Article Text

Abstract
Objective Extensive molecular heterogeneity of pancreatic ductal adenocarcinoma (PDA), few effective therapies and high mortality make this disease a prime model for advancing development of tailored therapies. The p16-cyclin D-cyclin-dependent kinase 4/6-retinoblastoma (RB) protein (CDK4) pathway, regulator of cell proliferation, is deregulated in PDA. Our aim was to develop a novel personalised treatment strategy for PDA based on targeting CDK4.
Design Sensitivity to potent CDK4/6 inhibitor PD-0332991 (palbociclib) was correlated to protein and genomic data in 19 primary patient-derived PDA lines to identify biomarkers of response. In vivo efficacy of PD-0332991 and combination therapies was determined in subcutaneous, intrasplenic and orthotopic tumour models derived from genome-sequenced patient specimens and genetically engineered model. Mechanistically, monotherapy and combination therapy were investigated in the context of tumour cell and extracellular matrix (ECM) signalling. Prognostic relevance of companion biomarker, RB protein, was evaluated and validated in independent PDA patient cohorts (>500 specimens).
Results Subtype-specific in vivo efficacy of PD-0332991-based therapy was for the first time observed at multiple stages of PDA progression: primary tumour growth, recurrence (second-line therapy) and metastatic setting and may potentially be guided by a simple biomarker (RB protein). PD-0332991 significantly disrupted surrounding ECM organisation, leading to increased quiescence, apoptosis, improved chemosensitivity, decreased invasion, metastatic spread and PDA progression in vivo. RB protein is prevalent in primary operable and metastatic PDA and may present a promising predictive biomarker to guide this therapeutic approach.
Conclusion This study demonstrates the promise of CDK4 inhibition in PDA over standard therapy when applied in a molecular subtype-specific context.
- pancreatic cancer
- extracellular matrix
- molecular mechanisms
- cell cycle
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Despite significant recent efforts into developing novel therapeutics, chemotherapy remains the standard of care for patients with advanced pancreatic cancer, despite only modestly improving overall survival.
We and others have previously shown that pancreatic cancer is a molecularly varied disease and requires development of personalised treatment strategies to improve survival rates.
Pancreatic cancer harbours frequent deregulation of the p16-cyclin D-CDK4/6-retinoblastoma (RB) protein pathway (CDK4 pathway), which could be therapeutically exploited.
What are the new findings?
Using a range of robust patient-derived xenograft and cell line models, as ‘avatars of matched patient pancreatic tumours’, we show exquisite sensitivity to CDK4/6 inhibitor combinations over standard therapy in subtypes of pancreatic cancer, associated with increased apoptosis, quiescence and reduced expression of markers associated with pancreatic stellate cell activity.
PD-0332991 facilitates remodelling of the extracellular matrix (ECM) to improve therapeutic efficacy of gemcitabine (GEM) in a subtype-specific manner and at multiple stages of disease progression: primary tumour growth, recurrence (second-line therapy) and the metastatic setting and may potentially be guided by a simple, clinically applicable biomarker (RB protein).
CDK4 inhibitor, PD-0332991 (palbociclib) hinders metastatic colonisation in the liver while sensitising cells to shear stress, commonly experienced by metastasising circulating tumour cells, an additional way in which CDK4 inhibition can directly impinge on metastasis in RB-high pancreatic ductal adenocarcinoma (PDA).
RB protein expression was identified as an independent prognostic factor for patient survival in two independent patient cohorts of pancreatic cancer and can be detected in a significant proportion of metastatic cases (67%). RB expression correlated between primary and matched metastatic human tissue.
CDK4 pathway inhibition alone or in combination with chemotherapy leads to multifaceted global inhibitory effects on tumour cells at different stages of PDA progression and reduction in key components within the tumour microenvironment (ECM), which collectively drive cancer cell proliferation, invasion and chemoresistance.
How might it impact on clinical practice in the foreseeable future?
Our findings, together with the recent clinical evidence on the efficacy of PD-0332991 in the treatment of breast cancer, support the further clinical companion biomarker-driven development of PD-0332991 as pancreatic cancer therapy, particularly in combination with chemotherapeutic, GEM.
The use of a tissue-based assay for RB expression could be adapted for clinical application in diagnostics. As high RB expression was detected in a significant proportion of patients with pancreatic cancer (~65%,~43% and 67% in cohorts of primary operable and metastatic PDA examined), it may enable stratification and enrichment for the potential responders to the CDK4/6 combination therapy.
These findings are timely and may be complementary and of direct relevance to the current non-biomarker-driven clinical trials testing PD-0332991 in pancreatic and other solid cancers (NCT02501902, NCT03065062).
Introduction
Pancreatic ductal adenocarcinoma (PDA) is a molecularly diverse disease, driven by aberrations in at least 10 core signalling pathways.1–4 In addition to revealing new biological insight into the molecular pathogenesis of PDA, these studies have created significant opportunities to develop novel, tailored treatments, as we have recently shown.5 6 The standard of care chemotherapeutic gemcitabine (GEM),7 in combination with nanoparticle albumin-bound paclitaxel (nab-paclitaxel),8 or FOLFIRINOX (oxaliplatin, irinotecan, fluorouracil and leucovorin)9 offers only modest increases in patient survival in unselected populations.10 Selective measures to personalise treatment in this aggressive disease are therefore urgently required.
The p16-cyclin D-CDK4/6-retinoblastoma (RB) protein pathway (CDK4 pathway) promotes G1/S-phase transition and is one of the key signalling pathways deregulated in PDA.1 2 11 Cyclin D complexes with cyclin-dependent kinases (CDKs) 4 and 6, driving RB protein phosphorylation and G1 phase progression.12 In cancer, CDK4 activity and subsequent uncontrolled proliferation can be enhanced by homozygous deletion of CDKN2A gene (which encodes p16INK4A protein), CDK4/6 mutations and/or cyclin D1 (CCND1) overexpression or amplification.13
Discovery of cyclin D/CDK4 as an oncogene has fuelled development of small-molecule CDK inhibitors (CDKi). Selective CDK4/6i, including PD-0332991 (palbociclib), are showing great potential in the treatment of ER+/HER2– metastatic breast cancer.14–16 Although no biomarker of treatment response has been found, a key requirement for sensitivity to CDK4/6i in breast cancer may be linked to ER– positivity and dependence of the ER+ subtype on cyclin D1 signalling, enhancing CDK4/6 activity to drive proliferation.12 17
In PDA, short-term antiproliferative activity of CDK4/6i has been described.18 Here, we investigated the therapeutic efficacy of CDK4/6i and clinically applicable combinations with long-term follow-up in realistic advanced preclinical patient-derived and genetically engineered models of PDA. We reveal a new role of CDK4 inhibition in regulating primary tumour growth and stromal desmoplasia in several independent mouse models of RB-high PDA, impairing colonisation of secondary sites (liver) and resistance to shear stress, frequently experienced by circulating tumour cells (CTCs), while delaying disease progression in metastatic RB-high PDA. Detailed analyses reveal a complex mechanism of action with PD-0332991 exerting dual effects on both tumour cells and the surrounding environment. Assessment of RB expression, candidate CDK4/6i companion biomarker, revealed high prevalence in primary operable and metastatic human PDA. Collectively, these preclinical findings may offer promising new opportunities in subtype-specific targeting of primary and metastatic PDA.
Methods
Patient-derived models
Our Kinghorn Cancer Centre (TKCC) patient-derived xenografts (PDXs; n=45) and patient-derived cell lines (PDCLs; n=19) have previously been whole genome sequenced.1 PDCLs were confirmed by short tandem repeat (STR) profiling as unique (cellbankaustralia.com). Culture conditions, fluorescent ubiquitination-based cell cycle indicator (FUCCI) and luciferase introduction into PDCLs and associated in vitro studies: cell cycle, migration, invasion, shear stress, contraction and apoptosis assays are detailed in online supplementary information 2.
Supplemental material
Animal studies
Survival studies were performed by subcutaneously implanting early third-passage PDX pieces (~4 mm3) into immunocompromised Balb/c-Fox1nuAusb (Nude) mice. PD-0332991, GEM and nab-Paclitaxel dosages were based on published studies,5 18 19 with treatment schedules, response measurement and setup for intrasplenic and orthotopic studies detailed in online supplementary information 2.
Patient materials and immunohistochemistry
Patient samples and data were acquired through the Australian Pancreatic Genome Initiative (APGI)/International Cancer Genome Consortium (ICGC) (n=200), New South Wales Pancreatic Cancer Network (NSWPCN; n=365) and the Royal North Shore Hospital metastatic cohort (RNSH; n=54), as described.1 5 20 Further cohort description and immunohistochemical procedures are outlined in online supplementary information 2.
Statistics
Statistical analyses were performed using GraphPad Prism (V7.0.1, GraphPad, La Jolla, California, USA) and Statview (V5.0, SAS Institute Inc., Cary, NC, USA). Correlation analyses were performed using Pearson correlation coefficient test. Comparison between clinicopathological correlates was performed using Fisher’s exact test, Student’s t-test and Χ2 test, where appropriate. Survival analyses were performed using the log-rank test. For multiple treatment group comparisons, significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Clinicopathological variables shown by univariate analysis to have p<0.25 were entered into a Cox proportional hazards regression model for multivariate analysis. To assess the agreement of RB status between primary and metastatic PDAs, Cohen’s Kappa and 95% CIs were reported.
Results
PD-0332991 sensitivity of pancreatic cancer patient-derived cell lines (PDCLs) correlates with high levels of total and phosphorylated RB protein
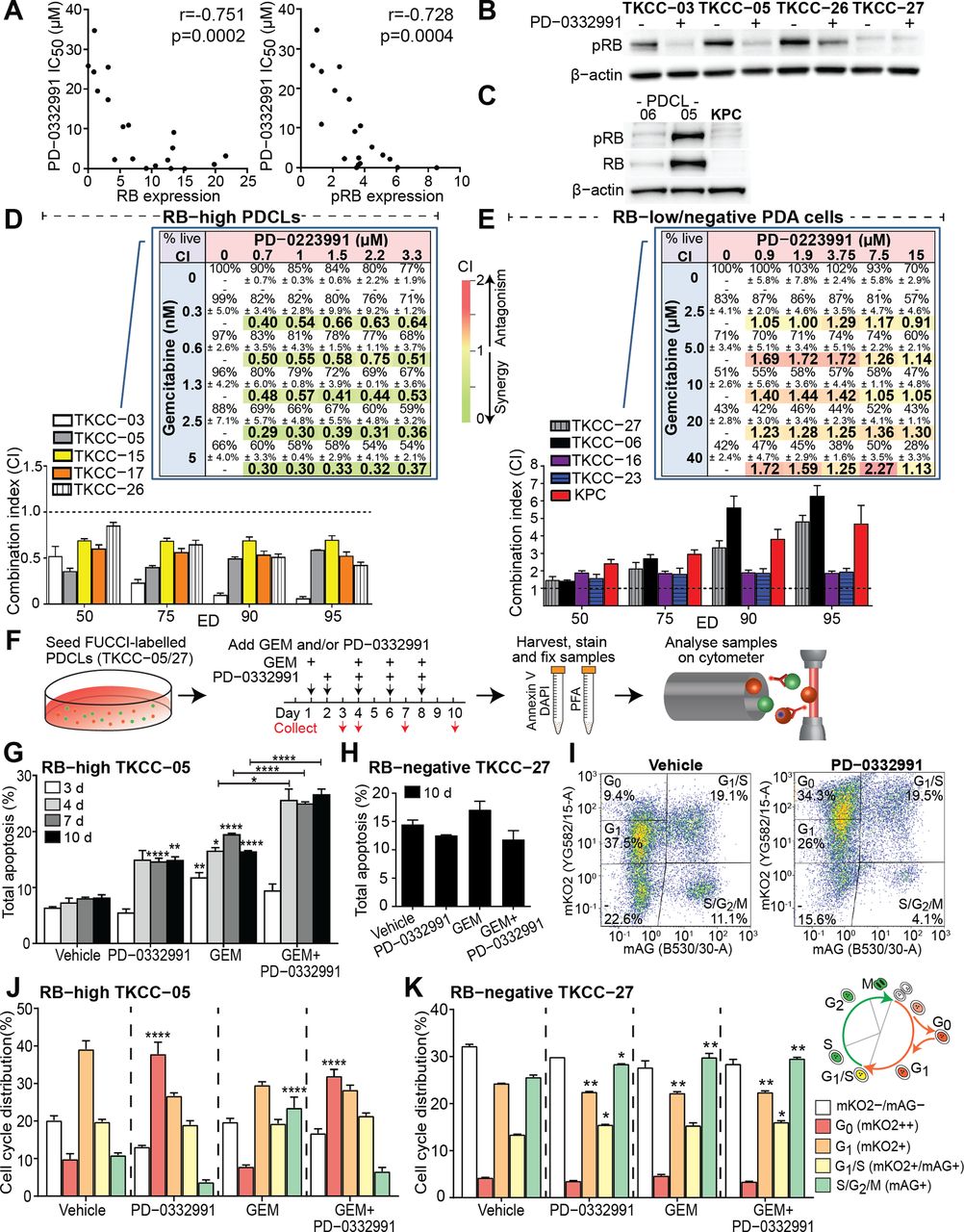
To systematically examine the efficacy of CDK4/6i PD-0332991 in PDA, we determined its toxicity in 19 whole genome-sequenced5 primary PDCLs and compared these data with several clinically used chemotherapeutics (see online supplementary table 1). PDCLs exhibited a broad range of PD-0332991 sensitivity (inhibitory concentration 50 (IC50) range 8 nM–35 µM), with a subset (26%) being highly sensitive (IC50 <1 µM). The ‘Genomics of Drug Sensitivity in Cancer’ (Wellcome Trust) database analysis revealed an association between RB1 mutations and pan-cancer PD-0332991 resistance (p=3.3×10–17, see online supplementary figure 1A). As RB1 mutations in pancreatic cancer are rare1 (1/19 PDCLs; see online supplementary table 1), we next correlated the baseline protein expression of key G1/S checkpoint components (see online supplementary figure 1B, supplementary table 2) with in vitro PD-0332991 response. Of note, total and phosphorylated RB (pRB; Ser780) levels were the most significant biomarkers predicting PD-0332991 sensitivity (figure 1A; r=−0.751; p=0.0002 and r=−0.728, p=0.0004, respectively), with no correlation between G1/S checkpoint mutations/expression and response to PD-0332991 or chemotherapeutics in pancreatic PDCLs (see online supplementary table 3, supplementary figures 1C–E).
Supplemental material

PD-0332991 potentiates gemcitabine (GEM) lethality in stratified pancreatic cancer patient-derived cell lines (PDCLs) and leads to increased apoptosis and G0 cell cycle quiescence. (A) Correlation of PD-0332991 sensitivity and expression of (left) retinoblastoma (RB) or (right) phosphorylated RB (pRB) in 19 PDCLs (normalised to β-actin levels and to low-expressing TKCC-16; included on multiple gels to account for any intergel/transfer variability). For densitometry analysis of normalised protein expression and correlation summaries please refer to supplementary tables 2 and 3. (B) Expression of pRB in RB-high (TKCC-03, TKCC-05, TKCC-26) and RB-negative (TKCC-27) PDCLs 24 hours post-PD-0332991 treatment (0.3 µM or 1 µM, respectively). (C) Western blot depicting negligible levels of pRB and RB proteins in pancreatic cancer cells isolated from the genetically engineered Pdx1-Cre; KrasLSL.G12D/+; p53R172H/+ (KPC) model of pancreatic cancer. Combination index (CI) of the GEM/PD-0332991 combination when agents were combined at a fixed ratio derived from their respective inhibitory concentration 50 (IC50) values in (D) RB-high and (E) RB-low/negative PDCLs. CI values determined at various effective doses (EDs) were calculated using CompuSyn program. CI >1 indicates antagonism, CI <1 synergy and CI=1 additive.23 Data are presented as mean± SEM; (n=4 independent experiments performed in triplicate). Inset: Heat maps showing viability and CI values of GEM/PD-0332991 combination in various ratios in candidate (D) RB-high (TKCC-03) and (E) RB-negative cells (TKCC-27; n=3 independent experiments performed in triplicate). (F) Fluorescent ubiquitination-based cell cycle indicator (FUCCI)-labelled TKCC-05 or TKCC-27 cells were treated with GEM (0.01 µM for both), PD-0332991 (0.3 µM or 1 µM, respectively) or combination and at selected time points (3–10 days) stained with Annexin-V-Cy5/4',6-diamidino-2-phenylindole (DAPI) and analysed using flow cytometry. Total apoptosis quantified across all examined time points and treatments for (G) RB-high TKCC-05 and (H) RB-negative TKCC-27 cells. (I) Representative TKCC-05-FUCCI cell cycle plots and gating strategy post-treatment (10 days) and control. Quantified cell cycle distribution across all examined treatments (10 days time point) in (J) RB-high and (K) RB-negative PDCLs. Inset: Illustration of FUCCI cell cycle oscillations. Data are shown as mean±SEM (n=4 independent experiments), with comparisons performed against relevant vehicle controls, unless specified. Significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test, where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. PDA, pancreatic ductal adenocarcinoma; PFA, paraformaldehyde.
Effective target modulation was confirmed in candidate RB-high PDCLs by western blotting for pRB post-PD-0332991 treatment (TKCC-03/-05; figure 1B), with no effect in RB-negative PD-0332991-resistant TKCC-27 cells (figure 1B, right). Interestingly, in RB-high, p16-positive, CDK6-amplified TKCC-26, pRB was reduced post-PD-0332991, suggesting that in PDA CDK6 amplification may support sensitivity to CDK4/6i, irrespective of p16 expression.
Chemotherapeutic GEM synergises with PD-0332991 in RB-high and CDK6-amplified pancreatic PDCLs
Combining PD-0332991 with standard of care therapy in PDA, GEM,7 in PDCLs and primary murine PDA cells (figure 1C) from the genetically engineered Pdx1-Cre, LSL-KrasG12D/+, LSL-Trp53R172H/+ (KPC) model21 22 produced robust cell line-specific efficacy, with combination index analysis23 showing a synergistic interaction between GEM and PD-0332991 in RB-high PDCLs (TKCC-03,-05,-15,-17,-26; figure 1D) and antagonism in RB-low/negative PDCLs (TKCC-06,-16,-23 and -27) and KPC cells (figure 1E). The robustness of synergy was confirmed by examining GEM/PD-0332991 treatment with 25 drug combinations tested on five PDCLs, with synergy repeatedly observed across variable drug ratios in RB-high and antagonism in RB-low/negative lines (figure 1D,E inset, see online supplementary figure 2).
PD-0332991-based monotherapy and combination therapy induce critical effects on RB-high tumour cells
To understand the cellular mechanism of anti-tumour activity of PD-0332991 and its synergy with GEM, we examined the effect of selected treatments on cell cycle and apoptosis in candidate RB-high/RB-negative PDCLs (figure 1F) transduced with FUCCI, a fluorescent, two-colour biosensor of cell cycle progression24 (see online supplementary figure 3). Biosensor function and target modulation were confirmed by live cell imaging of PD-0332991-treated FUCCI-PDCLs (see online supplementary figure 4), showing robust G0/G1 arrest specifically in RB-high TKCC-05, not in RB-negative TKCC-27 cells, confirmed by traditional analysis (see online supplementary figure 4E,F).
Next, PD-0332991 treatment of FUCCI-TKCC-05 significantly induced apoptosis (4–10 days; p<0.0001 and p<0.01, respectively; figure 1G, see online supplementary figure 5). In agreement with published work,25 GEM stimulated apoptosis. Importantly, GEM/PD-0332991 combination significantly increased the apoptotic effect of each monotherapy (figure 1G, see online supplementary figure 5C,E,G), not observed in RB-negative TKCC-27 (figure 1H, see online supplementary figure 5D,F,H), supporting the potentially cytotoxic mechanism of action of PD-0332991 in selected cell types. For comparison purposes, combination of PD-0332991 and clinically used GEM/nab-paclitaxel8 was found to significantly enhance apoptosis in RB-high TKCC-05 (compared with GEM/nab-paclitaxel; see online supplementary figure 5C), although GEM/PD-0332991 effects appeared more robust (across early/late time points, figure 1G).
To examine quiescence-associated alterations in cell cycle dynamics, we used an established flow cytometric methodology26 to analyse FUCCI and quantify G0 versus G1 accumulation post-PD-0332991-based treatments (figure 1I). Both PD-0332991 and GEM/PD-0332991 significantly increased the mKO2++ G0 quiescent fraction,26 compared with GEM or vehicle specifically in the RB-high setting (figure 1J,K; see online supplementary figure 6). Cell cycle analysis of drug-treated apoptotic cells revealed most significant increases in the G0 fraction post-PD-0332991-based therapy (see online supplementary figure 6E). Collectively, these findings provide evidence that activated apoptosis and G0 quiescence are key mechanisms behind the observed synergy between GEM and CDK4/6i in 2D.
CDK4/6 inhibition modulates the invasiveness of RB-high PDA cells and ECM organisation
A single study reported that following incubation with a high PD-0332991 concentration (5 µM), selected commercial PDA lines displayed increased migratory capacity.27 Because both 3D topology and mechanical cues provided by the stroma can shape and drive invasive tumour behaviour,28 we examined the effect of CDK4/6 inhibition on the invasive potential of RB-high and RB-negative PDA cells using an established 3D organotypic model of invasion6 29 (figure 2A). In 3D, PD-0332991 or GEM individually significantly impaired invasion of RB-high TKCC-05 cells, with PD-032291 treatment being more effective (figure 2B, left, middle). Moreover, GEM/PD-0332991 combination was markedly superior to each monotherapy (figure 2B, C). PD-0332991 did not affect the invasive capacity of RB-negative KPC cells in 3D (Figure 2D, E). Further, the observed inhibitory effects on RB-high TKCC-05 invasion were accompanied by decreased proliferation and increased apoptosis in PD-0332991 and GEM/PD-0332991-treated matrices (figure 2F-G, see online supplementary figure 7A,B). In contrast, PD-0332991 treatment slightly increased proliferation of invaded KPC cells, not affecting apoptosis (see online supplementary figure 7C,D). In 2D, GEM/PD-0332991 reduced migratory capacity of RB-high TKCC-05 cells (see online supplementary figure 8), although PD-0332991-mediated effects were more pronounced in 3D, highlighting potential intricacies of CDK4/6 targeting of tumour cells within a complex microenvironment.
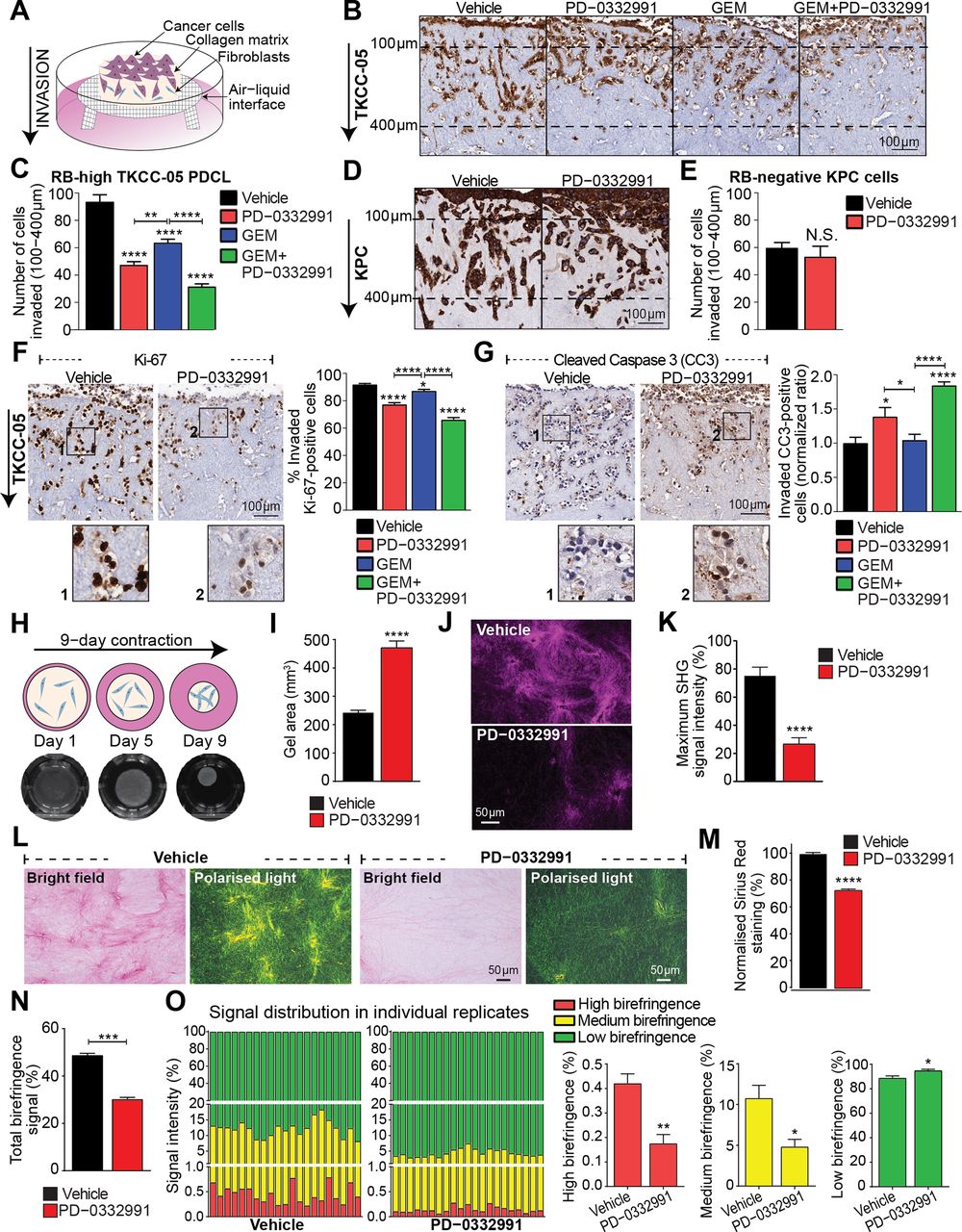
CDK4/6i inhibition impairs the invasive potential of retinoblastoma (RB)-high pancreatic ductal adenocarcinoma (PDA) cells and disrupts collagen matrix integrity. (A) Schematic representation of organotypic invasion assay with PDA cells. (B) Invasion of vehicle-treated and drug-treated enhanced green fluorescent protein (eGFP)-luciferase-labelled TKCC-05 cells through 3D organotypic matrices over 21 days, indicated by green fluorescent protein (GFP) immunohistochemistry. (C) Quantification of cell invasion (100 µm–400 µm). (D) Invasion of vehicle-treated and PD-0332991-treated Pdx1-Cre; KrasLSL.G12D/+; p53R172H/+ (KPC) cells through 3D organotypic matrices over 14 days, indicated by multi-cytokeratin staining and (E) quantified. (F) Representative images of Ki-67 staining as a marker of cell proliferation and quantification of the percentage TKCC-05 cells positive for Ki-67 in organotypic matrices following therapeutic intervention with PD-0332991-based approaches. (G) Representative images of cleaved caspase-3 (CC3) staining as a marker of cell apoptosis and quantification of TKCC-05 cells positive for CC3 (ratio normalised to control) in organotypic matrices post-treatment. (H) Schematic of a contraction assay (top), representative images of telomerase-immortalised fibroblast (TIF)–collagen control matrices (bottom) and (I) quantification of matrix area ±PD-0332991 at endpoint (9 days). (J) Representative maximum intensity projections of second harmonic generation (SHG) signal and (K) quantification of SHG signal intensity at peak in TIF–collagen matrices after 9 days of contraction ±PD-0332991. (L) Bright-field and polarised light imaging of Picro Sirius Red-stained TIF–collagen matrices±PD-0332991 and quantification of total collagen content measured as (M) the total signal intensity (bright-field) and (N) intensity of the signal acquired via polarised light. (O) Contribution and quantification of signal emitted from fibres with high, medium and low birefringence normalised to total signal acquired via polarised imaging of Picro Sirius Red-stained collagen matrices ±PD-0332991. Thick remodelled collagen fibres are highly birefringent (red-orange), whereas less remodelled fibres have a lower birefringence (green). Data for organotypic and collagen contraction studies are presented as mean±SEM (n=3 independent experiments, performed in triplicate matrices per condition, per repeat). Significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test, where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Unless specifically indicated, significance is compared against vehicle. GEM, gemcitabine; PDCL, patient-derived cell line.
Since extracellular matrix (ECM) deposition, fibrosis and signalling processes play a major role in regulating phenotype of pancreatic tumour cells and therapeutic response,6 30 we investigated the effect of CDK4/6i on ECM integrity using primary fibroblast-driven contraction assays,31 as recently achieved by our team (figure 2H).32 PD-0332991 significantly impaired the ability of fibroblasts to contract (figure 2I) and remodel fibrillar collagen, as shown by second harmonic generation (SHG) imaging (figure 2J-K; see online supplementary video 1), and bright-field/polarised light microscopy of Picro-Sirius Red staining (Figure 2L-O), indicating a significantly disorganised ECM network in PD-0332991-treated matrices. Impairment of matrix contraction was independent of changes in fibroblast proliferation (in 2D and 3D; see online supplementary figure 9). As these experiments revealed a significant anti-invasive, antiproliferative and ECM-modulatory activity of PD-0332991 in complex 3D settings, we therefore sought to assess whether PD-0332991 and combinations affect PDA progression and drug response in vivo.
Supplemental material
CDK4/6i monotherapy and combination therapy significantly improve survival of mice engrafted with RB-high patient-derived pancreatic tumours
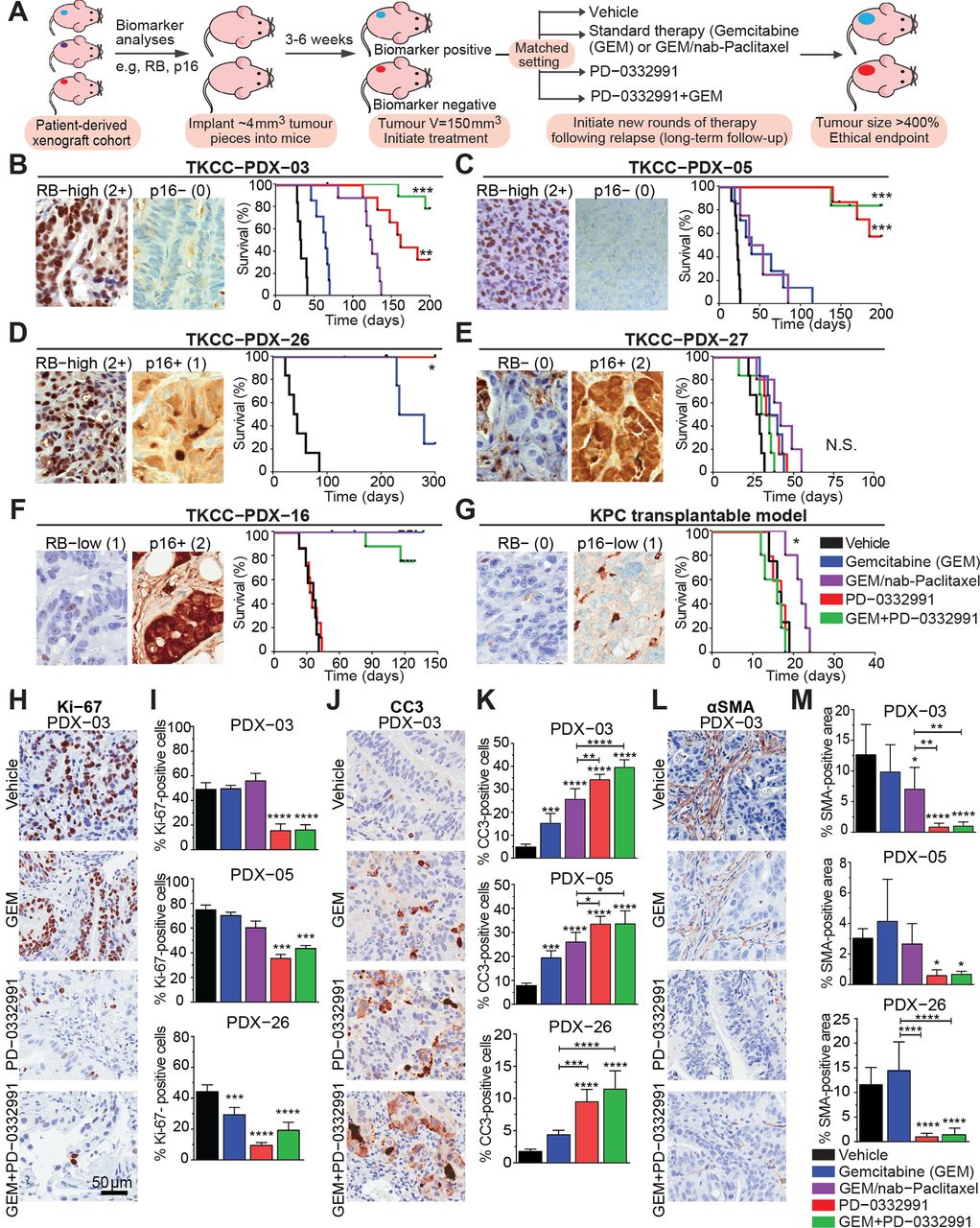
Therapeutic responsiveness in our early-passage PDXs was determined by measuring tumour growth, with experimental design (Figure 3A, see online supplementary figure 10A) enabling assessment of initial therapeutic responsiveness, and longitudinal analysis, comparing intrinsically resistant tumours with those that acquire resistance over time, as previously described.5 33 34 Strikingly, subtype-specific in vivo efficacy was observed, with all three RB-high PDXs examined showing significant long-term inhibition of tumour growth and improvement in overall survival following PD-0332991 monotherapy or GEM/PD-0332991 treatment, compared with standard of care (GEM and GEM/nab-Paclitaxel; Figure 3B-D, see online supplementary table 4A–C). Since GEM/nab-Paclitaxel was associated with cumulative intestinal toxicity when administered over 4–5 treatment cycles, efficacy of targeted therapies was compared with GEM only in the GEM-sensitive TKCC-PDX-26. In addition to improving survival, we observed significantly improved relapse-free survival (defined in online supplementary figure 10) following GEM/PD-0332991, compared with GEM (see online supplementary figure 10). In contrast, RB-negative TKCC-PDX-27, RB-low TKCC-PDX-16 and RB-negative KPC orthografts were resistant to PD-0332991-based therapy (Figure 3E–G, see online supplementary table 5A–C). ‘On-target’ modulation was confirmed by measuring inhibition of pRB and proliferation (Ki-67; see online supplementary figure 11A,B) in PD-0332991-treated RB-high PDX-05.

In vivo tailored response of pancreatic ductal adenocarcinoma (PDA) models to PD-0332991 and rationally designed treatment combinations. (A) Preclinical testing pipeline in subcutaneous models. Treatment commenced when tumours reached 150 mm3 (100%), where each mouse was randomised into outlined treatment groups: (a) gemcitabine (GEM) 120 mg/kg twice weekly intraperitoneally (IP) for 25 days, (b) PD-0332991 150 mg/kg daily gavage for 21 days, (c) GEM (120 mg/kg) twice weekly IP and PD-0332991 (150 mg/kg) daily gavage or (d) GEM (70 mg/kg) and nab-Paclitaxel (30 mg/kg) twice weekly IP for 25 days. In the combination arm, PD-0332991 was administered from week 2 for 21 days, 24 hours post-chemotherapy. Treatment response was measured from initiation of therapy (at maximal tolerable dose), through to the time resistance developed (characterised by progressive tumour growth in the presence of drug), and was based on our published work, with a 14-day minimal recovery time before additional treatment cycles. Details on treatment administration are further outlined in supplementary information 2. (B–D) Kaplan-Meier survival analyses of response to PD-0332991 and GEM/PD-0332991 in patient-derived xenografts (PDXs) stratified based on retinoblastoma (RB) status, presented as overall survival. (E–G) Overall survival of immunocompromised or syngeneic mice bearing the RB-negative/low TKCC-PDX-27, PDX-16 and subcutaneous Pdx1-Cre; KrasLSL.G12D/+; p53R172H/+ (KPC) tumours, respectively, treated with CDK4/6i monotherapy and combination, compared with standard therapies for PDA. (H) Representative images of Ki-67 immunohistochemistry performed on PDX-03 and (I) analysed for RB-high PDXs post-treatment (collected at endpoint). (J) Representative images of cleaved caspase 3 (CC3) staining, marker of apoptosis, in PDX-03 and (K) quantified for RB-high PDXs. (L) Representative images of α-smooth muscle actin (SMA) stromal expression post-therapy and (M) staining quantified for RB-high PDXs across all treatment groups (n=6 mice per treatment group). Data are shown as mean ±SD. Significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test, where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Unless specifically indicated, significance is compared against vehicle.
Analysis of PD-0332991 and GEM/PD-0332991-treated RB-high tumour remnants revealed significant, prolonged in vivo inhibition of proliferation and increased apoptosis (Figure 3H-K), also associated with decreased α-smooth muscle actin (SMA) expression (Figure 3L-M; see online supplementary figure 11C), a prototypical marker of activated pancreatic stellate cells (PSCs), which drive the extensive fibrosis typically observed in PDA.35 36 In PD-0332991-resistant tumours, only GEM/nab-Paclitaxel treatment showed any significant modulatory effects (see online supplementary figure 11D–F). In agreement with immunohistochemistry (IHC) analyses, the significant tumour regression observed in RB-high and CDK6-amplified PDXs post-treatment (30-day time point; see online supplementary figure 11G) further supports the idea that in addition to its cytostatic activity, PD-0332991 may exert cytotoxic effects on specific PDA subtypes in vivo.
PD-0332991-based therapeutic interventions inhibit spread in the liver while sensitising RB-high tumour cells to shear stress and reducing cell attachment
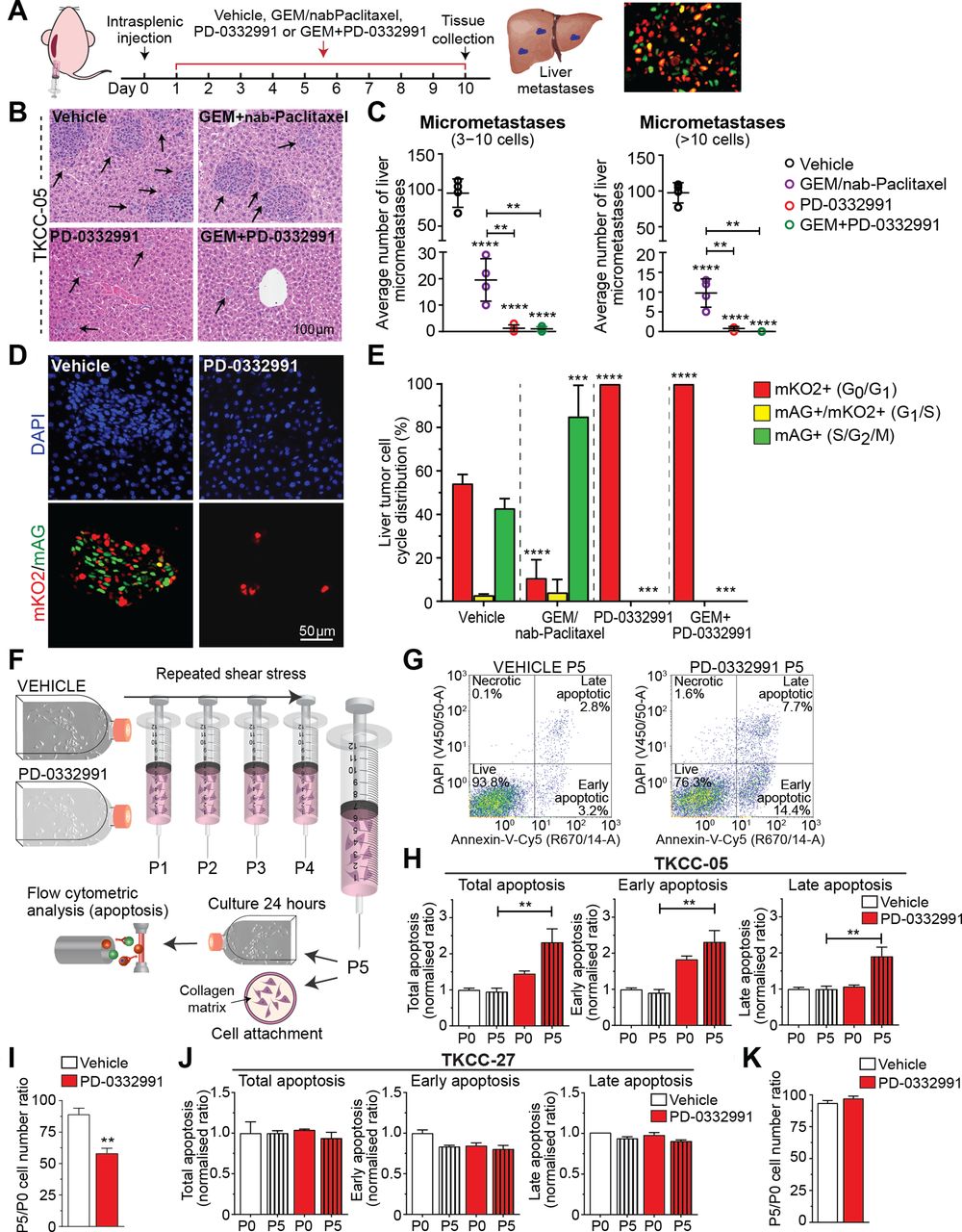
Next, we performed intrasplenic injections of RB-high TKCC-05-FUCCI cells in parallel with systemic PD-0332991 administration as a way of examining efficacy of PD-0332991-based therapies in limiting pancreatic cancer spread to distant sites (Figure 4A). Pathology analyses on serial sections of liver metastases (Figure 4B,C) confirmed that mice treated with PD-0332991 and GEM/PD-0332991 showed a marked reduction in metastatic spread compared with GEM/nab-Paclitaxel. FUCCI analyses of the same liver tissues further confirmed in vivo PD-0332991 activity and ability to effectively inhibit CDK4/6 in liver metastases (Figure 4D, E).
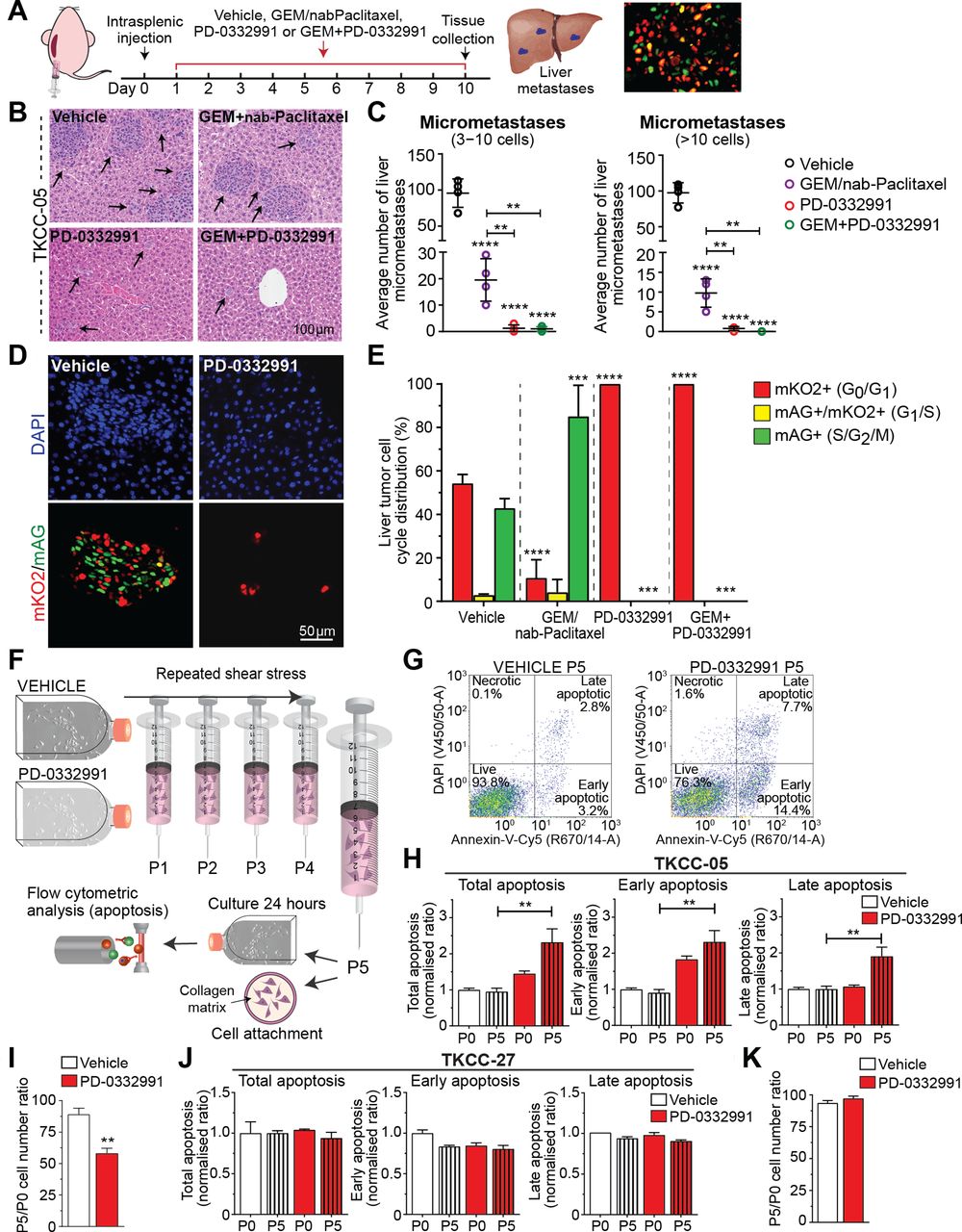
CDK4/6 inhibition decreases cell colonisation and improves chemotherapy response in metastatic sites and makes retinoblastoma (RB)-high tumour cells more vulnerable to shear stress. (A) Schematic representation of intrasplenic injection of RB-high TKCC-05-fluorescent ubiquitination-based cell cycle indicator (FUCCI) cells and associated treatment timeline. (B) Representative images and (C) quantification of liver micrometastasis and total metastasis normalised to liver surface area in H&E serial sections. Arrows point to metastases in the liver tissue. Data are shown as mean±SD (n=4 mice per group and five serial sections per organ (50 µm step)). (D) Representative pictures of metastases identified via imaging of FUCCI signal and (E) quantified cell cycle distribution in liver metastases from the TKCC-05-FUCCI intrasplenic model at the experimental endpoint. (F) Schematic of the fluid shear stress assay, adapted from Barnes et al [37]. (G) Representative TKCC-05-FUCCI apoptosis plots and (H) quantification of total, early and late apoptosis in TKCC-05 cells±PD-0332991 (0.3 µM) 24 hours following shear stress. P0 represents cells not subjected to shear stress and P5 represents cells subjected to five consecutive exposures to shear stress. (I) Quantification of cell attachment onto the collagen matrix, following shear stress of RB-high TKCC-05 cells±PD-0332991 treatment, measured 24 hours post-seeding. Y-axis values (P5/P0 ratios) are values for cells subjected to five exposures to shear stress divided by values for cells not exposed to shear stress. (J) Quantification of apoptosis in RB-negative TKCC-27 cells±PD-0332991 (1 µM) 24 hours following shear stress. (K) Quantification of cell attachment onto the collagen matrix, following shear stress of RB-negative TKCC-27 cells±PD-0332991 treatment. Data are shown as mean±SEM (n=3 biological repeats with two collagen-coated wells per condition). Significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test, where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Unless specifically indicated, significance is compared against vehicle. DAPI, 4',6-diamidino-2-phenylindole; GEM, gemcitabine.
To further understand the observed PD-0332991-mediated reduction of liver colonisation, we examined viability (apoptosis) and cell attachment of RB-high and for comparison, RB-negative PDA cells after exposure to controlled shear stress (100 µL/s, maximum force of 1950 dyn/cm2, Figure 4F),6 37 which CTCs are exposed to in vivo. PD-0332991 significantly induced post-stress cellular apoptosis and decreased cell attachment onto a collagen matrix, specifically in RB-high tumour cells (Figure 4G-I, J-K). Collectively, these experiments suggest a direct role of CDK4/6 targeting in sensitising CTCs already exposed to shear stress as a potential mechanism in which PD-0332991 may modulate metastasis in PDA.
PD-0332991-based therapies significantly improve survival and delay metastasis in metastatic RB-high PDA
To examine the effect of CDK4/6i on PDA tumour progression and metastasis, mice were orthotopically injected with luciferase-TKCC-05 tumours, imaged 1 week post-injection (see online supplementary figure 12A), randomised and treated with standard chemotherapies, PD-0332991 and PD-0332991-based combinations, until ethical endpoint (Figure 5, top).

CDK4/6i therapy induces in vivo quiescence at primary tumour site, impairs metastatic spread and improves gemcitabine (GEM) efficacy in a metastatic patient-derived retinoblastoma-positive model of pancreatic ductal adenocarcinoma (PDA). (A) Schematic illustrating in vivo testing in a metastatic patient-derived model (top). Briefly, 15 000 luciferase-labelled patient-derived cell lines (PDCLs) were injected into the pancreas of 6–8-week-old Nod/Scid/IL-2Rγnull (NSG) mice. Mice were randomised into treatment groups at 1 week post-injection, with two additional treatment arms included, involving triple combination of GEM (70 mg/kg), nab-Paclitaxel (30 mg/kg) twice weekly intraperitoneally administered either concomitantly with PD-0332991 (100 mg/kg) gavage from week 2 on a 5 day ‘on’, 5 day ‘off’ schedule for 20 days or with PD-0332991 administered as maintenance therapy by daily gavage after completion of GEM/nab-Paclitaxel treatment. Tumour burden was monitored weekly by bioluminescent imaging until ethical endpoint. Kaplan-Meier survival analyses of PD-0332991 monotherapy, in combination with GEM (left) or triple combinations with GEM/nab-Paclitaxel (right), compared with GEM/nab-Paclitaxel. Log-rank analysis, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. (B) Experimental setup for in vivo second-line therapeutic testing in metastatic PDA (top). In vivo efficacy of GEM/PD-0332991 combination following progression on GEM/nab-Paclitaxel (bottom). (C) Representative images of pancreas tumours (left) and associated liver metastases (right) from orthotopic fluorescent ubiquitination-based cell cycle indicator (FUCCI)-TKCC-05 tumours collected 50 days post-treatment with CDK4/6i-based treatment or standard therapy (GEM/nab-Paclitaxel). Vehicle controls were collected prior to the 50-day time point, as expected, due to a heavy tumour burden. Shown is representative nuclear staining (4',6-diamidino-2-phenylindole (DAPI), top), FUCCI (mKO2—red, mAG—green, overlay—yellow, CD31—blue; middle) and H&E section (bottom). Quantified cell cycle distribution of (D) primary tumours and (E) liver metastases from examined treatment groups, with (F) no metastases detected in PD-0332991 and GEM/PD-0332991-treated animals. (G) Representative overlay of maximum intensity projections of second harmonic generation (SHG) signal and FUCCI with quantification of SHG signal intensity at peak in orthotopic FUCCI-TKCC-05 tumours collected post-treatment. (H) Representative polarised light images of tumour tissue and quantification of total birefringence signal (black) and contribution to signal emitted from collagen fibres with high (red), medium (yellow) and low (green) birefringence acquired via polarised light imaging (n=4 animals per treatment group were used for analysis in (D, E, F, G and H). Significance was determined by one-way analysis of variance, followed by Tukey post hoc multiple comparisons test, where *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Critically, PD-0332991-based regimens were significantly more effective than standard therapies (Figure 5A, bottom, see online supplementary table 6) in RB-high metastatic PDA, with GEM/PD-0332991 proving the best therapeutic approach (median survival=117 days, p=0.0001; vs GEM/nab-Paclitaxel median survival=69 days). In addition, metastasis-free survival, monitored by in vivo imaging system (IVIS; see online supplementary figure 12B–C), was significantly increased by PD-0332991-based treatments (see online supplementary figure 13A, supplementary table 7). Given in vitro efficacy was observed when PD-0332991 was combined with GEM/nab-Paclitaxel (see online supplementary figure 5), we also examined this combination in vivo. While treatments were generally well tolerated in experimental animals, the two triple combinations examined using GEM/nab-Paclitaxel and PD-0332991 were associated with some toxicity, namely constipation, which was treated with paraffin oil and controlled by adjusting PD-0332991 dosage. Both regimens, involving concomitant PD-0332991 administration (concomitant) or aiming to assess the potential of CDK4/6i as maintenance therapy in between chemotherapy cycles (maintenance), demonstrated a significant therapeutic benefit over GEM/nab-Paclitaxel (Figure 5A, bottom right, see online supplementary tables 6 and 7), but were not superior to GEM/PD-0332991.
Following initial treatment with GEM/nab-Paclitaxel, a short-term response was followed by progression, measured as increased bioluminescence signal (see online supplementary figure 12C). To examine the therapeutic potential of GEM/PD-0332991 in slowing PDA progression in advanced disease settings, separate cohorts of relapsed animals were subsequently switched to GEM/PD-0332991 or as control, clinically used second-line therapy, capecitabine,38 39 with a superior therapeutic benefit measured with GEM/PD-0332991 (Figure 5B, see online supplementary tables 6 and 7). In concordance, analyses of orthotopic FUCCI-TKCC-05 tumours collected 50 days post-treatment revealed marked quiescence at the primary site (pancreas tumour), regardless of proximity to vessels (Figure 5C-E, see online supplementary figure 13B–C), and no liver metastases in animals treated with PD-0332991 or GEM/PD-0332991 (Figure 5C,F). SHG imaging and polarised light microscopy of Picro-Sirius Red-stained TKCC-05-FUCCI tumours confirmed that PD-0332991-based therapy reduces fibrillar collagen organisation (Figure 5G-H; see online supplementary figure 14A), leads to increased apoptosis and decreased expression of α-SMA in vivo (see online supplementary figure 14B,C).
These data indicate that CDK4/6 inhibition significantly delays pancreatic tumour progression by directly targeting tumour cells (G0 quiescence, apoptosis) and critical components of the microenvironment that have been shown to regulate chemoresistance (ie, decreased stromal activation, reduced fibrillar collagen organisation). As robust efficacy was observed in multiple, independent models of RB-high PDA, RB-based stratification could potentially facilitate a more tailored treatment approach for CDK4-targeting therapy in pancreatic cancer.
Nuclear RB is prevalent in primary operable and metastatic human PDA and has prognostic value
Next, we immunohistochemically examined the expression of RB in human PDA and whether its disruption has prognostic significance (Figure 6A). High RB expression was detected in 64.5% of primary tumour specimens from the APGI/ICGC cohort1 (n=200) and correlated with significantly improved survival (Figure 6B-C, see online supplementary table 8) and lower tumour grade (see online supplementary table 9). Multivariate analysis using a Cox proportional hazards model demonstrated RB loss as an independent poor prognostic factor for disease-specific survival (DSS) (HR 1.552, p=0.0148, table 1). In contrast, p16 expression, examined for comparison purposes, was not prognostic (see online supplementary figure 15A–D). Deregulation of RB signalling has been linked with improved chemoresponsiveness in breast cancer40; however when correlated, RB expression did not specifically associate with survival following adjuvant GEM (see online supplementary figure 15E,F). As expected, RB and p16 levels strongly correlated in the PDX (n=45) and patient tumours from which they were derived (p<0.001, see online supplementary table 10).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High retinoblastoma (RB) expression is prevalent in human pancreatic ductal adenocarcinoma (PDA) and is of prognostic relevance in independent cohorts of the disease. Examples of (A) (left) RB-negative primary tumour (0); (middle) weakly RB-positive tumour (1+) and (right) RB-high tumour (2+). Kaplan-Meier analyses of disease-specific survival based on RB immunohistochemistry in (B and C) the Australian Pancreatic Genome Initiative (APGI)/International Cancer Genome Consortium (ICGC) cohort and (D and E) the New South Wales Pancreatic Cancer Network (NSWPCN) patient cohort. Survival analyses were performed comparing (B and D) all three RB groups and (C and E) RB-high (2+) versus RB-low in both sets. p Values, log-rank. (F) Examples of (left) RB-high and (right) RB-low primary tumours and matched metastases. (G) The agreement of RB status between primary and metastatic tumours of PDA. The number of observed agreements is 11 (91.67% of the observation), with a Kappa score 0.75 (SE of Kappa 0.232, 95% CI 0.296 to 1.000), indicating the strength of agreement is ‘good’.
Multivariate analysis of retinoblastoma (RB) and key factors associated with disease-specific survival in independent clinical cohorts of resected pancreatic ductal adenocarcinoma.
The potential prognostic value of RB was subsequently validated in an independent patient NSWPCN cohort20 (n=314), where similar relationships between RB expression and survival were apparent in univariate (Figure 6D-E, see online supplementary table 8) and multivariate analyses (HR 1.364, p=0.02, table 1). As most patients present with advanced stage PDA, we also assessed RB status in metastatic disease (n=54; see online supplementary tables 11 and 12). RB protein was highly expressed in 67% of metastatic specimens. Concordance in RB expression between matched primary and metastatic tumours of PDA, available for 12 of the 54 metastatic samples, was observed in 11 cases (91.7%; Cohen’s Kappa score 0.75; Figure 6F-G) indicating the strength of the agreement as ‘good’.
Collectively, these data demonstrate that a significant proportion of primary operable and metastatic PDA express high levels of the prognostically relevant RB. This druggable, constitutively active pathway may represent an attractive therapeutic target, with immunohistochemical analysis of RB as a potentially simple prognostic and predictive biomarker to guide this therapeutic approach.
Discussion
Despite significant recent efforts into developing novel treatment combinations,41 chemotherapy remains the standard of care for most patients with advanced pancreatic cancer, despite only modestly improving survival.8 With rapid implementation in the clinical management of specific breast cancers, CDK4/6i are increasingly being tested in other solid tumours.42 43 Here, we examined whether CDK4 pathway inhibition in combination with chemotherapy represented a viable therapeutic approach for pancreatic tumours characterised by G1/S checkpoint aberrations, frequent in the molecular landscape of PDA.1 We demonstrate potent efficacy of single agent and combination CDK4/6i therapy in subtypes of PDA characterised by high RB expression, significantly improving long-term survival while delaying the development of metastasis in robust in vivo models of PDA. We also reveal that the potent efficacy of CDK4/6i monotherapy and combination therapy occurs via a complex mechanism involving deregulation of tumour cell signalling and disruption of the surrounding ECM that collectively drive cancer cell proliferation, invasion and metastasis6 30 44 while revealing a new role of CDK4/6 inhibition in governing tolerance to shear stress, an often overlooked area of premetastatic niche formation for CTCs.45 46
A major area of active investigation is the identification of biomarkers of therapeutic response to CDK4 inhibition. In preclinical models of melanoma and glioblastoma, homozygous loss, methylation or mutation of CDKN2A gene were shown to associate with sensitivity to CDK4/6i,47 48 although in the clinic, CDK4/6i sensitivity may not be strictly defined by p16 status.49 Here, we show that RB expression, but not G1/S checkpoint mutations or p16 levels, may present a robust stratifying biomarker for PD-0332991-based therapy in advanced preclinical models of PDA.
PD-0332991 has traditionally been thought to exert its anti-tumour effects by promoting cytostasis, also supported by limited data in PDA, whereby using 7-day assessment of proliferation as a read-out of PD-0332991 activity, early efficacy has been measured in almost all PDA explants examined.18 Deeper exploration of the long-term responsiveness to PD-0332991 and combination therapy, directly compared with standard of care treatments, in this study highlights the need for a personalised approach in the treatment of pancreatic cancer, where RB-high PDA tumours respond long term to PD-0332991-based therapy in vivo and RB-low/negative tumours do not. Moreover, building on recent studies in other cancers, which suggest a more complex mechanism of action for CDK4/6i, including inhibition of EMT signalling in breast cancer metastasis50 and improved anti-tumour immunity,51 our study provides additional new evidence that PD-0332991 displays multifaceted effects on tumour cells (quiescence, apoptosis), alters ECM organisation and reduces tolerance to shear stress (Figure 4), thereby having a global effect in this aggressive disease, not examined previously.
Using a combination of 2D, 3D in vitro and in vivo models, we specifically show that PD-0332991 significantly induces apoptosis in RB-high pancreatic tumour settings and also enhances the apoptotic effect of chemotherapeutic GEM, a well-tolerated and clinically used agent in PDA (figures 1-3). Monitoring the onset of liver metastasis in vivo revealed that PD-0332991 monotherapy and combination therapy significantly impaired liver colonisation in the RB-high setting (Figure 4A-E), delaying disease progression in a metastatic PDA model (Figure 5). Of note, CDK4/6i may potentially be a beneficial second-line therapy, following progression on GEM/nab-Paclitaxel in RB-high PDA, as our in vivo findings suggest (Figure 5B). Given the high rate of chemotherapeutic resistance in advanced patients with PDA treated with these chemotherapies and their subsequent extremely poor outcome, pending further preclinical study, CDK4/6i-based approaches may potentially be a viable second-line treatment for specific subtypes of advanced PDA.
CDK4/6 inhibition modulated ECM organisation in vitro and in vivo (Figures 2,5), with in vivo responses to PD-0332991-based therapy associated with decreased expression of α-SMA, a marker of activated PSCs and stromal activation. As our in vitro findings suggest that the effects on ECM are independent of the antiproliferative activity of PD-0332991, it is plausible to hypothesise that changes in ECM mechanics induced by PD-0332991 may directly disrupt PSC activation, the subsequent tumour cell–PSC crosstalk and ultimately further improve therapeutic sensitivity. Indeed, recent work has shown that matrix stiffness and organisation can directly regulate differentiation and migration of PSCs.52
PD-0332991 and GEM strongly synergised irrespective of p16 or p53 status (eg, whether pancreatic tumour harboured an inactivating TP53 mutation (C124*; PDX-26), mutation contributing to partial rescue (V173M; PDX-03) or higher than wild-type p53 activity (G245S; PDX-05).53 CDK4/CDK6 amplification or overexpression has been associated with in vitro CDK4/6i resistance,47 although CDK4-amplified liposarcomas are sensitive to PD-0332991.54 Interestingly, the CDK6-amplified PDX-26 and matched PDCL responded to PD-0332991-based treatment, suggesting that in PDA CDK6 amplification in the context of functional RB does not preclude effective CDK4 pathway inhibition. Although CDK6 is strongly functionally linked to CDK4, this enzyme may exert other kinase-independent functions in regulation of angiogenesis and/or cell differentiation,55 representing a potentially separate thus far unexamined role for CDK6 inhibition in PDA therapy.
Several smaller studies have investigated the prognostic role of RB in PDA, with inconsistent findings.56 Here, RB protein was highly expressed in a significant proportion of PDA (65%, APGI/ICGC; 43%, NSWPCN; 67%, metastatic cohort), suggesting that this could be a viable therapeutic target for tailored CDK4/6i-based treatments that act on this signalling network. Moreover, the observed concordance in RB levels in the small set of available matched primary and metastatic tissue lends hope that metastases may still be co-targeted with this type of therapy. Loss of RB protein was associated with poor patient outcome, with RB an independent prognostic factor for DSS in two sizeable independent PDA cohorts (table 1). RB immunohistochemistry was a clean nuclear stain without considerable background. In our hands, it appeared comparable with estrogen receptor (ER) staining in terms of interpretation of intensity within processed material. Pending further validation, the use of a tissue-based assay for RB expression could be adapted for clinical application in diagnostics to, combined with other markers, help identify those with poorest prognoses and also potentially identify a cohort of patients to trial CDK4/6i.
In conclusion, we provide preclinical proof-of-concept for the efficacy of a personalised treatment strategy using CDK4/6i to treat RB-high PDA. We reveal key mechanisms whereby CDK4-targeting therapy may regulate multiple cellular processes and environmental cues within the primary tumour, secondary site and during CTC transit, which collectively govern pancreatic cancer progression. We hope that with two non-biomarker-driven phase I/II trials of PD-0332991 in pancreatic cancer underway (NCT02501902, NCT03065062), these findings may have complementary and potentially significant therapeutic implications for the personalised treatment of PDA in future.
Acknowledgments
The authors would like to thank consumer representatives Professor Ros Pesman, Mr Benjamin Bravery and Ms Jan Mumford for their involvement in this project and a critical review of the manuscript. Dr Chou would also like to acknowledge Associate Professor Adrienne Morey for mentorship during PhD studies.
References
Footnotes
Contributors Study concept and design (MP, AJG, AC, SJC, PT); data acquisition (AC, DF, AMN, VTC, DW, AP, AS, RS, AD, KJM, JT); analysis and data interpretation (AC, DF, DW, AP, TRC, TJM, JT, CV, KJM, AB, OJS, JM); drafting of manuscript (AC, DF, MP); critical review and edit of manuscript (AJG, PT, AJ, SJC, JS, NW, SMG, LAC, AB, TRC, AVB, TJM, OJS, JM); patient cohort data (APGI, AJ, AVB, JS, AJG, JT, SMG); statistics (AC, MC, MP, DKC); obtained funding (MP, AJG, AC, SJC, PT, JS, AMN, AB, NW); study supervision (MP, AJG).
Funding This work was supported by the National Health and Medical Research Council of Australia (NHMRC) Project Grants 1081312 and 1105640, Cancer Australia Project Grants 1060522 and 1100722 with fellowship support from Cancer Institute NSW, Australian Research Council and NHMRC, scholarship support from NHMRC and Sydney Catalyst and philanthropic support from the Avner Pancreatic Cancer Foundation, The Philip Hemstritch Fellowship in Pancreatic Cancer; Len Ainsworth Pancreatic Cancer Fellowship, Estate of the late RT Hall Cancer Gene Discovery and Validation Program and Norman Green Support Grant.
Competing interests None declared.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval Ethics approval for acquisition of data and biological material was obtained from human research ethics committees from each participating institution (Sydney Local Health District Human Research Ethics Committee X11-0220). In vivo experiments were approved by Garvan/St Vincent’s animal ethics committee (14/11, 14/12).
Provenance and peer review Not commissioned; externally peer reviewed.