Article Text

Abstract
Background Inflammatory bowel disease (IBD) is driven by a seemingly aberrant immune response to the gut microbiota with disease development dictated by genetics and environmental factors. A model exemplifying this notion is our recent demonstration that colonisation of adherent-invasive Escherichia coli (AIEC) during microbiota acquisition drove chronic colitis in mice lacking the flagellin receptor TLR5 (T5KO). T5KO colitis persisted beyond AIEC clearance and requires TLR4 and the NLRC4 inflammasome. We hypothesised that AIEC instigates chronic inflammation by increasing microbial lipopolysaccharide (LPS) and flagellin levels.
Goal Examine if transient AIEC colonisation lastingly alters levels of LPS and flagellin and changes microbiota composition.
Methods Germ-free mice (wild type (WT) and T5KO) were inoculated with AIEC strain LF82 and placed in standard housing allowing a complex microbiota that eliminated AIEC in both mice strains. Faeces were assayed for the inflammatory marker, lipocalin-2, bacterial loads, and microbiota composition by pyrosequencing. Faecal LPS and flagellin bioactivity were measured via a cell-based reporter assay.
Results Transient AIEC colonisation, in WT mice, did not alter inflammatory markers, bacterial loads, microbiota composition, nor its pro-inflammatory potential. By contrast, transient AIEC colonisation of T5KO mice drove chronic inflammation which correlated with microbiota components having higher levels of bioactive LPS and flagellin. Such AIEC-induced elevation of LPS and flagellin persisted well beyond AIEC clearance, required AIEC be flagellated, and was associated with alteration in microbiota species composition including a loss of species diversity.
Conclusions AIEC, and perhaps other pathobionts, may instigate chronic inflammation in susceptible hosts by altering the gut microbiota composition so as to give it an inherently greater ability to activate innate immunity/pro-inflammatory gene expression.
- INFLAMMATORY BOWEL DISEASE
- CROHN'S DISEASE
- COLONIC MICROFLORA
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Ileal lesions of patients with Crohn's disease are frequently colonised by pathogenic adherent-invasive Escherichia coli (AIEC).
-
Mice lacking the flagellin receptor, toll-like receptor 5 (TLR5), are prone to developing spontaneous colitis or low-grade inflammation.
-
Transient colonisation by AIEC during microbiota acquisition results in chronic colitis in mice that lacked the flagellin receptor, TLR5.
What are the new findings?
-
Chronic intestinal inflammation in TLR5-deficient mice challenged with AIEC correlated with microbiota components having higher levels of bioactive lipopolysaccharide and flagellin.
-
AIEC-induced intestinal inflammation in TLR5-deficient mice is associated with alteration in microbiota species composition, including a loss of species diversity.
-
AIEC can instigate chronic inflammation in susceptible hosts by altering the gut microbiota composition so as to give it an inherently greater ability to activate innate immunity/pro-inflammatory gene expression.
How might it impact on clinical practice in the foreseeable future?
-
Increases in the inherent ability of microbiota components to activate innate immunity may play a central role in perpetuating chronic inflammation in patients with inflammatory bowel disease (IBD).
-
Our data support the approach of selectively inhibiting innate immune signalling in IBD even if the disease resulted from an inherent innate immune insufficiency.
-
Suggest possibility that strategies to protect against pathobiont during microbiota acquisition might be a means to prevent IBD in genetically susceptible persons.
Introduction
Crohn's disease (CD) and ulcerative colitis (UC), collectively referred to as inflammatory bowel disease (IBD) are multifactorial diseases in which immune response in the gastrointestinal tract leads to chronic inflammation.1––3 The aetiology of these disturbances is not well defined, but clearly involves both environmental and genetic factors that act in concert to eventuate in such chronic disturbances. Among environmental factors, gut microbiota has been described to play a central role, with changes in its general composition and with select bacterial strains that have been suggested to be opportunistic pathogens.3 ,4 For example, Escherichia coli bacteria have been assigned a putative role in CD. They are abnormally predominant in early and chronic ileal lesions of CD, and most E coli strains isolated from the ileal mucosa of CD patients adhere to intestinal epithelial cells (IEC).5––7 In addition to their ability to adhere, these E coli bacteria are able to invade IEC and belong to a new pathogenic group of E coli, designated adherent-invasive E coli (AIEC).8 On the host side, many genetic susceptibility loci have been identified. Such loci involve a panoply of genes that mediate immune regulation such as IL-23,9 or gene such as NOD2 and autophagy-related genes mutations10––12 that mediate innate defence and/or management of intracellular pathogens. While Nod2-deficient mice do not exhibit spontaneous colitis, they exhibit altered gut microbiota and are prone to developing severe colitis in response to the chemical colitogen dextran sodium sulfate.13 Moreover, the notion that an innate immune deficiency can result in chronic colitis is dramatically illustrated by the robust uniform colitis in mice with that unable to activate the canonical NF-κB pathway in gut epithelial cells.14 A subtler example of an innate immune deficiency that can result in colitis is mice engineered to lack the flagellin receptor, Toll-like receptor 5 (TLR5), in which a subset of mice develop spontaneous colitis.15
The dual role of genetics and microbiota composition is recapitulated in several mouse models of colitis in which incidence of spontaneous disease can be dramatically reduced by altering gut microbiota via either antibiotic treatment or via ‘rederivation’ by embryo transplant, which alters microbiota composition but not genetics of the offspring. The best characterised example of this concept is the IL-10 deficient mouse strain in which development of colitis has been associated with particular micro-organisms, such as Helicobacter species, whose functional role in promoting murine colitis is demonstrable by its ability to induce disease upon its colonisation of IL-10-deficient mice.16 ,17 We have made analogous observations in TLR5-deficient mice (T5KO). Specifically, we observed that development of colitis in T5KO mice was associated with increased γ-Proteobacteria, particularly in the post-weaning period.18 The role of the microbiota in driving colitis in these mice was supported by observations that antibiotics, rederivation, or maintenance in a germfree state eliminated disease.15 ,19 The potential role of Proteobacteria in this colitis model was further supported by our observation that colonisation with AIEC resulted in colitis in T5KO, but not in wild type (WT) mice. Ability of AIEC to trigger colitis in T5KO mice required ablation of the endogenous microbiota with antibiotics, or administering AIEC to mice as they began to acquire a gut microbiota—achieved via exposing germfree mice to AIEC and then placing them in specific pathogen-free (SPF) housing. Interestingly, while AIEC colonised WT and T5KO mice only transiently (up to 4 weeks), chronic colitis persisted in T5KO months later suggesting this microbe acted as an instigator, rather than a propagator, of colitis. If this concept proves applicable to humans, it suggests that, since patients are typically diagnosed long after initial symptoms of disease appears, bacteria that may have initiated disease development are no longer present and hence will avoid detection by the extensive efforts that have been initiated to identify them.
The goal of this study was to investigate how AIEC colonisation might instigate colitis in T5KO mice. Specifically, we used DNA pyrosequencing to investigate the extent to which transient AIEC colonisation might induce lasting changes in the microbiota in WT and T5KO mice. Moreover, we used a cell-based reporter assay to investigate if such changes altered the inherent proinflammatory potential. We report that AIEC altered microbiota composition in T5KO but not in WT mice. Such alteration correlated with increased potential of the microbiota to activate innate immune-mediated proinflammatory gene expression, thus suggesting a potential means by which transient colonisation by select microbes can instigate chronic inflammation.
Materials and methods
Mice
WT, T5KO and RagKO mice (back-crossed to C57BL/6 mice for 10 generations) were bred and maintained at Georgia State University, Atlanta, Georgia, USA. WT and T5KO mice were also bred and maintained in germ-free conditions. Animal protocols were approved by institutional animal care and use committee (Georgia State University, Atlanta, Georgia, USA), permit number A11024. Mice were housed in cages of uniform genotype and treatment.
In vivo bacterial infection
CD-associated AIEC strain LF8220 and its isogenic non-flagellated mutant LF82-ΔfliC21 were grown in Luria-Bertani (LB) medium overnight at 37°C without agitation. After centrifugation for 10 min at 3400 g, bacteria were resuspended in phosphate buffer saline (PBS).
Germ-free mice (WT and T5KO) were inoculated with 107 AIEC reference strain LF82, or non-flagellated mutant LF82-ΔfliC, by oral gavage at 4–6 weeks of age, and then placed in standard housing to allow establishment of a complex microbiota. This specific inoculum amount, which was also used in our previous studies, was chosen arbitrarily. The number of bacteria used is unlikely to be a key parameter since germ-free mice can be so efficiently colonised with a wide range of bacteria and we previously described that the intestinal bacterial loads do not differ significantly in response to a range of inoculating doses.18 Faeces were collected weekly.
Faecal AIEC quantification
Numeration was done by platting PBS-resuspended faeces on LB agar plates containing ampicillin (50 μg/mL) and erythromycin (25 μg/mL) since AIEC strain LF82 is resistant to these antibiotics, and no such resistant bacteria were found in non-infected mice. On the next day, colony forming units (CFU) were counted and the number of CFU/mg of faeces was evaluated. When 454 pyrosequencing was performed, sequence data was used to determine the proportion of sequences classified as LF82.
Quantification of faecal Lcn-2 by ELISA
Frozen faecal samples were reconstituted in PBS containing 0.1% Tween 20 to a final concentration of 100 mg/mL and vortexed for 20 min to get a homogenous faecal suspension. These samples were then centrifuged for 10 min at 14 000 g and 4°C. Clear supernatants were collected and stored at −20°C until analysis. Lcn-2 levels were estimated in the supernatants using Duoset murine Lcn-2 ELISA kit (R&D Systems, Minneapolis, Minnesota, USA).22
Faecal flagellin and lipopolysaccharide load quantification
We quantified flagellin and lipopolysaccharide (LPS) using human embryonic kidney (HEK)-Blue-mTLR5 and HEK-Blue-mTLR4 cells, respectively (Invivogen, San Diego, California, USA). We resuspended faecal material in PBS to a final concentration of 100 mg/mL and homogenised for 10 s using a Mini-Beadbeater-24 without the addition of beads to avoid bacteria disruption. We then centrifuged the samples at 8000 g for 2 min and serially diluted the resulting supernatant and applied to mammalian cells. Purified E coli flagellin and LPS (Sigma, St Louis, Missouri, USA) were used as positive controls for HEK-Blue-mTLR5 and HEK-Blue-mTLR4 cells, respectively. After 24 h of stimulation, we applied cell culture supernatant to QUANTI-Blue medium (Invivogen, San Diego, California, USA) and measured alkaline phosphatase activity at 620 nm after 30 min.
Quantification of faecal bacterial load
Total bacterial DNA was isolated from faeces using QIAamp DNA Stool Mini Kit (Qiagen). DNA was then subjected to quantitative PCR using universal 16S rRNA primers 8F=5′-AGAGTTTGATCCTGGCTCAG-3′ and 338R=5′-CTGCTGCCTCCCGTAGGAGT-3′ to measure total bacterial load, using a standard curve.
DSS-induced acute colitis
Mice were administered dextran sodium sulfate (DSS) (MP Biomedicals, Solon, Ohio, USA) at 3.0% in drinking water ad libitum for 5 days, and control mice were given water only. After such a treatment, faeces were collected, resuspended in PBS to a concentration of 100 mg/mL and homogenised for 10 s using a Mini-Beadbeater-24 without the addition of beads to avoid bacteria disruption. We then centrifuged the samples at 8000 g for 2 min. These faecal suspensions were used for intraperitoneal injection.
Intraperitoneal injection of faecal suspension to RagKO mice and quantification of sera IL-6 and CXCL1 by ELISA
Faecal suspensions were used for intraperitoneal injection to RagKO mice (50 μL). Two hours postinjection, mice were bled retro-orbitaly, and IL-6 and CXCL1 levels were estimated in the serum using Duoset murine ELISA kit (R&D Systems, Minneapolis, Minnesota, USA).
Faecal microbiota analysis by 16S rRNA gene sequencing
Bulk DNA was extracted from frozen extruded faeces using a PowerSoil-htp kit from MoBio Laboratories (Carlsbad, California, USA) with mechanical disruption (bead-beating). The 16S rRNA genes were PCR amplified from each sample using a composite forward primer and a reverse primer containing a unique 12-base barcode, designed using the Golay error-correcting scheme, which was used to tag PCR products from respective samples.23 We used the forward primer 5′-GCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3′: the italicised sequence is 454 Life Sciences primer B, and the bold sequence is the broadly conserved bacterial primer 27F. The reverse primer used was 5′-GCCTCCCTCGCGCCATCAGNNNNNNNNNNNNCATGCTGCCTCCCGTAGGAGT-3′: the italicised sequence is 454 Life Sciences’ primer A, and the bold sequence is the broad-range bacterial primer 338R. NNNNNNNNNNNN designates the unique 12-base barcode used to tag each PCR product, with ‘CA’ inserted as a linker between the barcode and rRNA primer. PCR reactions consisted of HotMaster PCR mix (Eppendorf), 0.2 µM of each primer, 10–100 ng template, and reaction conditions were 2 min at 95°C, followed by 30 cycles of 20 s at 95°C, 20 s at 52°C and 60 s at 65°C on an Eppendorf thermocycler. Three independent PCRs were performed for each sample, combined and purified with Ampure magnetic purification beads (Agencourt), and products visualised by gel electrophoresis. Products were quantified using Quant-iT PicoGreen dsDNA assay. A master DNA pool was generated from the purified products in equimolar ratios. The pooled products were sequenced using a Roche 454 Titanium pyrosequencer at the University of South Carolina (EnGenCore).
16S rRNA gene sequence analysis
For quality-filtering the raw data, we discarded sequences <200 or >470 bp, and sequences containing primer mismatches, uncorrectable barcodes, ambiguous bases, or homopolymer runs in excess of six bases. The sequences that passed the quality filters were analysed using the Quantitative Insights Into Microbial Ecology (QIIME) software package.24 Sequences were checked for chimaeras using UCHIME and assigned to operational taxonomic units (OTUs) using OTUpipe25 with a 97% threshold of pairwise identity, and then classified taxonomically using the Greengenes reference database and a confidence threshold of 60%.26 A single representative sequence for each OTU was aligned using PyNAST,27 then a phylogenetic tree was built using FastTree.28 The phylogenetic tree was used for computing the UniFrac distances between samples,29 and we selected 543 sequences per sample (rarefaction) and used this as a basis to compare abundances of OTUs across samples. Jackknifing principal coordinates analysis (PCoA) plots were used to assess the variation between treatment replicates. Rarefaction curves were built based on 454-based DNA sequencing of 16S rRNA gene libraries from faeces, and depicts the number of unique OTUs.
Statistical analysis
Significance was determined using Student t test or one-way analysis of variance (GraphPad Prism software). Differences were noted as significant *p≤0.05. Kolmogorov–Smirnov test was used to verify that all data was normally distributed.
A ‘nearest-shrunken centroid’ classification approach was performed to detect the genera that were particularly representative of each category (WT vs T5KO, both AIEC-infected).30 ,31 The amount of shrinkage was set to minimise the misclassification error. When family were used to classify samples, the accuracy of the classification was perfect (overall error rate = 0). These analyses allowed the identification of phyla/genera whose abundances significantly differed between categories. This analysis was performed using the Prediction Analysis for Microarrays (PAM) package within R software.
Results
Transient colonisation of T5KO mice by AIEC results in chronic gut inflammation that correlates with elevated gut microbiota levels of flagellin and LPS
We recently reported that monoassociation of germfree mice with AIEC strain LF82 results in mild colitis in TLR5-deficient (T5KO), but not WT mice, supporting the notion that T5KO mice spontaneous colitis may be driven, at least in part, by inability to manage flagellated pathobionts.18 However, the severity of colitis was much greater in germfree T5KO mice that were exposed to AIEC and simultaneously placed in SPF housing even though it resulted in rapid displacement of AIEC by other microbiota. Herein, we investigated the possibility that such colitis might reflect that transient AIEC colonisation might induce functionally significant alterations in the gut microbiota that promote chronic inflammation. Germfree WT and T5KO mice were inoculated with 107 AIEC, placed in SPF housing, and faeces were collected. First, we quantitated AIEC in faeces, which is known to reflect levels of colonisation of the intestine. In accord with our recent report, AIEC levels were elevated in T5KO, relative to WT mice, 3–5 days postinoculation (PI), but became undetectable in both strains by 10 days PI (figure 1A). In further accord with our recent study, AIEC-induced inflammation in T5KO persisted beyond the period in which AIEC could be detected. Specifically, levels of faecal lipocalin-2 (Lcn2), which we recently demonstrated as a reliable indicator of colitis in several mouse models of colitis including T5KO mice,22 were markedly elevated in T5KO mice throughout the 10-day period post-AIEC clearance that the mice were assayed (figure 1B).

T5KO mice infected with adherent-invasive Escherichia coli (AIEC) strain LF82 developed intestinal inflammation associated with increased flagellin and lipopolysaccharide (LPS) load. (A) AIEC colonisation was determined in WT and T5KO mice by platting on media containing selective antibiotics. Data are represented as means±SEM of N=5 mice per group. (B) Faecal Lcn-2 levels were measured in faeces by ELISA. Bars are means±SEM of N=5 mice per group. (C and D) Faecal flagellin (C) and LPS (D) load were determined using HEK reporting cells. Bars are means±SEM of N=5 mice per group. (E) Faecal bacterial load day 20 postinfection, determined by q-PCR. Bar represent the means, N=3–4 mice per group.
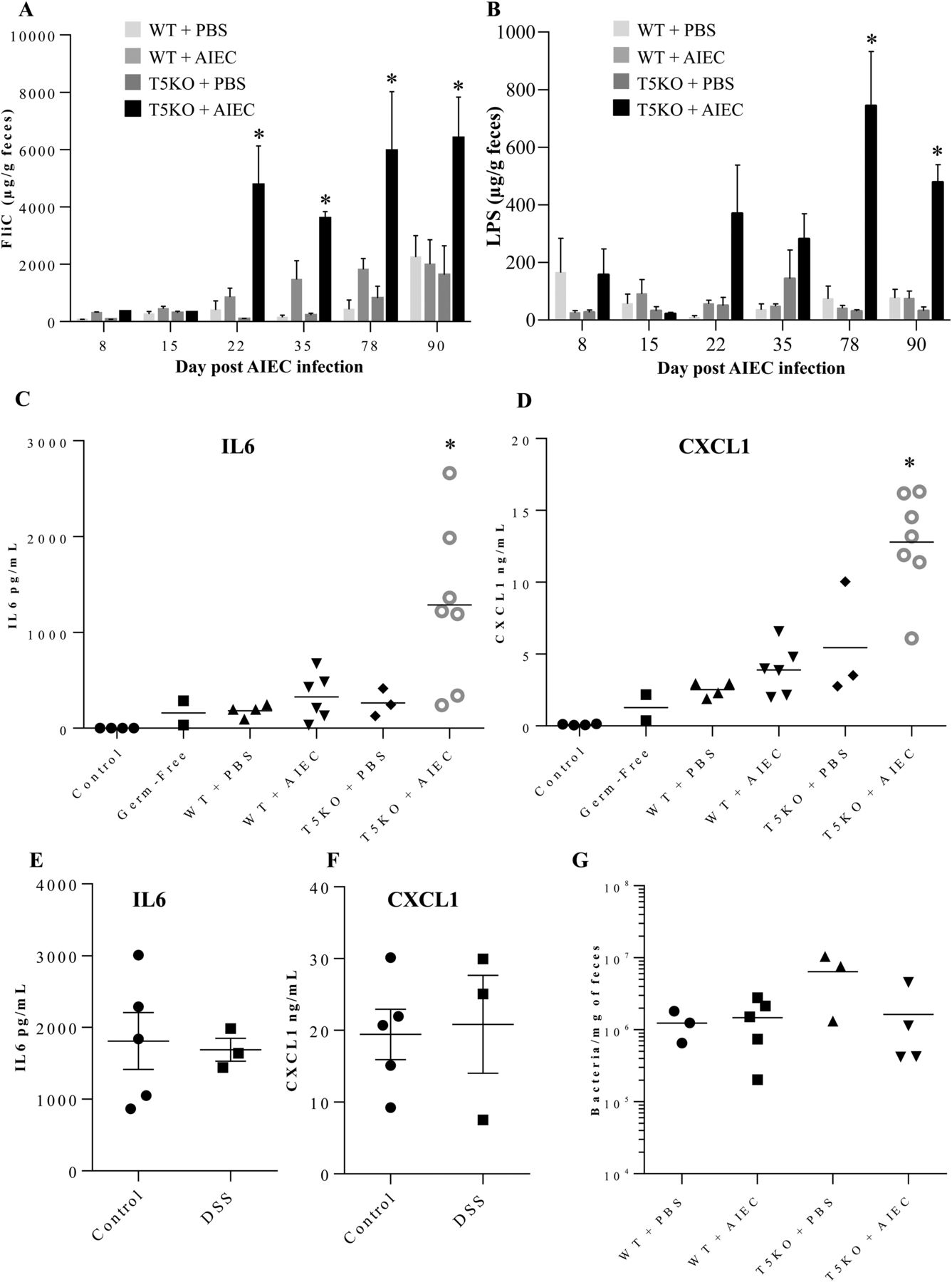
T5KO spontaneous colitis is dependent upon the LPS receptor TLR415 and the NLRC4 inflammasome,32 which is activated upon detection of flagellin. Thus, we hypothesised that the lasting colitis in T5KO mice induced by AIEC might involve alteration of the microbiota such that it might have an inherently greater potential to activate these pathways of innate immune activation. To investigate this possibility, we used a colorimetric-based reporter cell assay that measures relative levels of bioactive LPS and flagellin in mouse faecal extracts based on their abilities to activate cells expressing TLR4 or TLR5, which, like NLRC4, recognises conserved features of flagellin thought to be present in flagellins from numerous bacterial species. Controls for these assays are that the TLR4 and TLR5 reporter cells are activated by, respectively, LPS and flagellin, but not vice versa. Another control is that neither cell line is activated by faecal extract from germ-free mice (data not shown). Such reporter cell analysis revealed that the gut inflammation that followed transient AIEC colonisation correlated with increased levels of flagellin in T5KO mice by 10 days (figure 1C). Moreover, transient AIEC colonisation also resulted in statistically significant increased levels of LPS by 20 days post-AIEC colonisation (figure 1D). Such increased levels of bioactive flagellin and LPS exhibited by T5KO were not associated with significant differences in total levels of bacteria, as measured by q-PCR (figure 1E). Together, these results suggest that AIEC colonisation of T5KO mice causes alteration of microbiota composition and/or gene expression, resulting in it having inherently greater levels of LPS and flagellin, and thus, greater potential to activate TLR4 and NLRC4 in T5KO mice that had been exposed to this pathobiont prior to acquisition of a complex microbiota.
Our recent description of chronic colitis in T5KO mice transiently colonised by AIEC showed such inflammation persists at least 120 days following exposure to AIEC.18 Thus, we next analysed faecal samples from such mice to examine the extent to which such chronic colitis might also correlate with alterations in bioactive flagellin and LPS. Analogous to the results of the shorter-term experiment in figure 1, transient colonisation by AIEC as mice acquired a microbiota resulted in persistently increased levels of bioactive LPS and flagellin in T5KO but not WT mice (figure 2A,B). Next, we sought a means to assay the extent to which the reporter cell-based measures of LPS and flagellin bioactivity might represent ability of the microbiota to broadly activate innate immunity in a complex host, which, of course, has many innate immune receptors. Consequently, we injected the above-used faecal extracts from late time points (78–90 day) into RagKO mice (which have only an innate immune system) and, 2 h later, assayed serum levels of cytokines IL-6 and CXCL1 (mouse homologue of IL-8) by ELISA. Levels of these cytokines were all very low in untreated mice and went up only slightly in mice injected with faecal extracts from germfree mice (figure 2C,D). In accord with observations that healthy adult gut microbiota lack significant capacity to activate TLR4,33 only slight elevations in levels of these cytokines were observed upon injection of faecal extracts from conventionalised WT mice. While loss of TLR5 by itself, or addition of AIEC to WT mice, modestly enhanced the ability of faecal extracts to induce cytokines, a marked elevation in cytokines was observed in response to faecal extracts from chronically inflamed T5KO mice that had been earlier transiently colonised with AIEC (figure 2C,D). To investigate the extent to which such increased proinflammatory potential of the microbiota was a consequence of inflammation, this approach was applied to faeces from control and DSS-treated mice to induce intestinal inflammation (5-day treatment with 3% DSS). Interestingly, no difference was observed between faeces from control and DSS-treated mice, suggesting that the ability of faecal extracts to induce cytokines was not a simple consequence of the inflammatory state (figure 2E,F). Nor were such differences a consequence of altered total microbial loads, as all conditions resulted in similar total levels of faecal bacteria (figure 2G). Thus, transient presence of this pathobiont in T5KO mice results in long-term carriage of a microbiota with inherently greater proinflammatory potential.

Intestinal inflammation is dependent on adherent-invasive Escherichia coli (AIEC) infection and TLR5 deficiency. (A and B) Faecal flagellin (A) and lipopolysaccharide (B) load were determined using HEK reporting cells. Bars are means±SEM of N=3/5 mice per group. (C–F) Faecal suspension was injected intraperitoneally to RagKO mice, and IL6 (C and E) and CXCL1 (D and F) were measured in the serum 2 h postinjection. (G) Faecal bacterial load day 90 postinfection, determined by q-PCR. Bar represent the means, N=3/5 mice per group.
Pathobiont AIEC-induced inflammation in T5KO mice is associated with alterations in gut microbiota species composition
Next, we investigated the extent to which elevated levels of flagellin and LPS in AIEC-exposed T5KO mice reflect altered microbiota composition. Temporally collected faecal samples from WT and T5KO mice that had been exposed to AIEC or PBS were subjected to 454 pyrosequencing of 16S rRNA genes. Sequence data was analysed via the QIIME data processing pipeline.24 In agreement with our culture-based result that AIEC administered to germfree mice was cleared within a few weeks of placing these mice in SPF housing, 454 pyrosequencing of the microbiota indicated that the genus Escherichia, including E coli and other species, was in very low abundance in both WT and T5KO mice within 3 weeks (figure 3A). A modest bloom of Escherichia was seen in T5KO given PBS, and then placed in SPF in agreement with the notion that such bacteria are frequent albeit perhaps not ubiquitous member of the mouse microbiota. Analysis of microbiotas at the family level did not show any significant differences among the groups of mice assayed (data not shown) and, overall, differed from truly conventional lab mice (ie, those born and raised in SPF housing) in that they displayed a marked reduction in Bacteroidetes perhaps suggesting that methods of ‘conventionalising’ germfree mice can affect their microbiota composition. Nonetheless, analysis at the genus level by unweighted UniFrac and principal component (PC) analysis indicated that our placement of germfree mice into SPF housing eventuated in relatively stable microbiotas within a few months (figure 3). For WT mice, plotting of PC1 over time indicated that the transient colonisation by AIEC did not lastingly affect microbiota composition at the species level (figure 3B). Moreover, two-dimensional (2D) plotting of PC1 versus PC3 failed to show a pattern of clustering of WT mice that had been exposed to AIEC (figure 3C). Thus, transient colonisation by AIEC may not alter microbiota composition in WT hosts. By contrast, AIEC dramatically altered microbiota composition in T5KO mice well after the AIEC was cleared and the microbiota composition stabilised. Such difference was readily apparent by 3 months post-AIEC administration, when plotting PC1 versus time (figure 3D) and when 2D plotting PCs at the latest time points assayed (figure 3E). In accordance, comparison of microbiotas of WT and T5KO mice that had all received AIEC revealed a clear distinction in microbial composition emerging by about day 70, which is over a month after AIEC had largely been eliminated (figure 3F,G). Much of such difference in the PC analysis was driven by changes in relative abundance of Lachnospiraceae family of the Clostridiales order which, interestingly, are thought to produce flagellins that are immunodominant antigens in several T-cell-mediated colitis models and CD.34 ,35


T5KO mice infected with adherent-invasive Escherichia coli (AIEC) displayed modified intestinal microbiota. (A) AIEC colonisation were determined in WT and T5KO mice based on 454 pyrosequencing 16S rRNA analysis to determine their relative abundance in faeces. Data are represented as means±SEM of N=3/5 mice per group. (B) Mouse faecal bacterial communities from WT uninfected (PBS) or infected with 107 AIEC strain LF82 were clustered using principal coordinates analysis (PCoA) of the unweighted UniFrac distance matrix. Principal component 1 (PC1) is plotted for each time point (from day 1 to day 119). The time is expressed on the X-axis, and the percentage of the variation explained by the plotted principal coordinates is indicated in the Y-axis labels. (C) Mouse faecal bacterial communities from WT uninfected (PBS) or infected with 107 AIEC strain LF82 were clustered using PCoA of the unweighted UniFrac distance matrix. PC1 and PC2 are plotted and the percentage of the variation explained by the plotted principal coordinates is indicated in the Y-axis labels. (D and E) Mouse faecal bacterial communities from T5KO uninfected (PBS) or infected with 107 AIEC strain LF82 were clustered using PCoA of the unweighted UniFrac distance matrix (see figure 3B,C). (F and G) Mouse faecal bacterial communities from WT or T5KO mice infected with 107 AIEC strain LF82 were clustered using PCoA of the unweighted UniFrac distance matrix (see figure 3B,C). (H) Members of the microbiota that differ in abundance between WT and T5KO mice infected with 107 AIEC strain LF82. The heat map indicates the relative abundance of the top 13 bacterial operational taxonomic unit (OTU) accounting for the differences between WT and T5KO mice after AIEC infection. Columns show, for each sample, the abundance data of genera listed in the right part. The abundances of the genera were clustered using unsupervised hierarchical clustering (white, low abundance; red, high abundance). The phylum (P), class (C), order (O) and family (F) of each of the classifying OTUs are noted.
In addition to the specific composition of the microbiota, several studies indicate an important feature of a healthy microbiota is its species richness wherein greater diversity has been associated with health and a community with reduced species richness has been associated with disease such intestinal inflammation. Diversity can be assessed by rarefaction curves wherein the asymptote reflects level of diversity. In the absence of AIEC exposure, rarefaction curves from WT and T5KO mice were superimposable at both day 14 and day 119 (figure 4A,B). Further, rarefaction curves of WT and T5KO mice infected with AIEC revealed no difference in terms of microbiota diversity at early time points after AIEC clearance (day 14, figure 4C). In both WT and T5KO mice, there was an increase in diversity between day 14 and the later time point in accord with previous studies about microbiota acquisition. However, such transient colonisation by AIEC resulted in a significant reduction in diversity of the gut microbiotas in T5KO mice at the later time point (day 119, figure 4D), whereas no modification of total microbial loads was observed (figure 2G). Thus, transient colonisation of T5KO but not WT mice by AIEC, in early stages of microbiota development, results in altered gut microbiota composition, including reduction in diversity, that results in elevated levels of LPS and flagellin that gives the microbiota inherently greater proinflammatory potential and eventuates in development of chronic colitis in susceptible host.
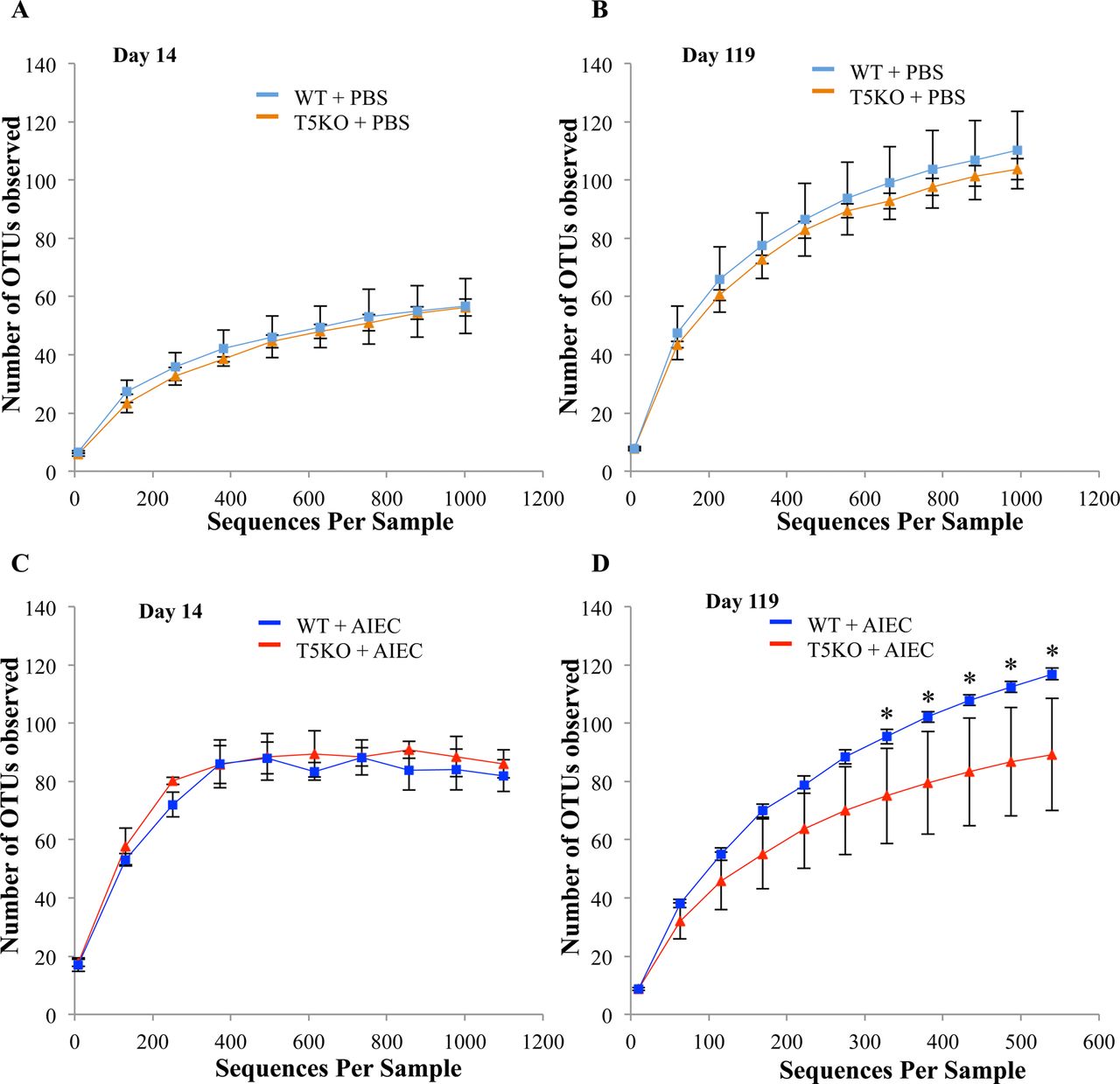
Adherent-invasive Escherichia coli (AIEC) infection decreases the richness and diversity of intestinal microbiota in T5KO mice. Rarefaction curves at day 14 (A–C) and day 119 (B–D) for uninfected (A and B) or AIEC infected (C and D) groups. The rarefaction curve is based on 454-based DNA sequencing of 16S rRNA gene libraries from faeces, and depicts the number of unique operational taxonomic units (OTUs). Data are represented as mean±SEM of N=3/5 mice per group. The slope of the line is directly related to the number of unique OTUs (ie, increased diversity) in a sample.
AIEC's dependence on flagellin to instigate colitis correlates with rapid and prolonged alteration in microbiota levels of flagellin and LPS
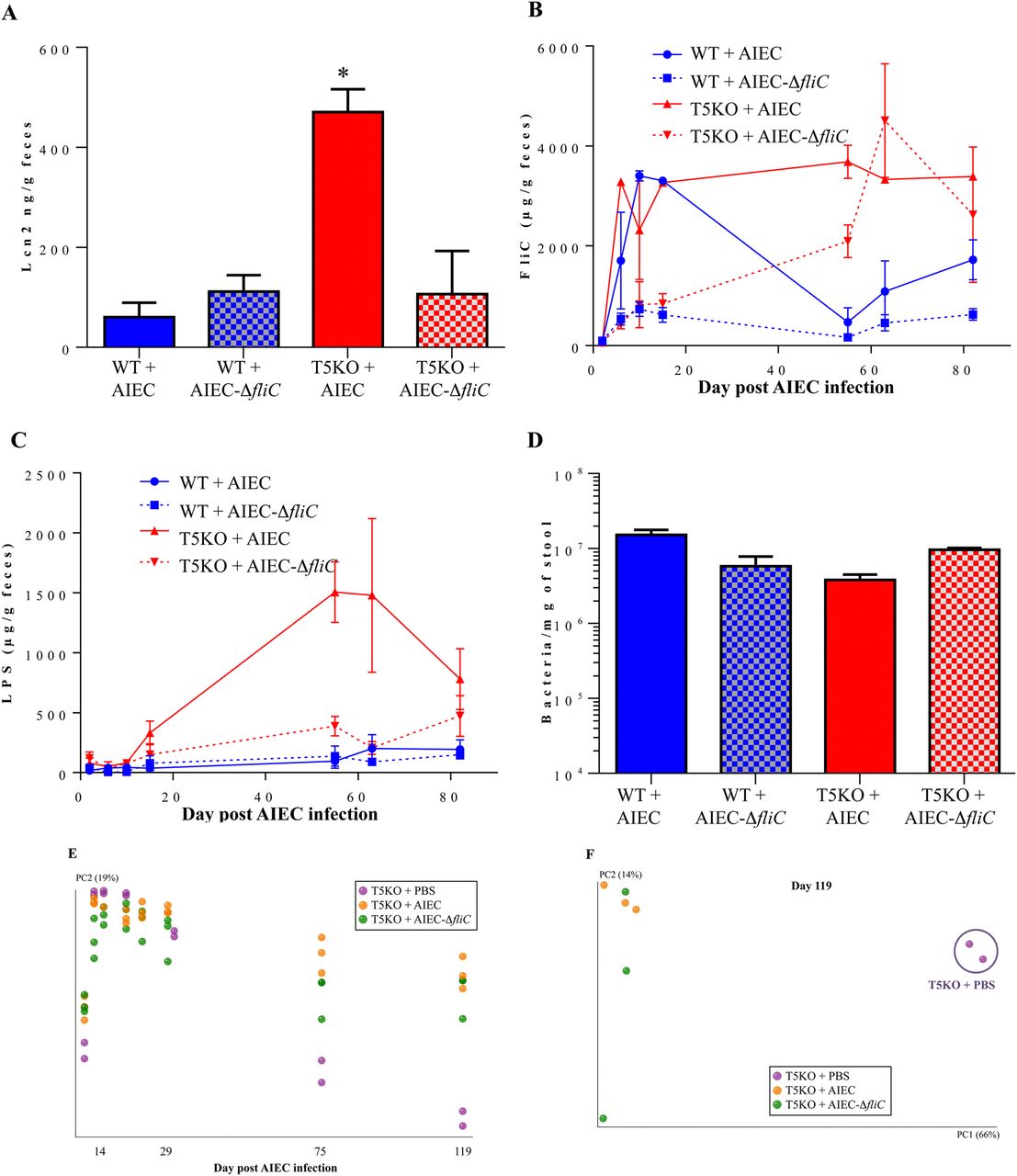
We recently reported that AIEC's ability to instigate chronic colitis in T5KO mice required the AIEC strain be flagellated.18 Thus, in order to better understand the inter-relationship between chronic inflammation, altered microbiota composition per se, and elevated levels of flagellin and LPS (which may or may not reflect composition), we investigated to what extent these parameters correlated in germ-free mice inoculated WT or aflagellate AIEC LF82-ΔfliC isogenic mutant. In accord with our recent report that germ-free WT and T5KO mice rapidly clear AIEC (WT or aflagellate strain), AIEC sequences were largely absent within a few weeks PI, but yet T5KO exposed to flagellated AIEC exhibited chronic inflammation as assessed by histological analysis or the faecal inflammatory marker LCN-218 (figure 5A, and data not shown). Use of reporter cells to measure flagellin levels revealed that, at early time points, that is, within 2 weeks PI, bioactive flagellin was readily detected in WT and T5KO mice that had been inoculated with flagellated AIEC but was present at only low levels in either mouse genotype inoculated with aflagellate AIEC (figure 5B). The levels of flagellin remained elevated in T5KO mice throughout the 120-day time course of study. By contrast, the flagellin levels decreased in WT mice and remained significantly lower than that of T5KO mice for the remainder of the study. In WT mice inoculated with aflagellate AIEC, flagellin levels remained low at all time points while similarly treated T5KO mice exhibited a slow climb in flagellin levels such that eventually became similar to that in T5KO mice exposed to flagellated AIEC. Levels of bioactive LPS were initially relatively low for all groups but, in T5KO mice, began to increase after a couple of weeks following inoculation with flagellated AIEC roughly paralleling their previous development of colitis (figure 5C). Such differences in flagellin and LPS were not paralleled by, and hence not a consequence of, differences in total bacterial load (figure 5D). The slow increase in flagellin seen in T5KO mice inoculated with aflagellate AIEC suggests a general inability of these mice to manage their gut microbiota even in the absence of overt colitis. Such gradual increase in flagellin loads may reflect inability to properly manage flagellated members of the microbiota.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adherent-invasive Escherichia coli (AIEC's) dependence on flagellin to instigate colitis correlates with rapid and prolonged alteration in microbiota levels of flagellin and lipopolysaccharide (LPS). (A) Faecal Lcn-2 levels were measured in faeces by ELISA. Bars are means±SEM of N=3 mice per group. (B and C) Faecal flagellin (B) and LPS (C) load were determined using HEK-reporting cells. Bars are means±SEM of N=3 mice per group. (D) Faecal bacterial load, determined by q-PCR. Bar represent the means, N=2/3 mice per group. (E) Mouse faecal bacterial communities from T5KO uninfected (PBS) or infected with 107 AIEC strain LF82 or LF82-ΔfliC isogenic mutant were clustered using principal coordinates analysis (PCoA) of the UniFrac unweighted distance matrix. Principal component 2 (PC2) is plotted for each time point (from day 1 to day 119). The time is expressed on the X-axis, and the percentage of the variation explained by the plotted principal coordinates is indicated in the Y-axis labels. (F) Mouse faecal bacterial communities from T5KO uninfected (PBS) or infected with 107 AIEC strain LF82 or LF82-ΔfliC isogenic mutant were clustered using PCoA of the UniFrac unweighted distance matrix. PC1 and PC2 are plotted and the percentage of the variation explained by the plotted principal coordinates is indicated in the Y-axis labels.
We next examined microbiota composition as previously described and we compared relative composition over time for T5KO mice untreated, or infected with either wild type LF82 bacteria or LF82-ΔfliC isogenic mutant. This analysis showed clear, distinct, clustering starting at day 75 postinfection between PBS-treated T5KO mice and AIEC-inoculated T5KO mice (figure 5E) regardless of whether the AIEC was flagellated or not. PCoA plots of unweighted UniFrac confirmed this observation, with a dramatic difference in the species level composition between PBS-treated T5KO mice and AIEC-inoculated T5KO mice at day 119 post-infection (figure 5F). These results suggest that loss of TLR5 makes mice prone to exhibiting pathobiont-induced alterations in microbiota composition regardless of whether they display overt inflammation. Moreover, they suggest that change in microbiota composition per se may not be as a pivotal factor as level of expression of immune modulating bacterial factors LPS and flagellin, of which early and prolonged levels of the latter were the best correlates of colitis in our conventionalised germfree mice.
Discussion
Alterations in select genes that mediate innate immunity predispose individuals to developing IBD. However, such alterations are generally not, by themselves, sufficient to result in IBD as most people carrying such risk alleles do not develop this disease.1 Many studies have suggested the possibility of an infectious trigger of IBD in susceptible hosts, but attempts to associate specific organisms with disease have mostly yielded negative results. Yet, the observation that the use of antibiotics in childhood is associated with development of IBD in adulthood suggests that key aspects of the host–microbial interactions that can eventuate in IBD may begin many years before disease clinically manifests, potentially obfuscating results from microbe-hunting studies in patients with IBD.36 Our recent study of T5KO mice provides an example of such potential complexity in that T5KO mice that developed chronic colitis displayed transient inability to control Proteobacteria in the post-weaning period.18 That such Proteobacteria played a role in driving colitis was supported by observation that administration of pathobiont Adherent Invasive E coli (AIEC) strain LF82 to T5KO mice as they acquired a microbiota-triggered colitis that persisted well after the administered AIEC was cleared. If applicable to humans, these results suggest that a transient infection by an opportunistic pathogen could, in a genetically susceptible host, result in chronic intestinal inflammation. However, how transient colonisation by a pathobiont might result in chronic inflammation is not clear, but could be envisaged to involve the transient presence of the perturbing microbe resulting in lasting changes in the mucosal immune system and/or the gut microbiota. Herein, we investigated the latter possibility in our model and found that, indeed, transient colonisation of T5KO mice by the CD-associated pathobiont AIEC strain LF82 resulted in lasting changes in the gut microbiota as assessed by measuring its species composition and inherent proinflammatory potential.
The observation that AIEC-induced alteration in microbiota composition in T5KO mice correlate with colitis is in accord with many human and animal studies that have associated altered microbiota with gut inflammation. However, the role of such altered microbiota in disease has remained elusive. Thus, our findings herein provide one relatively straightforward potential means by which an altered microbiota might drive inflammation. Specifically, that the development of colitis in AIEC-exposed T5KO mice, and its concomitantly associated altered microbiota, correlated with higher faecal levels of LPS and flagellin, as assayed by its ability to stimulate proinflammatory gene expression through their cognate innate immune receptors. Such higher levels of LPS and flagellin may result in increased activation of TLR4 and the NLRC4 inflammasome, thus explaining why the spontaneous colitis in T5KO mice depended upon presence of both TLR4 and NLRC4.15 Such functional differences in the microbiota are not necessarily restricted to flagellin and LPS per se, but may reflect a broad enhancement in its inherent innate immune activating potential. Indeed, in the absence of TLR4 or NLRC4, T5KO mice are still prone to develop colitis upon induction of immune dysregulation, such as blockade of signalling by the anti-inflammatory cytokine IL-10. Thus, while LPS and flagellin are two major activators of proinflammatory mediators, there is likely a role for other microbial products in driving gut inflammation. It will be important for future studies to determine if such boosting of the microbiota's inherent proinflammatory potential is specific to T5KO mice, a general consequence of a number of innate immune deficiencies, or common to gut inflammation in general. It will also be important to determine the extent to which increased microbiota proinflammatory potential may be a consequence of inflammation. Concerning this notion, our data presented here using faeces from DSS-treated mice suggest that the ability of faecal extracts to induce cytokines may be independent of inflammation state. Regardless, increases in the inherent ability of microbiota components to activate innate immunity may play a role in perpetuating chronic inflammation in IBD. This would support the approach of selectively inhibiting innate immune signalling in IBD even if the disease resulted from an inherent innate immune insufficiency.
While it is inherently difficult to distinguish the extent to which alterations in microbiota are a cause and/or consequence of disease, the changes seen in the microbiotas of colitic T5KO mice are likely to play a role in driving gut inflammation in that transfer of microbiota from colitic T5KO to germ-free T5KO promotes development of colitis (Gewirtz/Chassaing, unpublished observation). However, defining the roles of specific bacterial species remains quite difficult. Nonetheless, analysis of the specific OTUs whose abundance are altered in AIEC-administered T5KO relative to similarly treated WT mice proved intriguing. Specifically, of the 13 OTU whose abundance differed the most between AIEC-treated WT and T5KO mice, 12 were of the Clostridiales order (of the Clostridia class), of which eight were of the Lachnospiraceae family members. Considering that Lachnospiraceae flagellins have been reported to be dominant targets of adaptive immunity in both T-cell-mediated models of murine colitis and CD,30 ,31 it is tempting to speculate that changes in their relative abundance are involved in the chronic inflammatory process. However, given that some OTU increase in abundance while others decrease, and that little is known concerning flagellin genes/expression by individual OTUs, deciphering the roles of the specific OTUs will be challenging. Moreover, considering that WT germ-free mice given microbiota from colitic T5KO mice do not develop colitis (Gewirtz/Chassaing, unpublished observation) and, analogously, WT germ-free mice given flagellated E coli LF82 do not develop colitis (our results herein), suggest that the mere presence of flagellated (and LPS containing) bacteria, even in a select developmental window, is not sufficient to drive colitis, but rather such factors and a host defect in controlling/containing the challenge may be required.
In further accord with the notion that mere presence of LPS and flagellin is sufficient to drive a developing microbiota-mucosal immune relationship to result in chronic colitis, we note that administration of a flagellated commensal E coli during microbiota acquisition does not result in colitis even in T5KO mice.18 Thus, there is likely a role for specific virulence factors in AIEC LF82 and perhaps other AIEC isolates. Indeed, strain LF82 is one of a numerous AIEC strains that exhibit considerable genetic heterogeneity. Consequently, it will be important for future research to define the extent to which our findings with LF82 apply to other AIEC isolates. In any case, we note that AIEC LF82 is far better equipped to colonise the human rather than murine intestine in that its adherence to the intestine is mediated by the type 1 pili receptor, namely CEACAM6, which is present in humans but not mice.37 ,38 Thus, we do not propose a central role for AIEC in development of colitis per se but rather put forth the concept that pathobiont-altered microbiota composition rather than any of these specific species differences that might be relevant to human disease. One overarching mechanism could be the lowering of overall microbiota diversity, a feature induced by AIEC in T5KO mice and seen in the IBD-associated microbiota in several studies.39 Again, that such reduction of diversity, or indeed any lasting change in microbiota composition, was not seen in WT mice suggests that AIEC colonisation per se was not sufficient to induce these changes. Rather, we hypothesise that, in a genetically susceptible host, the presence of a pathobiont in a developing microbiota can result in lasting changes in microbiota composition that may eventuate in chronic inflammation.
Acknowledgments
We thank Dr Arlette Darfeuille-Michaud (Université d'Auvergne) for providing E coli LF82 and aflagellated isogenic mutant.
References
Footnotes
-
Contributors BC, FAC and ATG designed the study. BC, OK, FAC and ATG conducted the experiments, and analysed the data. BC, REL and ATG wrote the manuscript.
-
Funding This work was supported by NIH grant DK061417. BC is a recipient of the Research Fellowship award from the Crohn's and Colitis Foundation of America (CCFA).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.