Article Text

Abstract
Objectives The aim of this study was to explore the association of serum fibrosis marker levels with the risk of clinical and histological disease progression in a large cohort of patients with chronic hepatitis C (CHC).
Methods 462 prior non-responders to peginterferon and ribavirin enrolled in the randomised phase of the Hepatitis C Antiviral Long-term Treatment against Cirrhosis (HALT-C) Trial had baseline and annual serum samples tested for hyaluronic acid (HA), N-terminal peptide of procollagen type 3, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) and YKL-40. All patients underwent a pretreatment liver biopsy and follow-up biopsies at years 2 and 4. Histological progression was defined as a ≥2 point increase in Ishak fibrosis score in patients without cirrhosis. Clinical outcomes included development of decompensation, hepatocellular cancer, death or an increase in the CTP (Child–Turcotte–Pugh) score to ≥7.
Results Mean patient age was 49.5 years and 39% had histological cirrhosis at entry. Baseline HA, YKL-40 and TIMP-1 levels combined with other laboratory parameters were all significantly associated with clinical outcomes in the 69 (15%) patients with disease progression (p<0.0001). The best multivariate model to predict clinical outcomes included baseline bilirubin, albumin, international normalised ratio (INR) and YKL-40 levels. All of the baseline serum fibrosis marker levels were also significantly associated with histological fibrosis progression that developed in 70 (33%) of the 209 patients with cirrhosis (p <0.0001). However, baseline HA and platelet counts were best at predicting histological progression (area under the curve (AUC)=0.663).
Conclusion Pretreatment serum fibrosis marker levels are significantly increased in patients with CHC at risk of clinical and histological disease progression. If validated in additional cohorts, measurement of these markers could help identify patients with CHC who would benefit from more frequent and intensive monitoring.
Trial Registration Number NCT00006164.
- Cirrhosis
- non-invasive markers
- liver fibrosis
- natural history
- viral hepatitis
- fibrogenesis
- Hepatitis C
- liver biopsy
- liver cirrhosis
- staging
Statistics from Altmetric.com
- Cirrhosis
- non-invasive markers
- liver fibrosis
- natural history
- viral hepatitis
- fibrogenesis
- Hepatitis C
- liver biopsy
- liver cirrhosis
- staging
Significance of this study
What is already known about this subject?
Progression of hepatic fibrosis is a key determinant of the natural history of CHC which is a leading cause of liver-related morbidity and mortality worldwide.
Serum fibrosis markers have been proposed to assess disease severity non-invasively in patients with CHC but have not been validated against clinical and/or histological disease progression in a large group of well-characterised patients with CHC prospectively followed.
What are the new findings?
An algorithm consisting of baseline YKL-40 and other routine laboratory parameters was strongly associated with the likelihood of clinical disease progression in 462 patients with CHC with advanced fibrosis and prior non-response to antiviral treatment that were followed over 3.5 years.
Consistent increases in serum YKL-40 and TIMP-1 levels over time in patients with clinical disease progression compared with non-progressors demonstrates the potential utility of these markers to monitor patients with CHC non-invasively.
Baseline hyaluronic acid levels combined with platelet counts were associated with the risk of histological fibrosis progression.
How might it impact on clinical practice in the foreseeable future?
Measurement of serum fibrosis marker levels in combination with routine laboratory parameters may help clinicians identify which patients with CHC need closer follow-up and monitoring
Introduction
The morbidity and mortality associated with chronic hepatitis C virus (HCV) infection is projected to increase substantially over the next two decades as the proportion of patients with cirrhosis, decompensation and hepatocellular carcinoma (HCC) increases.1 2 Management guidelines recommend obtaining a liver biopsy in potential antiviral treatment candidates to grade and stage the severity of liver disease and assist with decision making.3–6 However, due to the risks and sampling variability of liver biopsy, accurate and reliable non-invasive means to assess patients with chronic hepatitis C (CHC) at increased risk of developing worsening hepatic fibrosis are needed.7 8 Numerous biochemical indices, serum fibrosis marker algorithms and liver elasticity measurements have been proposed to assess disease severity non-invasively in patients with CHC.9–13 However, most of these modalities have not been tested or validated in a large group of patients with CHC who were prospectively followed for histological and/or clinical liver disease progression.
Administration of ‘maintenance’ interferon to patients with CHC who failed to respond to a course of antiviral treatment may slow the rate of liver disease progression.14 15 As a result, several large randomised controlled studies were initiated in prior non-responders to determine if maintenance interferon could reduce the rate of clinical decompensation and histological disease progression.16–19 In the Hepatitis C Antiviral Long-term Treatment against Cirrhosis (HALT-C) Trial, the rate of histological disease progression and clinical outcomes were similar in the 1050 patients with advanced fibrosis that were randomised to receive peginterferon or no additional treatment and followed over 3.5 years.18 Additional studies were carried out at four HALT-C Trial clinical sites to determine if serum fibrosis marker levels would correlate with initial disease severity and disease progression over time. In our analysis, we detected significant associations between pretreatment serum hyaluronic acid (HA), tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), YKL-40 and N-terminal peptide of procollagen type 3 (PIIINP) levels and baseline disease severity.10 In addition, all of these marker levels significantly declined at week 72 compared with pretreatment baseline in subjects who achieved a sustained virological response following 48 weeks of full dose peginterferon and ribavirin treatment, suggesting that these analytes reflect active fibrogenesis and fibrolysis.20 In the current study, pretreatment baseline serum fibrosis marker levels were tested as potential predictors of clinical and histological disease progression during the randomised phase of the HALT-C Trial. We also hypothesised that the levels of these analytes would increase over time in subjects who experienced clinical or histological disease progression compared with patients who did not progress.
Methods
Patient population
Randomised HALT-C Trial patients had detectable serum HCV RNA and bridging hepatic fibrosis (ie, Ishak fibrosis score ≥3) or cirrhosis on liver biopsy obtained within 12 months of enrolment and had failed to achieve a sustained virological response to a prior course of peginterferon and ribavirin.18 Initially, subjects were retreated with peginterferon alfa-2a and ribavirin for 24 weeks in the ‘lead-in phase’ of the study. Subjects who remained viraemic at week 20 were eligible for randomisation at week 24 to maintenance peginterferon versus no treatment for 3.5 years; subjects with undetectable HCV RNA at week 20, as determined by PCR assay (Roche Molecular Systems, Branchburg, New Jersey, USA; COBAS Amplicor v 2.0, sensitivity of 100 IU/ml) continued in the ‘responder arm’ of the study and completed a 48 week course of combination antiviral treatment. Patients who experienced an on-treatment breakthrough or post-treatment virological relapse following the responder arm were also eligible for enrolment. Finally, express patients treated with at least 12 weeks of peginterferon and ribavirin without viral clearance were eligible for randomisation. All HALT-C Trial participants entering the randomised phase at the University of Michigan, University of Massachusetts/University of Connecticut, Massachusetts General Hospital and Virginia Commonwealth University had serum collected at baseline and study months 12, 24, 36 and 48 following randomisation. Serum isolated from whole blood samples was frozen immediately at −80°C and stored at a central repository (SeraCare, Gaithersburg, Maryland, USA). The study was approved by local Institutional Review Boards and all patients provided written informed consent.
Laboratory and clinical assessment during the randomised phase
Lifetime alcohol consumption was estimated with a modification of the Skinner survey.18 Routine baseline laboratory values (ie, serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin, bilirubin and platelet count) were obtained at local hospital laboratories. Insulin resistance based on HOMA-IR (homeostasis model assessment of insulin resistance) was calculated as HOMA=((insulin× glucose)/22.5)×0.5551. A baseline liver biopsy was scored for the degree of hepatic fibrosis and inflammation defined by the Ishak scoring system, and the degree of hepatic steatosis was estimated as grade 0–4.21 22 Splenomegaly was defined by a spleen length >13 cm on sonography.
All patients were seen every 3 months during the randomised phase for laboratory and clinical assessment. In addition, annual liver ultrasounds were obtained to screen for HCC, and serum α-fetoprotein levels were obtained every 3 months. Clinical end points for the study included an increase in CTP (Child–Turcotte–Pugh) score to ≥7 on two separate occasions 3 months apart, variceal bleeding, ascites, spontaneous bacterial peritonitis, hepatic encephalopathy, HCC or death. For the subgroup of patients with non-cirrhotic fibrosis at baseline, histological progression was defined as a ≥2 point increase in the Ishak fibrosis score.18
There were 624 patients enrolled in the randomised phase of the HALT-C Trial at the four participating sites. The baseline features of the 462 patients with sufficient serum samples for analysis in the current study were similar to those of the 162 excluded patients, including the frequency of cirrhosis and likelihood of disease progression over time. However, the excluded patients were more likely to be Caucasian (86% vs 73%) and less likely to be Black (9% vs 24%), p=0.0001.
Serum fibrosis marker assays
Stored serum samples were tested for TIMP-1 (normal range: 80–500 ng/ml) (Quantikine, R & D Systems, Minneapolis, Minnesota, USA), YKL-40 (normal range: 24–125 μg/l) (Metra YKL-40, Quidel, San Diego, California, USA) and PIIINP (normal range: 2–4 μg/l) (UniQ, Orion Diagnostica, Espoo, Finland) using commercially available ELISAs as described previously.10 Samples that exceeded the upper limit of quantitation were retested using a 1:10 dilution. Serum HA levels were determined by an automated liquid-phase immunoassay using the LiBASys Analyser (Wako Diagnostics, Richmond, Virginia, USA) (normal range: 10–100 ng/ml).10 Serum was available for testing in 421 patients at month 12, 404 patients at month 24, 380 patients at month 36 and 364 patients at month 48.
Statistical analyses
Log transformation of non-normally distributed variables was undertaken when needed. When considering all four serum fibrosis markers to predict clinical and histological progression, first a model adjusting for all non-invasive predictors of clinical progression (variables for which the p value was <0.20 as per table 1), as well as race, age, gender and body mass index (BMI) was fit. Then, from this model, only variables for which the p value was <0.05 were kept, and this became the final clinical/laboratory multivariable model of each serum fibrosis marker. Then, we adjusted for all histological variables (eg, cirrhosis and hepatic steatosis) and generated the final model including only significant variables. In addition, a multivariate model that included all of the baseline serum fibrosis marker levels as well as other factors significantly associated with the outcome was constructed to determine the best overall model. For time to first clinical outcome, Cox proportional hazards regression was used and data were censored at the patient's last follow-up visit or at 1400 days (ie, 3.8 years) after randomisation, whichever occurred first. For time to histological progression, complementary log–log regression analysis was used. The Akaike information criterion (AIC) statistic was used to compare the goodness of fit of individual models with each other, and the model with the lowest AIC was considered the best.23 To demonstrate that the model score is associated with outcome, patients were split into three groups (low, medium and high risk) according to the 75th and 90th percentiles of the distribution of scores from the multivariate model. To assess changes in individual analyte levels over time, random effects models (via proc mixed of SAS; SAS Institute, Cary, NC, USA) were used. Specifically, the models included the random effect of time using a repeated statement, as well as the interaction of time with treatment group. All analyses were performed at the data coordinating centre (New England Research Institutes, Watertown, Massachusetts, USA) with SAS statistical software version 9.2 (SAS Institute).
Pretreatment baseline characteristics of the 462 HALT-C Trial patients
Results
Overall study population
The mean age of the 462 HALT-C patients was 49.5 years, 70% were male, 73% were Caucasian and 49% were randomised to receive low-dose peginterferon (table 1). Overall, 28% had diabetes mellitus, the mean BMI was 29.9 kg/m2 and 39% had histological cirrhosis.
Baseline predictors of clinical outcomes
During a mean follow-up of 51 months, a primary clinical outcome developed in 69 (15%) of the 462 patients, including a CTP score increase (n=40), HCC (n=7), ascites (n=32), encephalopathy (n=15), variceal bleeding (n=8) and death (n=24). Because the rate of outcomes was similar in the peginterferon-treated and untreated control patients, the two groups were combined for this analysis. As anticipated, patients that developed a primary clinical outcome had baseline features suggestive of more severe liver disease with a higher likelihood of having cirrhosis on biopsy, higher bilirubin and INR levels, and lower albumin and platelet counts (table 1). In addition, the pretreatment levels of HA, YKL-40, PIIINP and TIMP-1 were all significantly higher among the subjects who eventually developed a primary clinical outcome compared with those without clinical progression. The overall rate of clinical outcomes was 27.5% in patients with baseline cirrhosis and 6.8% in patients with non-cirrhotic fibrosis.
Multivariate models of clinical outcomes
The utility of individual serum fibrosis marker levels in combination with other baseline parameters was determined using multivariate regression analysis. Although baseline PIIINP levels were significantly associated with clinical outcomes on univariate analysis (table 1), only baseline bilirubin, INR and albumin levels remained associated in a multivariate model that included PIIINP (Supplementary table 1). In contrast, when baseline TIMP-1 levels were combined with other baseline variables, a multivariate model consisting of baseline TIMP-1, bilirubin, INR and albumin levels was generated. Similarly, when baseline HA levels were combined with other significant variables from table 1, a multivariate model consisting of HA, bilirubin, INR and albumin levels was generated. Finally, when baseline YKL-40 levels were combined with other variables, a multivariate model consisting of YKL-40, bilirubin, albumin and INR levels was generated.
In addition to the above multivariate models, a final model that included all four baseline serum fibrosis marker levels entered together with other significant parameters from table 1 was constructed. This exercise confirmed a final model consisting of baseline total bilirubin, albumin, INR and YKL-40 levels for predicting clinical outcomes which had the lowest AIC compared with the other multivariate models. Inclusion of diabetes mellitus and baseline Ishak fibrosis and hepatic steatosis scores did not improve the performance of the model, with the AIC remaining unchanged (data not shown). The equation describing the risk of developing a clinical outcome is clinical risk score=0.883×(total bilirubin)+0.809 (INR >1.0)−1.63×(albumin)+0.89×(log YKL-40).
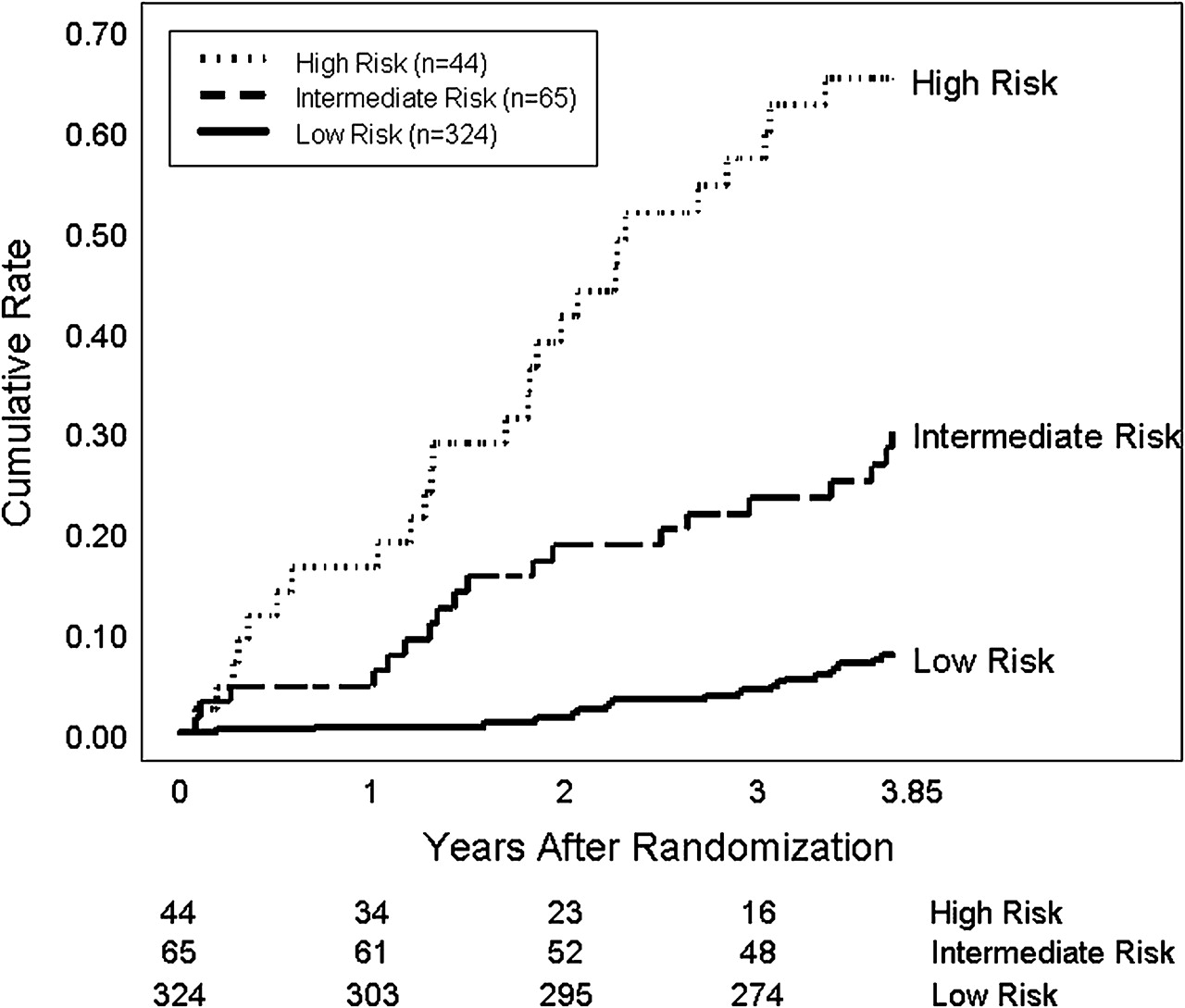
The output of this model can be divided into low (score less than −2.5), medium (score between −2.5 and −1.7) and high risk (score greater than −1.7) for the development of a clinical outcome. According to figure 1, only 8% of the 324 HALT-C Trial patients with a low risk score would be expected to experience a primary clinical outcome compared with 30% of the 65 patients with an intermediate risk score and 65% of the 44 patients with a high risk score. For example, a patient with a total bilirubin of 0.7 mg/dl, INR of 1.1, albumin of 3.8 g/dl and a YKL-40 level of 550 ng/ml has a risk score of −2.32 and would be at intermediate risk for the development of a clinical outcome during follow-up.

Risk of developing a primary clinical outcome during follow-up. The 324 HALT-C Trial patients with a risk score of under −2.5 have a low risk of a clinical outcome (8%) while the 44 patients with a risk score greater than −1.7 are at the highest risk of a clinical outcome (65%) and would warrant the most intensive monitoring.
The model derived from our data set (Model 1) was compared with a model that incorporates serum PIIINP, TIMP-1 and HA levels into an algorithm called the ‘Enhanced liver fibrosis score’ or ELF score.24 The AIC of Model 1 which reflects its overall goodness of fit for predicting outcomes was 710, while the AIC for the ELF score was 760, indicating that Model 1 explains more of the variation in clinical outcomes than the ELF score.
Serum fibrosis marker levels over time
Serial YKL-40 levels in patients who progressed clinically were compared with levels in patients who did not progress using random effects modelling. As seen in figure 2A, the YKL-40 levels increased in both groups of patients over time (p=0.0026) and were significantly higher in the progressors (p<0.0001). In addition, minimal overlap was observed in the values of progressors compared with those who did not progress, suggesting that assessment of this fibrosis marker over time may prove useful in identifying patients at high risk for disease progression.

Serum fibrosis marker levels in HALT-C patients with and without clinical progression. (A) Serum YKL-40 levels significantly changed over time in the 69 patients with disease progression and in the 393 without disease progression (p=0.0026). YKL-40 levels also differed significantly in the progressors compared with the non-progressors (p <0.0001), but the changes over time did not differ by outcome group (p=0.21). (B) Serum tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) levels also changed significantly over time (p<0.0001), were significantly different in progressors compared with non-progressors (p <0.0001) and the changes over time did differ by outcome group (p=0.0023). (C) Serum N-terminal peptide of procollagen type 3 (PIIINP) levels changed significantly over time (p <0.001), were significantly different in the two outcome groups (p <0.0001) and the changes over time did differ by outcome group (p <0.0001). (D) Serum hyaluronic acid (HA) levels significantly changed over time (p<0.0001), were significantly different in the two outcome groups (p<0.0001) and also differed over time by outcome group (p=0.0111).
The changes in baseline PIIINP, TIMP-1 and HA levels over time were also analysed using random effects modelling. As seen in figure 2B–D, the mean PIIINP, TIMP-1 and HA levels all changed significantly over time both in patients who progressed clinically and in those that did not (p<0.0001). In addition, the mean values of PIIINP, TIMP-1 and HA were consistently higher in the patients who progressed compared with patients who did not (p<0.0001). Finally, the changes over time of PIIINP, TIMP-1 and HA were greater in the progressors compared with the non-progressors (p<0.0001).
Peginterferon treatment and serum fibrosis marker levels
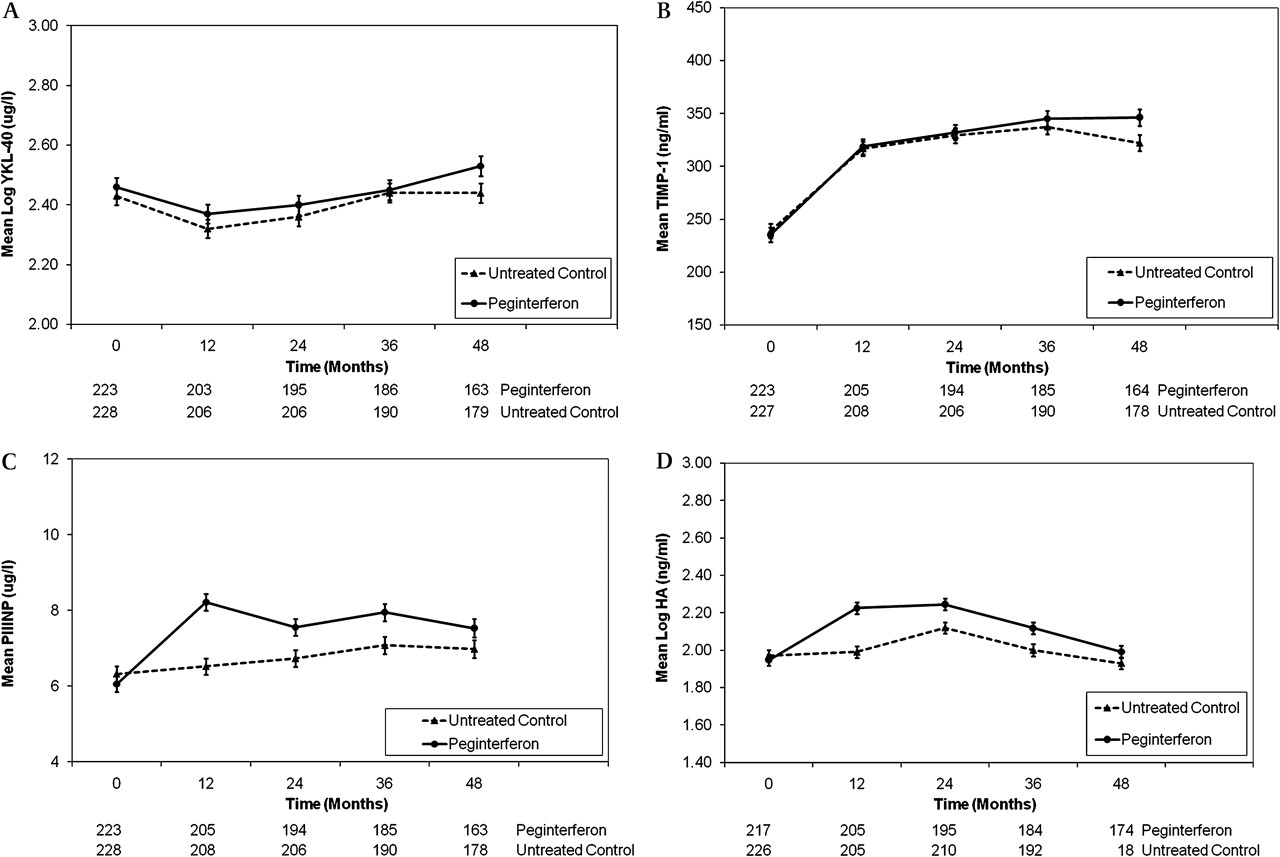
Serum fibrosis marker levels during the randomised phase of the HALT-C Trial were analysed by treatment group to determine if the patients receiving low-dose peginterferon had different levels compared with the untreated controls.20 Both YKL-40 and TIMP-1 levels changed significantly over time in the peginterferon-treated and untreated control patients (p <0.0001 for both) but did not differ significantly between the two study groups (YKL-40 p=0.230; TIMP-1, p=0.34) (figure 3A,B). In contrast, while PIIINP and HA levels changed significantly over time in treated and untreated patients (p <0.0001), the increase in levels of these two markers was significantly higher in the peginterferon-treated patients (p <0.0001 for both) (figure 3C,D). In addition, PIIINP levels in treated patients were lower at month 54 compared with month 48 (7.1 vs 7.5 ng/ml) while PIIINP levels were higher in the untreated patients at month 54 vs month 48 (7.6 vs 6.9 ng/ml). In addition, at month 54, HA levels in the treated patients decreased compared with month 48 (1.99 vs 2.18 ng/ml) and HA levels also decreased in the untreated patients (1.93 vs 2.24 ng/ml). Thus, a non-specific increase in both serum PIIINP and HA levels was observed in the peginterferon-treated patients compared with the untreated patients during the randomised phase of HALT-C but to a lesser extent than that seen in the lead-in phase.20

{kind=link}
{kind=link}
{kind=link}
Serum fibrosis marker levels over time by treatment group. (A) Serum YKL-40 levels changed significantly over time in both groups of patients (p<0.0001), but the changes did not differ by treatment group (p=0.23). (B) Serum tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) levels changed significantly over time (p<0.0001), but the changes did not differ by treatment group (p=0.34). (C) Serum N-terminal peptide of procollagen type 3 (PIIINP) levels increased significantly over time in the two treatment groups (p<0.0001). The levels were higher in the peginterferon-treated patients (p=0.0015) and the changes over time were greater in the peginterferon-treated patients (p<0.0001). (D) Serum hyaluronic acid (HA) levels also changed significantly over time in the two patient groups (p<0.0001), were higher in the peginterferon treated patients (p=0.0007) and the changes over time were greatest in the peginterferon group (p<0.0001).
Baseline predictors of histological progression
Worsening of fibrosis defined as an increase in the Ishak fibrosis score of ≥2 points at month 24 or 48 compared with baseline was a primary end point in the HALT-C Trial for patients with non-cirrhotic fibrosis.18 Amongst the 280 patients with a pretreatment Ishak fibrosis score of <5, 209 had a follow-up biopsy adequate for analysis. These 209 patients had similar pretreatment characteristics to the 71 excluded patients, except for older age (50.0 vs 47.6 years) and lower serum AST/ALT ratios (0.84 vs 0.91). During follow-up, 70 (34%) patients had worsening hepatic fibrosis while 139 patients had stable or unchanged Ishak fibrosis scores. In addition, a clinical outcome developed in 3 of the 70 (4.3%) patients with histological progression but in only 2 of the 139 (1.4%) histologically stable patients.
As anticipated, the 70 patients with worsening hepatic fibrosis had baseline laboratory features suggestive of more severe liver disease including significantly lower platelet counts and a trend towards higher bilirubin levels (table 2). In addition, baseline HA, YKL-40, PIIINP and TIMP-1 levels were all significantly higher in subjects who histologically progressed compared with those who did not. Furthermore, patients with histological progression had more severe hepatic steatosis at baseline than those who did not progress, but they were equally likely to receive peginterferon versus no treatment.
Pretreatment characteristics of the 209 HALT-C Trial patients included in the histological progression analysis
Multivariate models of histological progression
Using complementary log–log regression modelling of individual serum fibrosis marker levels in combination with other laboratory parameters, baseline YKL-40 and TIMP-1 levels did not remain associated with histological progression. However, both baseline PIIINP and HA levels remained significantly associated with histological progression when they were individually combined with other laboratory parameters. When all four serum fibrosis marker levels were combined with other baseline laboratory parameters, a final multivariate model consisting of baseline platelet and HA levels was identified (Supplementary table 1). The risk of fibrosis progression is estimated as risk= −0.3285×platelets+0.8831×logHA. The area under the receiver operator characteristic curve (AUROC) for the model was 0.663. Inclusion of baseline hepatic steatosis scores did not improve the model performance (data not shown).
Discussion
Serum fibrosis markers have been proposed as a simple and convenient means to estimate the severity of histological fibrosis in patients with liver disease. Although the markers may be effective in differentiating patients with cirrhosis from those with minimal to no fibrosis, the utility of these markers in discriminating between individual stages of fibrosis is limited.25 Developing accurate and reliable prognostic biomarkers that are linked to clinically important milestones in liver disease progression, such as decompensation and worsening CTP scores, is also an area of active investigation.24 26
The HALT-C Trial provided a unique opportunity to test several serum fibrosis markers as individual or combined predictors of the risk of clinical and histological disease progression. Serum PIIINP, HA, TIMP-1 and YKL-40 levels were selected for this study because previous studies had linked them to disease severity and progression in patients with CHC.27–30 In particular, these serum fibrosis markers were reduced in sustained virological responders compared with relapsers/non-responders, suggesting that they are closely linked with hepatic fibrogenesis.20 28 29 In addition, previous longitudinal studies have linked serum and liver tissue expression of YKL-40 with the risk of fibrosis progression.30
On univariate analysis, baseline levels of HA, PIIINP, YKL-40 and TIMP-1 were all strongly associated with the risk of a clinical outcome during follow-up (table 1). Multivariate regression modelling demonstrated that each marker retained significance when combined with other baseline laboratory parameters, except for serum PIIINP levels. Interestingly, HA, TIMP-1 and YKL-40 levels are all associated with clinical progression when combined with baseline albumin, bilirubin and INR levels individually, but the model which included YKL-40 performed the best with the lowest AIC. Application of our model to individual patients could provide stratification of the risk of clinical decompensation into low, medium and high risk, and potentially help clinicians determine which patients need closer follow-up and monitoring (figure 1). Our study findings are consistent with other studies demonstrating the short-term prognostic utility of markers of liver synthetic function (albumin and INR) and excretory function (bilirubin) in patients with CHC and advanced fibrosis.31–33 However, our data also demonstrate that the addition of baseline HA, TIMP-1 or YKL-40 levels to these routine laboratory parameters provides important incremental ability to predict clinical outcomes in patients with CHC.34 35 Furthermore, auxiliary analyses considering time-varying covariates indicate similar results, suggesting that HA, TIMP-1 and YKL-40 levels might not need to be monitored frequently over time.
The identification of baseline YKL-40 levels as an independent predictor of clinical outcomes is a novel and potentially important finding. Baseline YKL-40 levels have been associated with clinical outcomes in patients with alcoholic liver disease.36 YKL-40 is a growth factor for fibroblasts and endothelial cells and is expressed in areas of active hepatic fibrogenesis.37 38 In addition, YKL-40 levels strongly correlate with histological markers of stellate cell activation and the risk of rapid fibrosis progression in liver allograft recipients with recurrent hepatitis C.39 Furthermore, functional polymorphisms in YKL-40 gene expression are associated with the severity of liver fibrosis in patients with CHC, further strengthening the potential importance of this protein in hepatic fibrogenesis.40 Although serum YKL-40 levels are increased in patients with various solid organ tumours, the HALT-C Trial excluded patients with known cancer at enrolment, and a review of the clinical outcomes indicates that only six deaths in our cohort were attributed to a non-hepatic malignancy. Therefore, our data add to the growing body of literature supporting the potential utility of YKL-40 as a clinically useful biomarker for hepatic fibrosis.
Serum YKL-40 levels increased over time to a greater extent among patients in whom a clinical outcome developed compared with patients who did not progress clinically (figure 2). In addition, serum PIIINP and TIMP-1 levels also significantly increased over time and were consistently higher in patients who clinically progressed compared with patients who did not. These data suggest that serial assessment of these markers may prove useful to clinicians following patients with CHC. However, our previous study had also demonstrated a non-specific increase in PIIINP and HA levels in patients receiving full-dose peginterferon and ribavirin treatment.20 Therefore, the impact of low-dose peginterferon on serum fibrosis marker levels was explored. Both the YKL-40 and TIMP-1 levels significantly increased over time, but no significant difference was noted in these levels between those who did and did not receive peginterferon (figure 3). In contrast, the serum PIIINP and HA levels also changed significantly over time but were consistently higher in the peginterferon-treated patients compared with the untreated patients. These data confirm our previous observations that peginterferon may have systemic effects on other tissues that lead to an increase in these markers over time.41 In support of this, the PIIINP levels decreased after stopping treatment at month 54 compared with month 48 but remained unchanged or increased in the untreated patients.
Overall, our model explained more of the variation in outcomes than the ELF algorithm, with a lower AIC indicative of an improved ability to predict clinical outcomes. In addition, our model performed better than a model derived from the overall HALT-C cohort that did not include serum fibrosis markers (AIC of 710 vs 715).33 These differences may be due to the fact that our model was derived from this subset of HALT-C Trial patients, our use of commercial assay kits versus an automated platform, and the co-linearity of the serum fibrosis marker levels with each other. The addition of liver histological features such as baseline fibrosis and steatosis did not improve the model's performance.34 35 However, our model was substantially better at predicting clinical outcomes compared with the baseline Ishak fibrosis score alone (AIC of 710 vs 760). Limited resources precluded us from comparing our serum fibrosis marker panel with that expected with the Fibrotest assays or other proposed algorithms and, unfortunately, liver elastography was not available for use when the HALT-C Trial was initiated.27
The HALT-C Trial also provided a unique opportunity to explore the role of serum fibrosis marker levels in predicting the likelihood of histological disease progression over time. Baseline levels of YKL-40, PIIINP, TIMP-1 and HA were all associated with the risk of fibrosis progression on univariate analysis (table 2). However, a multivariate model of baseline HA and platelet counts was most strongly associated with histological progression. Low platelet counts are well known to be associated with more severe hepatic fibrosis, a reflection of portal hypertension and hypersplenism as well as reduced thrombopoietin production.9 42 Similarly, baseline and serial platelet levels were reported to be associated with a higher likelihood of developing varices and death in patients with alcoholic cirrhosis.43 The increase in serum HA levels associated with worsening hepatic fibrosis has been attributed to reduced HA clearance by hepatic sinusoids and increased HA production by hepatic stellate cells.44 However, the clinical utility of this model may be limited due to the anticipated frequency of both false-positive and false-negative results expected with the observed AUROC. In addition, only patients with CHC with advanced fibrosis were enrolled into the HALT-C study, which may limit the generalisability of our findings.
Strengths of our study include the large number of well-characterised patients who underwent serial testing for the analytes of interest. In addition, the patients were treated in the setting of a controlled clinical trial, and clinical disease progression was defined by prospectively identified, objective end points that were reviewed and confirmed by an independent expert committee. Lastly, histological progression was defined by a two-point increase in Ishak fibrosis score rather than one fibrosis stage as used in other studies. However, the potential for sampling error and understaging of fibrosis remains possible.7 8 The generalisability of our findings to other patients with CHC and prior non-response to peginterferon and ribavirin may be limited due to the strict inclusion criteria of the HALT-C Trial. Finally, logistical constraints precluded us from obtaining direct portal pressure measurements in enrolled patients to compare with our serum fibrosis marker data. Still, this study represents the largest and longest prospective assessment of serum fibrosis markers in association with liver disease progression in patients with CHC reported to date.
In summary, pretreatment levels of TIMP-1, YKL-40 and HA in combination with baseline albumin, bilirubin and INR enhance the ability to identify patients with CHC who are at increased risk of disease progression. In our final model that includes pretreatment serum YKL-40 levels, the risk of disease progression could be stratified into low, medium and high risk groups. The observed increase in YKL-40 levels and TIMP-1 levels in patients who progress clinically compared with patients who do not further demonstrates the potential utility of these fibrosis markers in tracking patients with CHC at risk for disease progression. However, validation of our exploratory models in independent patient cohorts that are longitudinally followed is needed as well as in a group of patients with CHC with a broader distribution of fibrosis severity. If validated, measurement of serum fibrosis marker levels in conjunction with standard laboratory parameters may help clinicians identify which patients with CHC need closer follow-up and monitoring.
References
Supplementary materials
Web only data
Files in this Data Supplement:
Footnotes
Linked articles 219162.
This is publication #49 of the HALT-C Trial.
Funding This study was supported by the National Institute of Diabetes & Digestive & Kidney Diseases (contract numbers are listed below). Additional support was provided by the National Institute of Allergy and Infectious Diseases (NIAID), the National Cancer Institute, the National Center for Minority Health and Health Disparities and by General Clinical Research Center and Clinical and Translational Science Center grants from the National Center for Research Resources, National Institutes of Health (grant numbers are listed below). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Additional funding to conduct this study was supplied by Hoffmann-La Roche, through a Cooperative Research and Development Agreement (CRADA) with the National Institutes of Health. In addition to the authors of this manuscript, the following individuals were instrumental in the planning, conduct and/or care of patients enrolled in this study at each of the participating institutions as follows: University of Massachusetts Medical Center, Worcester, Massachusetts (contract N01-DK-9-2326), Gyongyi Szabo, Barbara F Banner, Maureen Cormier, Donna Giansiracusa; University of Connecticut Health Center, Farmington, Connecticut (grant M01RR-06192), Gloria Borders, Michelle Kelley; Massachusetts General Hospital, Boston, Massachusetts (contract N01-DK-9-2319, grant M01RR-01066; grant 1 UL1 RR025758-01, Harvard Clinical and Translational Science Center), Raymond T Chung, Andrea E Reid, Atul K Bhan, Wallis A Molchen, Cara C Gooch; University of Michigan Medical Center, Ann Arbor, Michigan (contract N01-DK-9-2323, grant M01RR-00042, grant 1 UL1 RR024986, Michigan Center for Clinical and Health Research), Joel K Greenson, Pamela A Richtmyer, R Tess Bonham; Virginia Commonwealth University Health System, Richmond, Virginia (contract N01-DK-9-2322, grant M01RR-00065), Mitchell L Shiffman, Melissa J Contos, A Scott Mills, Charlotte Hofmann, Paula Smith; National Institute of Diabetes and Digestive and Kidney Diseases, Division of Digestive Diseases and Nutrition, Bethesda, Maryland, James E. Everhart, Leonard B Seeff, Patricia R Robuck, Jay H Hoofnagle; New England Research Institutes, Watertown, Massachusetts (contract N01-DK-9-2328), Kristin K Snow, Margaret C Bell, Teresa M Curto; Armed Forces Institute of Pathology, Washington, DC, Fanny Monge, Michelle Parks; Data and Safety Monitoring Board Members: (Chair) Gary L. Davis, Guadalupe Garcia-Tsao, Michael Kutner, Stanley M. Lemon, Robert P. Perrillo.
Competing interests We are enclosing two sections relating to disclosures of potential conflicts of interest. Disclosures that pertain to the industrial sponsors who have partnered with the NIDDK to support this study, which are also listed in the text of the manuscript, are listed in section A. In addition, many of the HALT-C Trial investigators have other associations with industry relating to the area of hepatitis C, and, to achieve the highest level of disclosure, we list these for you as well in section B. Authors with no financial relationships related to this project are: DN, JLD, ZDG, ECW, GLS, Section A. The following are disclosures that pertain to the industrial sponsors who have partnered with the NIDDK to support this study, which are also listed in the text of the manuscript. Financial relationships of the authors with Hoffmann-La Roche, Inc., are as follows: RJF is on the speaker's bureau; HLB receives research support; RKSt is a consultant, receives research support, ASL is a consultant. Section B. In addition, many of the HALT-C Trial investigators have other associations with industry relating to the area of hepatitis C, and, to achieve the highest level of disclosure, we list these for you as well. RJF: Bristol-Meyers Squibb–Speaker's bureau and consultant. Abbott Pharmaceuticals–Consultant. Bayer/Siemens–Consultant. JLD: Vertex Pharmaceuticals–receives research support. Serves on a Data Monitoring Committee for Schering-Plough Research Institute and Human Genome Sciences. HLB: Merck—receives research support. Novartis Pharmaceuticals–consultant and receives research support. Lundbeck Pharmaceuticals—consultant and on speakers' bureau. Vertex Pharmaceuticals–receives research support. RKS: WAKO Diagnostics–consultant and receives research support. Schering-Plough Corporation–consultant and speaker's bureau. Glaxo Smith Kline–speaker's bureau. Vertex Pharmaceuticals–Advisory Board. ZDG: Research support from Schering-Plough and Novartis. ASL: Schering-Plough Corporation–consultant and receives research support. Vertex Pharmaceuticals–consultant. Novartis Pharmaceuticals–receives research support.
Ethics approval This study was conducted with the approval of the ethics committess at all of the participating sites.
Provenance and peer review Not commissioned; externally peer reviewed.