Article Text

Abstract
Pain mechanisms in patients with chronic pancreatitis are incompletely understood and probably multifactorial. Recently, evidence from experimental human pain research has indicated that in many of these patients pain processing in the central nervous system is abnormal and mimics that seen in neuropathic pain disorders. The current review focuses on several lines of evidence supporting this hypothesis. Hence, the spontaneous and postprandial pain in chronic pancreatitis may reflect the characteristic pain features seen in patients with neuropathic pain. Biochemical and histopathological findings in tissues from patients with chronic pancreatitis are similar to those observed in patients with other nerve fibre lesions. Experimental studies have shown that patients with chronic pancreatitis show signs of spinal hyper-excitability counter-balanced by segmental and descending inhibition. Changes in the brain with cortical reorganisation to gut stimulation and increased activity in specific electroencephalographic features characteristic for neuropathic pain are also seen in patients with chronic pancreatitis. Finally, principles involved in the treatment of pancreatic pain have many similarities with those recommended in neuropathic pain disorders. In conclusion, a mechanism-based understanding of pain in chronic pancreatitis may have important implications for the treatment.
Statistics from Altmetric.com
One of the most important symptoms in chronic pancreatitis (CP) is constant or recurrent abdominal pain that is present in 80–90% of patients during the course of the disease.1 Pancreatic pain presents characteristically with severe dull epigastric pain, often radiating directly to the back. The pain is often recurrent, intense and long-lasting and may be associated with malnutrition, narcotic addiction, and major socio-economic problems. Pain mechanisms are incompletely understood and probably multifactorial. In some cases the reason for the pain is obvious, such as extrapancreatic (eg, peptic ulcer or bile duct and duodenal stenosis due to extensive pancreatic fibrosis and inflammation) or intrapancreatic (eg, pseudocysts) complications. However, in most patients the source of the pain remains unknown. In these cases the following pathophysiological mechanisms have been suggested: (1) increased intrapancreatic pressure either within the pancreatic duct or in the parenchyma causing tissue ischaemia; (2) inflammation in the pancreas; and (3) alterations in pancreatic nerves, including an increase in nerve fibre diameter and evidence for neurogenic inflammation.2–4 Genetic factors probably also play a role in a patient’s pain experience.4 Because the pain mechanisms are poorly understood, treatment is often empirical and insufficient. Animal models have contributed to the understanding of pain pathogenesis in chronic pancreatitis.5–7 Although highly relevant to our understanding of pain mechanisms in general, the data should be interpreted cautiously. Hence there are major differences between pain studies in different species and strains. Furthermore, mechanisms related to the relatively short-lasting evoked inflammation in most animal studies on the one hand and the long-lasting pain in humans with CP on the other are probably very different. Recently, evidence from experimental human pain research has indicated that pain processing in the central nervous system (CNS) is abnormal and in many cases may mimic that seen in neuropathic pain disorders.8–10 Neuropathic pain is defined as “pain after a lesion or disease of the afferents in the peripheral or central nervous system that normally signals pain”.11 Neuropathic pain is prevalent in all diseases in which lesions of the nerves are present, and after surgery neuropathic-like pain can be seen in 20–40% of patients.12 Although other pain mechanisms may also be of importance, the following lines of evidence point towards neuropathic pain mechanisms in patients with CP:
Clinical features of the pain
Biochemical and histological findings
Spinal changes, with neuronal hyper-excitability and amplification of the incoming afferent activity
Changes in the brain–gut axis
Clinical and experimental profile of the drugs used to treat the pain
Although the characteristics listed above are not specific for neuropathic pain mechanisms, taken together they do support this theory. By focussing on recent neurophysiological experiments in humans, these topics will be reviewed in the current paper with the aim of improving our understanding and treatment of pain in CP.
NEUROPATHIC PAIN MECHANISMS AND CLINICAL FEATURES
Most nerve afferents mediating pain from the pancreas belong to the splanchnic nerves that pass through the coeliac ganglion and enter thoracic dorsal root ganglia. The vagal nerve with cell bodies in nodose ganglia normally does not participate in pain signalling but can indirectly modify spinal pain processing, as shown by in vivo electrical stimulation.13 ,14 Cholinergic modulation of pancreatic inflammation has also been demonstrated (for a review, see Fregni et al15). Hence the nervous system plays a major role in the inflammatory response as well as in pain pathogenesis and any abnormalities in the sensory system will invariably affect the disease process.
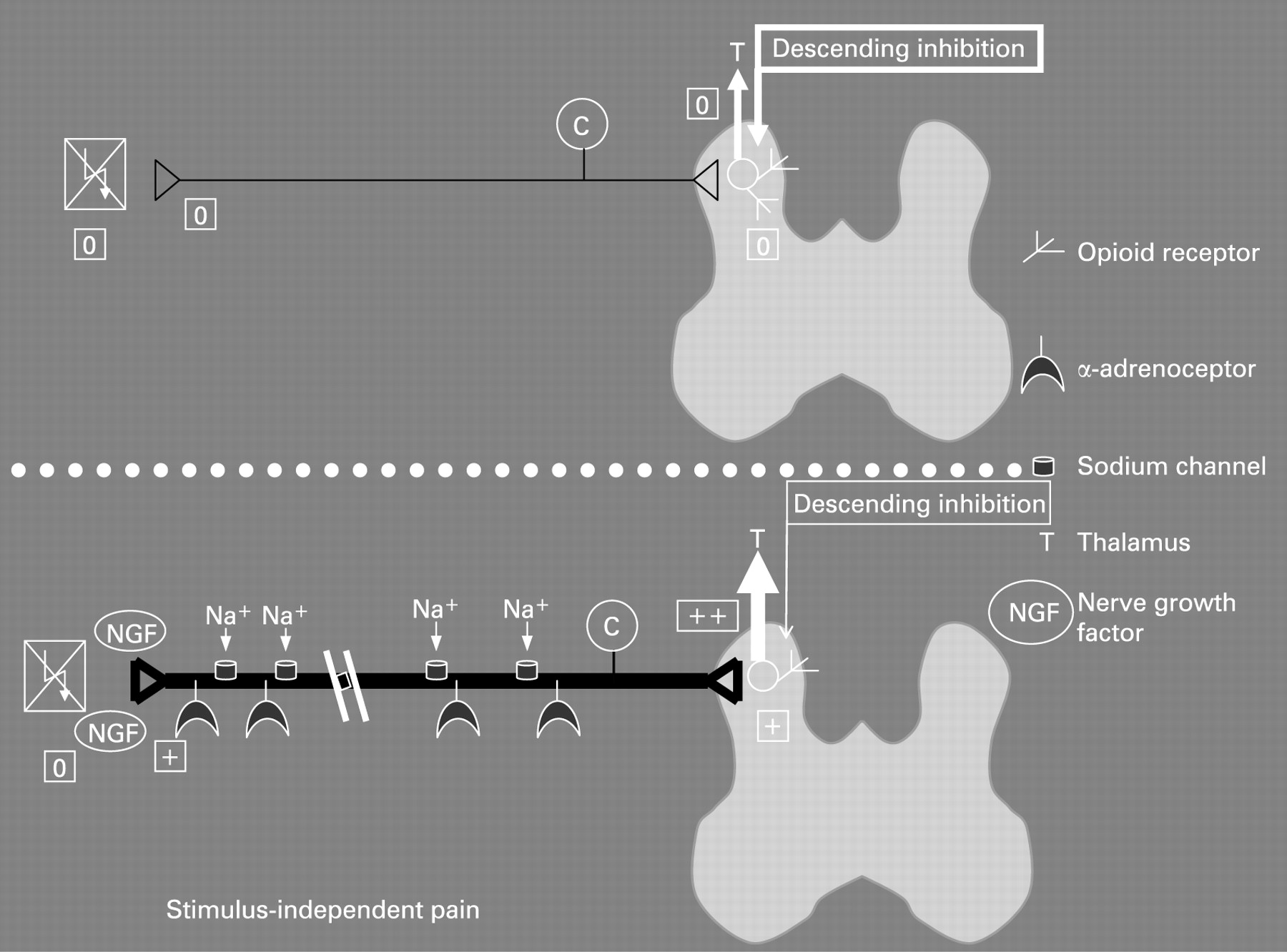
Neuropathic pain typically presents as a stimulus-independent shooting or burning pain together with stimulus-dependent increased sensations.16 The stimulus-independent pain is the result of both peripheral and central changes (see figs 1 and 2 for a schematic overview). In the peripheral nervous system the nerve lesion results in spontaneous firing of the axons due to the accumulation of sodium channels and α-adrenoceptors, together with membrane changes in the primary afferent neurons. Furthermore, sprouting of sympathetic axons into the dorsal horn takes place, rendering the nerves sensitive to autonomic reflexes and circulating adrenaline, for example, irrespective of any peripheral input to the nerves. In the spinal cord, neuroplastic changes and alterations in the membrane properties result in spontaneous firing of second-order neurons. These neurons also show integration of the response to repeated stimulation, resulting in increased firing (a proxy of the so-called “wind-up” phenomenon seen in animal experiments, as described later in this paper). Normally, the increased afferent barrage of nociceptive information to the brain is counterbalanced by a “feed-back” inhibitory control system based on descending nerve pathways originating in the brain stem. Changes in this feed-back system may also alter the excitability and result in spontaneous pain. Central down-regulation of opioid receptors may also be of importance.

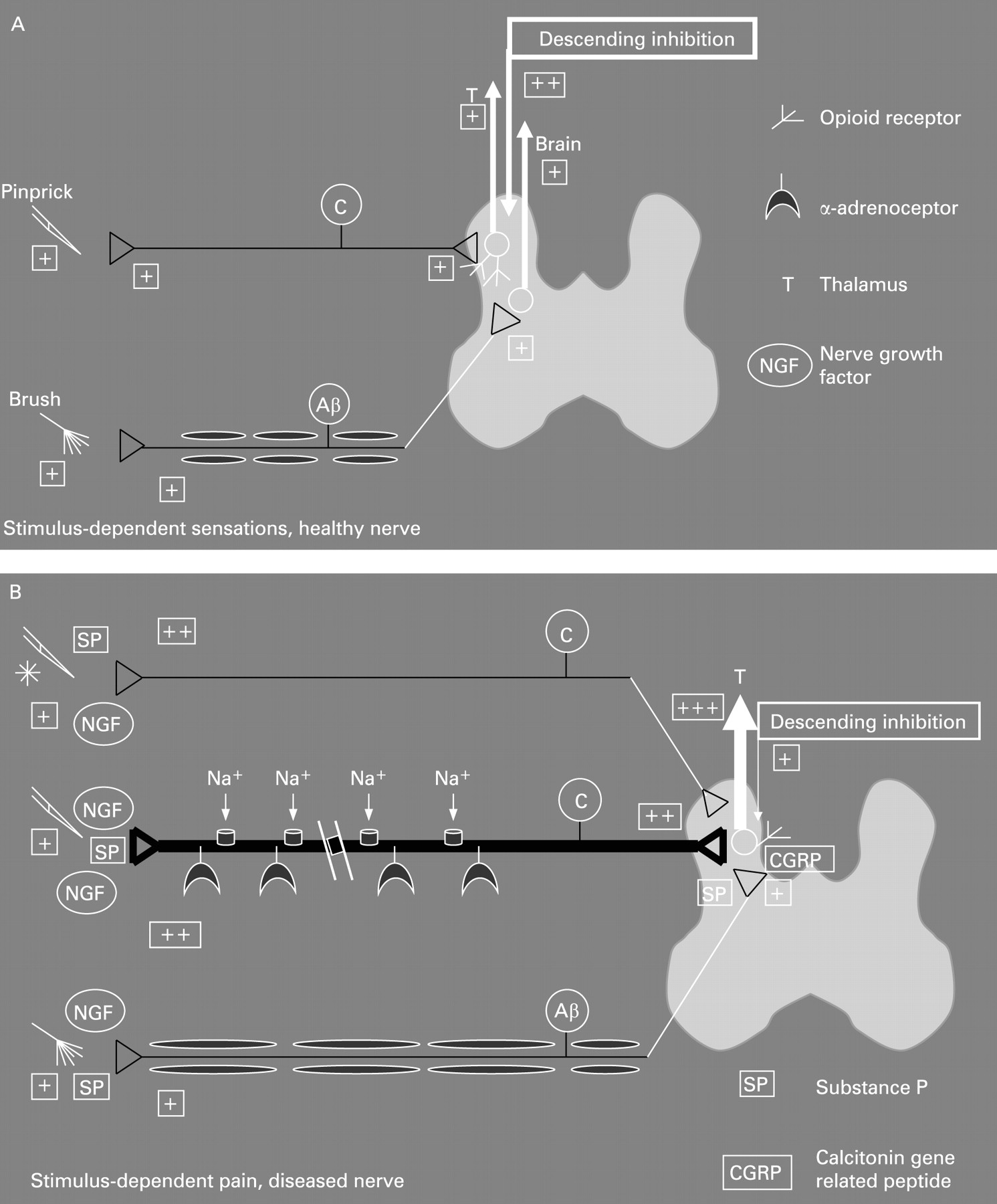
The stimulus-dependent pain is also a result of the peripheral and central changes. The cardinal features of classical neuropathic pain in the skin, such as seen in diabetes and in post-herpetic neuralgia, are allodynia (a painful response to stimuli not normally painful) to cold and brush stimuli and hyperalgesia (increased pain to a normal painful stimulus) to punctuate stimuli.16 In the periphery the increased neural activity results in neurogenic inflammation. The neurotransmitters released are typically spread to adjacent nerves, rendering them more sensitive. The central changes are a result of increased nociceptor drive on second-order neurons leading to central hyper-excitability and firing at a lower threshold. Furthermore, there is a phenotypic switch where Aβ fibres (which are normally responsible for non-painful sensations such as touch) start to release substance P and calcitonine gene-related peptide (CGRP). These mediators will stimulate specific nociceptive receptors in the spinal cord, which leads to activation of second-order neurons involved in pain processing. The outcome is allodynia to touch (brush allodynia). In the spinal cord, the touch-sensitive Aβ fibres normally terminate in the deeper lamina, whereas neurons in superficial lamina receive nociceptive information mainly from unmyelinated C fibres and thinly myelinated Aβ fibres. In neuropathic pain, however, the deep Aβ fibres sprout to terminate in the “nociceptive specific” lamina 2 and come into contact with neurons that normally transmit pain, again resulting in tactile allodynia.
The pain in patients with CP is often burning and intermittent or shooting, features also seen in stimulus-independent neuropathic pain.16 ,17 In patients with CP the allodynia due to touch and movements seen in neuropathic pain from somatic structures may be reflected during postprandial pain. After a meal the pancreas may be compressed from expansion of the stomach and/or affected by postprandial secretion of pancreatic juice. This may lead to increased pressure in the pancreatic tissue, in particular if calculi and/or duct stenosis are present. The increased pressure and/or shearing forces will lead to compression of the pancreatic nerves, and in neurogenic inflammation this results in stimulus-dependent allodynia and hyperalgesia via the mechanisms described above. Postprandial pain may, of course, also result from peptic ulcer and complications such as stenosis of the duodenum, and if present these should be treated appropriately.
BIOCHEMICAL AND HISTOPATHOLOGICAL FINDINGS
Recent animal studies in which CP has been induced by intravenous injection of dibutyltin dichloride or intraductal injection of trinitrobenzene sulfonic acid have shown behavioural, serological and histological characteristics similar to those seen in patients with CP, including damage of nerve fibres.5 ,6 ,18 Acute pancreatitis is characterised by intra-acinar activation of proteolytic enzymes and fusion of zymogen granules with lysosomal vacuoles containing cathepsin B, contributing to enzyme activation and resulting in autodigestion of the pancreas.19 Moreover, enzyme activation stimulates the release of inflammatory mediators, which contribute to fat tissue and haemorrhagic necrosis in the pancreas.20 Alcoholic CP, the most common subtype of CP in Western countries, is the result of recurrent attacks of acute alcoholic pancreatitis.21 It has been shown in alcoholic and autoimmune CP tissue specimens and in cell culture studies that the profibrotic cytokines platelet-derived growth factor B and transforming growth factor β1 are upregulated in macrophages and ductal cells and stimulate myofibroblasts and activated pancreatic stellate cells to synthesise extracellular matrix.22–25 These changes result in the occurrence of inflammation together with marked fibrosis in the pancreas, representing the histological hallmarks of CP.20 Furthermore, it was shown 20 years ago26 that pancreatic pain was related to inflammation surrounding pancreatic nerve fibres, indicating a neuropathic component in this pain. The diameter of intrapancreatic nerve fibres was increased and the mean area of tissue served per nerve was decreased, possibly due to growth and branching of the nerves.26 Several neuropeptides, growth factors and corresponding receptors active in neurogenic inflammation are upregulated and involved in pain generation in CP. A detailed description of this topic is far beyond the scope of this review. (For comprehensive reviews, the reader is referred to Fasanella et al4 and Vera-Portocarrero and Westlund.17) However, some of the mechanisms causing neurogenic inflammation in CP will be mentioned and are illustrated schematically in fig 3.

As shown in fig 3, one of the key receptors in neurogenic inflammation is the transient receptor potential vanilloid 1 (TRPV-1). This non-selective cation channel, preferentially located on sensory nerve fibres, has been investigated in experimental pancreatitis in rats,27 ,28 in human pancreatic cancer, and in the normal pancreas.29 Some inflammatory mediators, especially those with agonistic effects on G protein coupled receptors (ie, bradykinin30 and trypsin31), are able to indirectly sensitise TRPV-1 and thereby lower the threshold for activation in the sensory nerves.28 Recent data indicate that hydrogen sulfide-mediated activation of T-type Ca2+ channels is also involved in pancreatitis-related pain.32 It was shown that pancreatic hydrogen sulfide targets T-type Ca2+ channels, which most probably are located on the peripheral endings of intrapancreatic sensory nerves. Inhibition of the endogenous hydrogen sulfide pathway inhibited the pain behaviour in caerulein-evoked experimental pancreatitis. Interestingly, a similar pro-nociceptive role of hydrogen sulfide was demonstrated in the mouse colon.33 The resulting activation of sensory nerves in the pancreas stimulates the synthesis of substance P (SP) and CGRP in dorsal root neurons. SP and CGRP are transported antidromically along the axon to the site of the lesion, where they stimulate inflammatory cells to synthesise further amounts of inflammatory mediators. SP acts primarily via the neurokinin 1 (NK1) receptor, and in alcoholic CP it was shown that expression of the NK1 receptor was increased in mononuclear and polynuclear cells, fibroblasts and nerves.34 At the vascular level, the actions of SP are mostly vasoconstrictive.17 As the NK1 receptor has also been detected in the epineural layers of nerves, where the small arteries feeding the endoneural vasculature are located,35 it may also play a direct role in the generation of pain in CP by causing vasoconstriction and thereby ischaemia of intrapancreactic nerves.36 SP has been found to directly stimulate the synthesis of interleukin 8 (IL8) in macrophages.37 IL8 also generates hyperalgesia by stimulation of post-ganglion sympathetic neurons, and increased expression of IL8 has been detected in inflammatory cells in alcoholic CP.37
Evidence for a neuropathic component of pain generation in CP was further provided by the finding that growth associated protein 43 is increased in pancreatic nerve fibres in alcoholic CP.38 This protein was shown to be re-expressed after neuronal lesions,39 introducing it as a marker of neuronal growth and plasticity. Moreover, the neurotrophin nerve growth factor and its high affinity receptor tyrosin kinase receptor A are upregulated in alcoholic CP.40 Nerve growth factor is possibly a regulator of the synthesis of SP and CGRP41 and modulates nociception in peripheral nerve fibres.42 Another neurotrophin with impact on neuropathic pain in CP is brain-derived neurotrophic factor, which is increased in ductular complexes, degenerating acinar cells, and enlarged nerve fibres as well as in intrapancreatic ganglia in CP.43 During peripheral inflammation, brain-derived neurotrophic factor is upregulated in dorsal root ganglia in rats.44 Recent studies have also identified artemin45 and the cannabinoid system46 as possible mediators of neural changes and pain perception in CP. Artemin belongs to the family of glial-derived neurotrophic factors and enhances survival, proliferation and regeneration of neurons. Both artemin and its receptor are increased in CP tissue.45 Moreover, the expression of the cannabinoid receptor 1 is upregulated in human acute pancreatitis, a finding that is accompanied by an increase in the endocannabinoid anandamide.46 In another study, cannabinoid receptors located on peripheral nociceptive endings have been shown to negatively modulate nociceptors and inhibit release of neuropeptides.47
These findings point towards a complex interplay of inflammatory cells, nerve fibres, neuropeptides, and corresponding receptors, resulting in the phenomenon of peripheral (and subsequent central) sensitisation of the nervous system.
Summary box 1
Constant and/or recurrent abdominal pain is present in 80–90% of patients with chronic pancreatitis
The spontaneous and postprandial pain in chronic pancreatitis may reflect the characteristic stimulus independent and stimulus dependent pain features seen in patients with neuropathic pain
Biochemical an histopathological features in tissues from patients with chronic pancreatitis mimic those observed in tissues from patients with other nerve fibre lesions
The principles involved in the treatment of pancreatic pain have many similarities with those used in neuropathic pain disorders
SPINAL CHANGES WITH HYPER-EXCITABILITY AND AMPLIFICATION OF THE INCOMING NEURAL ACTIVITY
In most diseases characterised by chronic pain, sensitisation of the nervous system is a cardinal feature. This is also the case in neuropathic pain disorders. This sensitisation results in plastic changes in the neurons at spinal (and supraspinal) sites. It has been proposed that these plastic changes in the dorsal horn of the spinal cord correspond to the mechanism in “long-term potentiation”.48 Through a cascade of molecular changes at the cell synapses, long-term potentiation results in an increase in synaptic transmission efficacy, thus leading to an increase in synaptic strengthening which further increases electrical activity and sensory information transmission. This sensitisation may ultimately result in an autonomous state in which the central nervous system reports pain even in the absence of peripheral noxious input.49 Sensitisation includes both peripheral and central components. Peripheral nociceptor sensitisation underlies the hyperalgesia that develops immediately at the site of injury. However, in CP, acute inflammation of the gland has normally ceased, and central rather than peripheral sensitisation is thought to account for most of the symptoms experienced by the patients. In animal studies, central sensitisation is characterised by increased spontaneous activity, decreased firing threshold, and expansion of the receptive fields of dorsal horn neurons.50 ,51 The increase in excitability of spinal cord nociceptive neurons amplifies the signal coming from the periphery, resulting in allodynia and hyperalgesia. The alterations in functional structure may result in central plasticity and “pain memory,” which after some time may be consolidated and independent of the original peripheral input.50 ,51 Human experimental models have been used to sensitise the gut and mimic the findings in animal studies.52 Studies using electrical and mechanical stimuli have indicated that acid perfusion of the oesophagus results in peripheral as well as central sensitisation of the nervous system.53 ,54 In conditions with chronic pain there is substantial evidence for persistent central changes that outlive the initial disease. Hence, when the peripheral stimulation (such as inflammation due to CP) has subsided, sensitised second-order neurons continue to fire, and sub-threshold regulatory stimuli are still perceived as painful.
Importantly, CP is often complicated by diabetes, and it is estimated that 60–70% of patients with diabetes suffer from mild to severe forms of nervous system damage including autonomic neuropathy.55 Using a multimodal experimental approach, Frøkjær et al56 recently demonstrated that gastrointestinal sensory nerves in patients with diabetes are affected throughout all layers of the gut. The neuronal damage will invariably result in changes in the neuronal pain matrix, including interactions between peripheral and central pain mechanisms, and thereby the sensory processing in CP. Moreover, activation of pain related structures in the brain might trigger behavioural responses resulting in negative changes in eating, daily activity and sleep patterns that could worsen the pain.15 ,57
Although simplified, referred pain is a result of visceral and somatic fibres converging on the same second-order neuron (for details see Arendt-Nielsen et al58). Hence spinal hyper-excitability will be manifested by enlargement of and changes in localisation and sensitivity of the referred pain area.59 In patients with CP, Buscher et al60 investigated the sensitivity in the referred pain areas of the abdomen. Although findings were not completely consistent between genders, they showed increased sensation to pressure (predominantly a deep muscle stimulus). On the other hand, there were higher thresholds to electrical skin stimulation in dermatome T10, the “viscerotome” that shares innervation with the pancreatic nerves, whereas the sensation in other dermatomes (C5, T4, L1 and L4) was normal. The authors suggested that this local hyposensitivity was related to segmental inhibition caused by a local feedback system in the spinal cord. As with the deep stimulus, changed sensation in the somatic referred pain area (sharing central termination with activated visceral nerves) has also been shown experimentally in healthy subjects as well as in patients with diseases such as appendicitis and choledocholithiasis.61 ,62 Therefore, such data give evidence for central hyper-excitability in CP.
In experimental studies the pancreas is difficult to access and there are potentially harmful complications during endoscopic procedures. Therefore it has not yet been possible in humans to investigate directly whether allodynia or other characteristic features of neuropathic pain are present during manipulation of the pancreas. However, visceral pain is typically diffuse and difficult to localise, which to a large extent can be explained by the widespread termination of visceral afferents into second-order neurons in multiple segments of the spinal cord.63 Due to this overlap in central termination of visceral afferents, disease mechanisms affecting the pancreas are typically also activated during pain stimulation of nearby organs like the duodenum and oesophagus.64 Dimcevski et al9 ,10 showed that the referred pain area to experimental electrical stimulation of the oesophagus in CP was increased compared with that in healthy controls (fig 4). Because increases in the size of the referred pain area after experimental stimulation have also been shown in other patient groups with organic diseases (eg, erosive and non-erosive oesophagitis, peptic ulcer disease) this strengthens the validity of the findings.65 ,66 Changes in the referred pain area, however, are not specific for any specific pain disorder, but merely reflect the importance of central changes such as seen those in neuropathic pain.

Another feature that is predominant in central sensitisation is wind-up (in humans this is termed temporal summation or central integration). Temporal summation is a frequency dependent, gradual response of “wide-dynamic” spinal neurons to repetitive stimuli, which may lead to increased excitability if the stimulus is sufficiently strong.67 ,68 Hence, afferent fibres that are repeatedly stimulated give a progressive increase in second-order neuronal responsiveness. Dimcevski et al found an increase of temporal summation in the skin,64 muscle and oesophagus10 in patients with CP (fig 4). Although not specific for any pain disorder, facilitated temporal summation is prevalent in neuropathic pain patients,69–73 and thus these findings support the important role of neuropathic pain mechanisms in CP.
The dissociation between local sensory disturbances in the area of nerve damage and spontaneous pain per se suggests that the pain is neuropathic in nature. However, other mechanisms are also of importance. The second-order neurons in the spinal cord are subject to descending control from higher brain centres.74–77 One of the first systems described was diffuse noxious inhibitory control (DNIC), defined as “painful heterotopic stimuli that can cause suppression of nociceptive activity in dorsal horn neurons”. DNIC was shown to involve a spinal–supraspinal–spinal feedback loop producing a descending inhibitory control.78 Apart from being inhibitory, descending pain control from the brainstem can also be facilitating, and the balance between the inhibiting and activating systems may determine the overall level of excitability (and hence pain) of the neuron network in the dorsal horn.79 ,80 In an animal model of persistent pancreatic pain Vera-Portocarrero et al81 reported that descending facilitation from the rostral ventromedial medulla plays a critical role in the maintenance of pancreatic pain. When they selectively destroyed cells in this brain centre, it attenuated pancreatic-induced hypersensitivity. Several cortical centres are connected to the periaquaductal grey and the raphe magnus nucleus in the medulla, which again exert control over the spinal cord. The balance between the excitatory system and the inhibitory modulation is therefore a major determinant for the final interpretation of the pain, and descending influence from brainstem structures seems to be the major way by which the brain controls pain perception.75 ,76 ,82–84 Recent evidence supports the notion that in patients with functional pain disorders, such as irritable bowel syndrome, the inhibitory control system functions insufficiently. This may result in an increased barrage of noxious stimulation reaching pain centres in the brain.85 On the other hand, in patients with organic disorders the descending control system is probably fully activated to prevent hyperalgesia evoked by the afferent activity arising in the diseased tissue. Hence, hypoalgesia to experimental visceral stimulation was seen in patients with diseases such as oesophagitis, Crohn’s disease and peptic ulcers.65 ,86 ,87 Therefore in patients with CP there will probably be an activation of the descending inhibitory system, whereby the pain system tries to counterbalance the incoming nociceptive information from the pancreas. However, activation of descending control systems probably affects the spinal cord diffusely, resulting in widespread hypoalgesia to stimulation from other body regions. Correspondingly, in an experimental study in which the skin, oesophagus and duodenum were stimulated, generalised hypoalgesia was found in patients with CP in comparison with healthy controls (see fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Evidence indicating increased descending inhibition in CP was also seen in a study based on evoked brain potentials resulting from painful stimulation of the oesophagus. Evoked brain potentials are the summated outcome of a series of time-locked electroencephalographic (EEG) responses to external stimuli.88 ,89 Reduction in latency of the evoked brain potentials may be related to hyper-excitability within central pain pathways evoked by the chronic pain attacks in the patients.90 This is most probably due to central hyper-excitability and opening of faster conducting latent connections. Correspondingly, in patients with CP the latency to upper gut stimulations was decreased.9 However, the latency to sigmoid stimulations was decreased (Drewes et al, unpublished data) (fig 5). The reason for these findings is probably that, although the pain in CP evokes hyper-excitability in a widespread area of the spinal cord, this is predominant in the thoracic region. Hence the excitability in remote lumbar segments receiving afferents from the sigmoid colon could be dampened by the descending control systems exerting diffuse inhibition of the incoming activity at all segmental levels of the spinal cord. However, electrical stimulation may activate both vagal and splanchnic pathways.91 Because activation of the splanchnic/spinal pathways probably activates an endogenous pain inhibitory pathway, whereas vagal stimulation may result in facilitation, this can make interpretation of the data difficult.13
The most recent neurophysiological evidence for neuropathic pain mechanisms in CP is based on studies of the brain. Several experimental and clinical studies indicate that deafferentation, chronic pain, and hyperalgesia are associated with functional reorganisation of the cortex. In particular, there is evidence for neuroplastic changes and reorganisation of the brain in patients with amputations and neuropathic pain.92 Importantly, the degree of cortical reorganisation correlates with the subjective pain rating, and the cortical changes can be reversed by analgesic interventions (for a review, see Wiech et al93). In recent experiments carried out by our group, analysis of the brain sources to upper gut stimulation showed that the insula activity was reorganised in CP patients.9 The insula plays an important role in the integration of the visceral sensory and motor activity together with the limbic system. It is connected to many limbic structures as well as to the motor cortex and basal ganglia. Thus the changes in the insula region in patients with CP may reflect severe disturbances in the coordination and sensory processing of visceral pain. There were also more discrete changes in the cingulate cortex where the neuronal source was more posterior in the patients. The findings indicate that pain in chronic pancreatitis leads to changes in cortical projections of the nociceptive system, supporting a neuropathic component in pancreatic pain.
Finally, is has been proposed that changes of power in the theta (3.5–7.5 Hz) EEG band may be a hallmark of patients with different kinds of neuropathic pain.94–96 Recently, Sarnthein and co-workers showed that patients with neuropathic pain disorders exhibited higher spectral power of the resting EEG especially in the theta range,97 which normalised after successful treatment. It was hypothesised that the theta activity reflects a disturbance in higher cognitive functions such as those engaged in neuropathic pain. In a recent study we examined the EEG during painful stimulation of the oesophagus in patients with CP.98 In comparison with controls, there was increased activity in the theta band in CP patients, and such findings also support the neurogenic pain hypothesis.
Interestingly, Fregni and co-workers15 recently proposed that the cortical neural network activated by pancreatic inflammation may act via a feed-back mechanism and modulate the immune system, thereby influencing the healing process and decreasing (or increasing) pancreatic inflammation. The neuroplastic changes may become self-perpetuating, and account for the disabling chronic pain. The authors proposed that the cyclic variation of pain levels and flare-ups may not only be a simple response to inflammation but, on the contrary, could reflect CNS modulation of the inflammatory process. Non-invasive brain stimulation can enhance or suppress activity in the targeted regions, and repetitive transcranial magnetic stimulation has been effective in modifying the dysfunctional brain activity associated with pain. In a pilot study, Fregni et al demonstrated increased activity in the secondary somatosensory cortex (located close to the insula) in patients with CP. Magnetic stimulation, which suppresses the activity in the targeted brain region, resulted in relief of the visceral pain.15 Although these observations need to be replicated in larger studies, they stress the importance of studies exploring the gut–brain axis in CP.
Summary box 2
Spinal changes due to hyper-excitability of dorsal root neurons accompany neuropathic pain and are reflected in integration of the response to repeated stimuli and enlargement of the referred pain areas in response to stimulation of the upper gut
The spinal changes are counterbalanced by segmental and descending inhibition, which may change sensibility in the gut and in somatic “viscerotomes”
Changes in the brain with cortical reorganisation of the response to gut stimulation and increased activity in specific electroencephalographic features are characteristics of neuropathic pain and are also seen in chronic pancreatitis
PROFILE OF DRUGS USED TO TREAT THE PAIN IN CHRONIC PANCREATITIS
Treatment of CP depends on the clinical course and abnormal imaging findings (for reviews, see Mitchell et al99 and Steer et al100). Thus, pseudocysts and ductal obstruction may lead to endoscopic, extracorporeal shock-wave lithotripsy and/or surgical treatment, whereas resection of a dominant inflammatory mass (typically in the pancreatic head) may relieve the patient of the pain. It is of utmost importance to exclude autoimmune pancreatitis if an inflammatory mass is present because this relatively new entity of chronic pancreatitis responds to steroid treatment.101 Neurolysis and nerve blocks have mainly been used in the past, whereas methods based on endoscopic ultrasound and thoracoscopic splanchnicectomy are now increasingly used. Alternative treatments have included transcutaneous electrical nerve stimulation, spinal cord stimulation, and transcraniel magnetic stimulation.4 ,15 However, in most patients medical treatment is the mainstay of pain management (for reviews, see Andren-Sandberg et al1 and van Esch et al49). Treatment of the pain in chronic pancreatitis is difficult, and although the World Health Organization analgesic ladder recommends starting with non-narcotic analgesics followed by weak narcotics (with or without non-opioids and adjuvant therapies), it is often necessary to use strong opioids.1 ,49 The cornerstone in the treatment of neuropathic pain relies on multifunctional drugs, targeting different areas in the CNS, such as tricyclic antidepressants, gabapentin/pregabalin, tramadol and, eventually, opioids (for a review, see Finnerup et al102). Drugs shown to be effective in neuropathic pain also seem to be efficient in CP. Tricyclic antidepressants and pregabalin, which are recommended in neuropathic pain, have in our experience been of value as adjuvant therapy in difficult patients. Interestingly, gabapentin was shown to potentiate the effect of morphine in an animal model of pancreatitis.103 In animals, opioids have been shown to influence the nociceptive processing in experimental pancreatitis.6 Although narcotics have many side effects and carry a risk of addiction, several papers report that opioids are among the best analgesics in the treatment of neuropathic pain (for a review, see Cruccu104). Wilder-Smith et al105 demonstrated that tramadol, a weak opioid drug with several non-opioid effects in the central nervous system, in high doses was superior to traditional opioids in the treatment of pain in CP. Among the strong opioids, oxycodone has shown to be effective in neuropathic pain.106 Correspondingly, in an experimental study, Staahl and co-workers8 administered oxycodone and morphine in equipotent dosages to patients with pain due to CP. They showed that oxycodone was superior to morphine in attenuating pain evoked from somatic structures (skin and muscle) as well as from the oesophagus. The superior effect of oxycodone on pain from somatic structures was not seen in a similar study in healthy subjects,107 and probably the disease process in CP was responsible for this difference. Hence oxycodone, although a strong μ-agonist, also has some effects on the κ-opioid receptor. Animal experiments have shown both a peripheral and a spinal upregulation of the κ-opioid receptors in the presence of inflammation.108 This upregulation will affect the pain system in several tissues, as has been shown in the CP patients.109 The potential effect of a receptor differentiated effect was supported in a clinical study involving a peripherally restricted κ agonist (ADL 01-0101). This agonist showed analgesic effects in patients with pain due to CP.110 Hence in contrast to a pure μ-agonist such as morphine, opioids with more complex effects such as tramadol and oxycodone may be of value in CP. Experimental studies should, however, be supported by large-scale clinical studies.
CONCLUSION
The pathogenesis of pain in CP is poorly understood. In a subset of patients, extrapancreatic diseases such as peptic ulcer and intrapancreatic complications such as duct obstruction or pseudocysts can explain the pain and should be treated appropriately. Endoscopic or surgical treatments are still the mainstay in selected cases if an inflammatory mass is present or there is substantial evidence for increased pressure in the pancreatic duct or gland.111–113 When an inflammatory mass is present, autoimmune pancreatitis and inflammatory pseudotumor have to be considered.114 However, in the majority of patients the cause of the pain is not obvious. Although the different lines of “evidence” presented in the current paper are not specific for neuropathic pain, they point towards this mechanism as a major explanation for the pain seen in many patients. If neuropathic pain dominates the clinical picture, surgical procedures may increase the neuronal lesions and worsen the pain. Furthermore, pharmacological treatment of neuropathic pain differs from that of other pain disorders, with focus on multifunctional drugs having different effects on the CNS, such as tricyclic antidepressants, tramadol and ion-channel inhibitors. Future studies should therefore aim at developing methods to characterise neuropathy in individual patients before they are subjected to treatment, and at addressing the prevalence of neuropathic pain in a large series of CP patients. Furthermore, the effect of drugs effective against neuropathy should also be evaluated systematically in well defined patient cohorts.
REFERENCES
Footnotes
Funding: The study was supported from “SparNord Fonden”, “Det Obelske Familiefond” and The Danish Health Research Council.
Competing interests: None.