a Marie Sklodowska-Curie Individual fellowship (SingCelCD 843042), by Inserm-Plan Cancer (SCILD) by Cancéropôle and INCa (2020-1-EMERG-33-II-1).
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.