Article Text

Abstract
The promise of personalised medicine is now a clinical reality, with colorectal cancer genetics at the forefront of this next major advance in clinical medicine. This is no more evident than in the recent advances in testing of colorectal cancers for specific molecular alterations in order to guide treatment with the monoclonal antibody therapies cetuximab and panitumumab, which target the epidermal growth factor receptor. In this review, genetic mechanisms of colorectal cancer and how these alterations relate to emerging biomarkers for early detection and risk stratification (diagnostic markers), prognosis (prognostic markers) and the prediction of treatment responses (predictive markers) are examined.
- Colon cancer
- biomarkers
- EGFR
- KRAS
- K-Ras
- BRAF
- microsatellite instability
- msi
- chromosome instability
- cetuximab
- panitumumab
- personalised medicine
- colorectal cancer genes
- molecular biology
- molecular oncology
- tumour markers
Statistics from Altmetric.com
- Colon cancer
- biomarkers
- EGFR
- KRAS
- K-Ras
- BRAF
- microsatellite instability
- msi
- chromosome instability
- cetuximab
- panitumumab
- personalised medicine
- colorectal cancer genes
- molecular biology
- molecular oncology
- tumour markers
Introduction
The promise of personalised medicine is now a clinical reality, with colorectal cancer genetics at the forefront of this next major advance in clinical medicine. This is no more evident than in the testing of colorectal cancers for specific molecular alterations in order to guide treatment with the monoclonal antibody therapies cetuximab and panitumumab, which target the epidermal growth factor receptor (EGFR).1–3 Indeed, the discovery that acquired KRAS mutations are a robust predictive marker of resistance to cetuximab and panitumumab4 5 has led to clinically validated and cost-effective testing strategies to direct these drugs to appropriate patients. This discovery resulted from a detailed understanding of colorectal cancer genetics, including the role of KRAS mutations in colorectal carcinogenesis, as well as knowledge of the EGFR signalling pathways.6 The success of KRAS mutation testing in predicting treatment response is just the beginning of the use of genetic markers for directing the care of patients with colorectal cancer. Many other genetic markers in colorectal cancer show promise for their use in treatment selection, prognosis and early cancer detection. In this context, knowledge of the underlying genetic mechanisms of colorectal tumorigenesis and the potential of specific genetic lesions for clinical decision making is expected to become part of the working knowledge of care providers managing colon cancer patients. However, despite the promising advances in the molecular pathology of colorectal cancer that are highlighted in this review, it is important to emphasise that clinicopathological staging of tumour tissue is still the cornerstone of prognostication and treatment selection. The modern tumour–node–metastasis (TNM) classification system is recommended, although the original Dukes staging system is still used by some clinicians and is taught to pathologists in training.7 The pathological features with greatest prognostic power are depth of tumour invasion, burden of lymphovascular invasion (estimated by the number of lymph nodes infiltrated by cancer) and presence of distant metastases. Efforts to correlate genetic alterations with histological features have had limited success, although microsatellite instability is a molecular feature that shows modest correlation with certain histological features such as cribriform architecture and medullary histology.8 Thus, molecular testing is usually required for accurate assessment of specific gene mutations or genomic instability that provide prognostic and predictive information beyond clinicopathological features.
In this review, we examine genetic mechanisms of colorectal cancer and how these alterations relate to emerging biomarkers for early detection and risk stratification (diagnostic markers), prognosis (prognostic markers) and the prediction of treatment responses (predictive markers) (table 1, box 1). The genetic features of colorectal cancer that are currently most clinically useful will be emphasised in this review, and a detailed description of the molecular genetics and molecular biology of the germane genetic and epigenetic alterations will be provided. We will conclude by reviewing the potential role of genetic markers in the selection of targeted colorectal cancer treatments that are in preclinical development or in phase I and II trials.
Selected biomarkers that have been evaluated in colorectal cancer
Box 1 Summary points
Chromosome instability (CIN) and microsatellite instability (MSI) are distinct mechanisms by which colorectal cancers arise, and that are associated with unique molecular features.
Key pathways that drive colorectal cancer are WNT signalling, transforming growth factor β (TGFβ) signalling and epidermal growth factor receptor (EGFR) signaling. Ras/Raf/MAPK and phosphatidylinositol 3-kinase (PI3K) pathways are both stimulated by EGFR. Currently, only downstream mediators of EGFR have a clinical role as biomarkers.
KRAS mutations in codon 12/13 are a highly validated predictive marker for resistance to monoclonal antibody drugs that target EGFR; BRAF V600E mutation is likely to be a second predictive marker. Additional resistance markers including PIK3CA mutations and PTEN protein loss are being evaluated.
MSI+ cancers have a better prognosis and CIN+ cancers do worse; 18q LOH+ tumours also have a worse prognosis but are frequently associated with CIN. Downstream mediators of EGFR are under study for prognostication.
The role of colorectal cancer molecular biomarkers in clinical decision making is likely to expand as more targeted drugs become available.
Molecular mechanisms of colorectal carcinogenesis
The adenoma/carcinoma progression sequence
Colorectal cancer arises as the result of the accumulation of acquired genetic and epigenetic changes that transform normal glandular epithelial cells into invasive adenocarcinomas. Steps that transform normal epithelium into benign neoplasia (adenoma), followed by invasive carcinoma and eventually metastatic cancer are described in the classic tumour progression model proposed by Fearon and Vogelstein (figure 1).6 Since this model was originally proposed, our understanding of the molecular pathogenesis has advanced considerably and led to numerous revisions of the Vogelstein and Fearon model. For instance, the original model proposed that only tubular and tubulovillous adenomas had the potential to progress to invasive adenocarcinoma. It is now recognised that serrated polyps including sessile serrated adenomas (SSAs) and traditional serrated adenomas (TSAs) also have the potential for malignant transformation.10 11 These polyps are an alternative pathway to malignancy whereby a subset of hyperplastic polyps progress to serrated neoplasms (SSAs or TSAs) and a fraction of these serrated neoplasms progress to cancer. Premalignant serrated polyps more frequently arise in the proximal colon12 and are associated with microsatellite instability and aberrant DNA methylation at CpG islands, whereas conventional tubular adenomas arise via biallelic inactivation of the APC tumour-suppressor gene and display chromosome instability.13 Furthermore, other molecular lesions, such as BRAF V600E mutations, are characteristically found more often in tumours arising via the serrated neoplasia pathway.13

The adenoma–carcinoma progression sequence. Colorectal carcinogenesis progresses by at least two well-recognised pathways. The chromosome instability (CIN) pathway is characterised by classic tubular adenoma histology and the early acquisition of APC mutations that lead to deregulated WNT signalling, frequent activating mutations of the KRAS oncogene at the early adenoma stage, loss of heterozygosity at chromosome 18q (18qLOH) in late adenomas and TP53 mutations that facilitate the transition to frank malignancy. In contrast, tumors that harbour microsatellite instability (MSI) frequently acquire BRAF mutations and are not associated with 18qLOH or TP53 mutations. Sporadic MSI cancers appear to arise commonly via the serrated neoplasia pathway, in which sessile serrated adenomas are the most frequently observed precancerous lesions.
Genomic and epigenomic instability and chromosomal alterations
Genomic and epigenomic instability distinguishes neoplastic from normal colonic epithelium and is a hallmark feature of colorectal carcinogenesis.14 15 At least four kinds of genomic or epigenetic instability have been described in colorectal cancers: (1) chromosomal instability (CIN); (2) microsatellite instability (MSI); (3) CpG island methylator phenotype (CIMP); and (4) global DNA hypomethylation. Overlap between these categories and imprecise use of these terms has led to confusion and confounds interpretation of the literature.16 Thus, in this section, we will first define the different types of genomic and epigenetic instability in colorectal cancer and will delineate in general terms how these mechanisms are clinically relevant.
CIN. The most common form of genomic instability is chromosome instability, which is found in as many as 85% of colorectal cancers.17 Chromosome instability, which can be recognised by the presence of aneuploidy, is defined as the presence of numerical chromosome changes or multiple structural aberrations and is typically assessed by DNA flow cytometry.18 Despite the frequent occurrence of CIN in colorectal cancer, the mechanisms that give rise to this form of genomic stability and the role of aneuploidy in tumour progression remain poorly understood. However, there is some evidence that CIN promotes cancer progression by increasing clonal diversity.19–21 Importantly, from a clinical perspective, large meta-analyses have demonstrated that CIN is a marker of poor prognosis in colorectal cancers.18 22
MSI. Microsatellite unstable tumours, which account for ∼15% of colorectal cancers, are generally regarded as being mutually exclusive of CIN tumours because they display a normal karyotype and exhibit unique genetic features, although there does appear to be a subset of tumours that show both CIN and MSI.16 MSI colorectal cancer has been defined by the presence of at least 30% unstable loci in a panel of 5–10 loci consisting of mono- and dinucleotide tracts selected at a National Cancer Institute consensus conference.23 Currently, many clinical laboratories assess MSI using a panel of five mononucleotide markers (BAT-25, BAT-26, NR-21, NR-24 and MONO-27) that were selected for high sensitivity and specificity.9 A subset of tumours with only 10–29% unstable loci has been designated as a form of microsatellite tumours designated ‘MSI-low’. Although there is evidence that MSI-low cancers have distinct features compared with MSI (also referred to as ‘MSI-high’, or ‘MSI-H’) and microsatellite stable (MSS) tumours, there is considerable controversy regarding whether MSI-low is a unique molecular subclass of colorectal cancer.16 17 24 Patients with colorectal cancer with MSI tumours have been shown to have a better prognosis compared with patients with CIN tumours,18 25 and probably respond differently to adjuvant chemotherapy compared with patients with MSS cancer.26–28
In contrast to CIN, the mechanisms underlying MSI are relatively well understood and involve inactivation of genes in the DNA mismatch repair (MMR) family, either by aberrant methylation or by somatic mutation.21 Furthermore, individuals with Lynch syndrome (hereditary non-polyposis colorectal cancer, HNPCC) almost exclusively develop MSI colorectal cancers because they have germline mutations in the MMR genes, which include MLH1, MSH2, MSH6 and PMS2. In contrast, sporadic MSI colorectal cancers most often have loss of MMR activity as the result of silencing of MLH1 by aberrant methylation.21 29 It is also now recognised that sporadic MSI tumours are associated with the serrated neoplasia pathway and frequently carry BRAF V600E mutations, while cancers resulting from germline mutations in MMR genes (Lynch syndrome) do not have mutated BRAF.30 31 Thus, the presence of a BRAF mutation in an MSI tumour effectively excludes the possibility that the tumour arose as the consequence of Lynch syndrome (figure 2).

Testing strategies for Lynch syndrome (hereditary non-polyposis colorectal cancer (HNPCC)). A multistage approach to facilitate the cost-effective diagnosis of Lynch syndrome is outlined. Patients with a high clinical suspicion of Lynch syndrome are first screened by immunohistochemistry (IHC) studies of the tumour tissue to assess for loss of mismatch repair (MMR) protein expression and by MSI testing of the tumour DNA (Tier 1 Screening Tests). Patients with tumours that show microsatellite instability (MSI) with loss of MSH2, MSH6 or PMS2 by IHC undergo germline DNA mutation analysis of the gene corresponding to the missing protein. In contrast, patients with MSI tumours that lack MLH1 are further assessed, with assessment of the tumoir for MLH1 promoter methylation and mutant BRAF V600E (Tier 2 Screening Test) because most sporadic MSI colon cancers have methylated MLH1 and Lynch syndrome MSI cancers rarely harbour BRAF mutations. When there is no evidence of MLH1 promoter methylation or BRAF mutation, mutation analysis of the MLH1 gene is performed to identify patients with Lynch syndrome with mutations in this gene.
CIMP. Epigenetic instability in colorectal cancer is manifested as both hypermethylation of gene promoters that contain CpG islands (the CpG Island methylator phenotype, CIMP), and global DNA hypomethylation. Mechanisms that give rise to CIMP are not yet clear, although the strong association between BRAF V600E mutations and CIMP colorectal cancer suggests a role for activated BRAF in the pathogenesis of the methylator phenotype and a link between sporadic MSI and CIMP.32 33 However, in vitro studies of mutant BRAF in colorectal cancer cell lines have not demonstrated a direct cause and effect relationship between BRAF and CIMP.34 Furthermore, although CIMP tumours do appear to represent a distinct subset of colorectal cancer, the clinical utility of this designation is hindered by lack of a universally accepted definition of the methylator phenotype. CIMP is usually defined as methylation of at least three loci from a selected panel of five gene-associated CpG islands. Because this panel is not always the same across studies, attempts are being made to facilitate standardisation of CIMP markers for clinical use.33 35 Some authors have proposed two classes of CIMP, CIMP-low and CIMP-high, depending on the number of methylated marker loci detected.32 Another group suggested that CIMP colorectal cancers be divided into two distinct classes (called CIMP1 and CIMP2) based on the results of unsupervised cluster analysis of a large panel of methylation markers.36 Finally, considerable overlap between CIMP and sporadic MSI tumours adds to the challenge of incorporating CIMP status into clinical trials and clinical decision making.33 Retrospective studies suggest CIMP will ultimately be shown to be a predictive marker for colorectal cancer, but the data are not adequate at this time to recommend its clinical use.36 37 Thus, the discovery and classification of CIMP tumours has advanced our understanding of the molecular pathology of colorectal cancer but has not yet impacted clinical care.
In addition to aberrant gene methylation, a global decrease in methylation has also been identified in many colorectal cancers and is tightly associated with CIN tumours.38 39 Further research is necessary to determine if measurement of global DNA hypomethylation in colorectal cancer has any role in the clinical setting.
Role of specific genetic alterations and signal pathway deregulation as biomarkers
Just as important as genomic and epigenomic instability for the pathogenesis of colorectal cancer is the accumulation of mutations in specific genes and the resulting deregulation of specific signalling pathways that control the hallmark behaviours of cancer: cell proliferation, differentiation, apoptosis, immortalisation, angiogenesis and invasion. The best-studied pathways that are deregulated in colorectal cancer are the WNT–β-catenin signalling pathway, the transforming growth factor β (TGFβ) signalling pathway, the EGFR–mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K) pathway.5 16 Selected deregulated pathways in colorectal cancer and targeted treatments in clinical use or in clinical trials are summarised in table 2.
Pathways commonly deregulated in colorectal cancer and targeted drugs in clinical use (in bold) or in clinical trials
Key tumour-suppressor genes that do not necessarily mediate their effects through signal pathway deregulation, such as TP53, and recurrent cytogenetic aberrations such as 18q loss of heterozygosity (LOH) are also well studied in colorectal cancer and affect the malignant transformation of colon epithelial cells through specific effects on the behaviour of the cells (figure 1). The use of these molecular alterations in the management of patients with colorectal cancer will also be discussed in more detail below.
WNT pathway
Mutations in the adenomatous polyposis coli (APC) gene occur in up to 70% of sporadic colorectal cancers and are the cause of the familial adenomatous polyposis (FAP) cancer predisposition syndrome. APC mutations can be found at the earliest stages of neoplasia and are predominantly associated with the classic tubular adenoma pathway and CIN cancers (figure 1).6 40 41 The APC protein negatively regulates WNT signalling via targeting β-catenin for ubiquitin-mediated proteasomal degradation. Disruption of the APC protein results in increased WNT signalling through stabilisation of nuclear β-catenin. Activating mutations in the β-catenin gene (CTNNB1) that protect the protein from APC-mediated degradation are also observed in colorectal neoplasia, although they are found more frequently in adenomas (12.5%) than invasive cancer (1.4%), suggesting that CTNNB1-mutant tumours do not frequently progress to carcinoma.42 Despite the critical and nearly universal role of WNT pathway activation in colorectal carcinogenesis, there is currently no clinical use for APC or CTNNB1 mutations for treatment selection, prognosis or early cancer detection (table 1). There has been intense effort to develop small molecule inhibitors of this pathway, but these efforts are still confined to the preclinical arena. If these agents eventually reach the clinic, the assessment of APC mutations or activated β-catenin (by the detection of nuclear localisation of β-catenin by immunostaining) is likely to have a role in directing the selection of patients who will respond to these agents.
TGFβ pathway
Deregulation of TGFβ signalling, which is generally considered a tumour-suppressor pathway in the colon, occurs in the majority of colorectal cancers.43 Inactivating mutations have been observed in receptor genes (TGFBR2 and TGFBR1), postreceptor signalling pathway genes (SMAD2, SMAD4) and TGFβ superfamily members (ACVR2).17 44–46 Functionally significant mutations in TGFBR2 are detected in as many as 30% of all colorectal cancer and are associated with the malignant transformation of late adenomas. TGFBR2 mutations are most common in MSI tumours, but also occur in ∼15% of MSS tumours (figure 1).46–48 SMAD4 is located on 18q in the region commonly deleted in colorectal cancer, and is associated with adenoma formation and adenoma–carcinoma progression in mouse models, supporting a role for SMAD4 as a tumour-suppressor gene.49 Furthermore, loss of SMAD4 expression as detected by immunostaining has been reported in >50% of colon cancers and is associated with lymph node metastases.50 There is still not any definite clinical role for any genetic markers in the TGFβ signalling pathway; however, there is some evidence that SMAD4 expression levels may be associated with prognosis and response to 5-fluorouricial (5-FU), and there is ongoing investigation of 18qLOH as a predictive marker, which is discussed further in the next section.51 52
18qLOH
Loss of the long arm of chromosome 18 (18qLOH) is the most frequent cytogenetic alteration in colorectal cancer and is observed in up to 70% of tumours.6 22 Two genes thought to have a role in the tumourigenic effects of this loss are deleted in colorectal carcinoma (DCC) and SMAD4. Additional mediators of the TGFβ pathway, including SMAD2 and SMAD7, are also in the 18qLOH region, suggesting that 18qLOH promotes tumourigenesis at least in part through deregulation of TGFβ signalling. It appears that deletion at 18q is associated with a worse prognosis; however, efforts to definitively link 18qLOH to prognosis are limited by a lack of consistent results across studies and heterogeneous detection methods.22 Ongoing clinical trials (eg, NCT00217737, also designated ECOG 5202) are assessing the utility of 18qLOH for treatment selection.
TP53
Mutations in the tumour-suppressor gene TP53 occur in about half of all colorectal cancers and promote the malignant transformation of adenomas (figure 1).6 Like APC, TP53 is a key tumour suppressor that has been extensively studied in colorectal cancer but currently has no predictive or prognostic role in the clinical setting.16
Mediators of EGF signalling
EGFR/RAS/RAF/RAF/MAPK
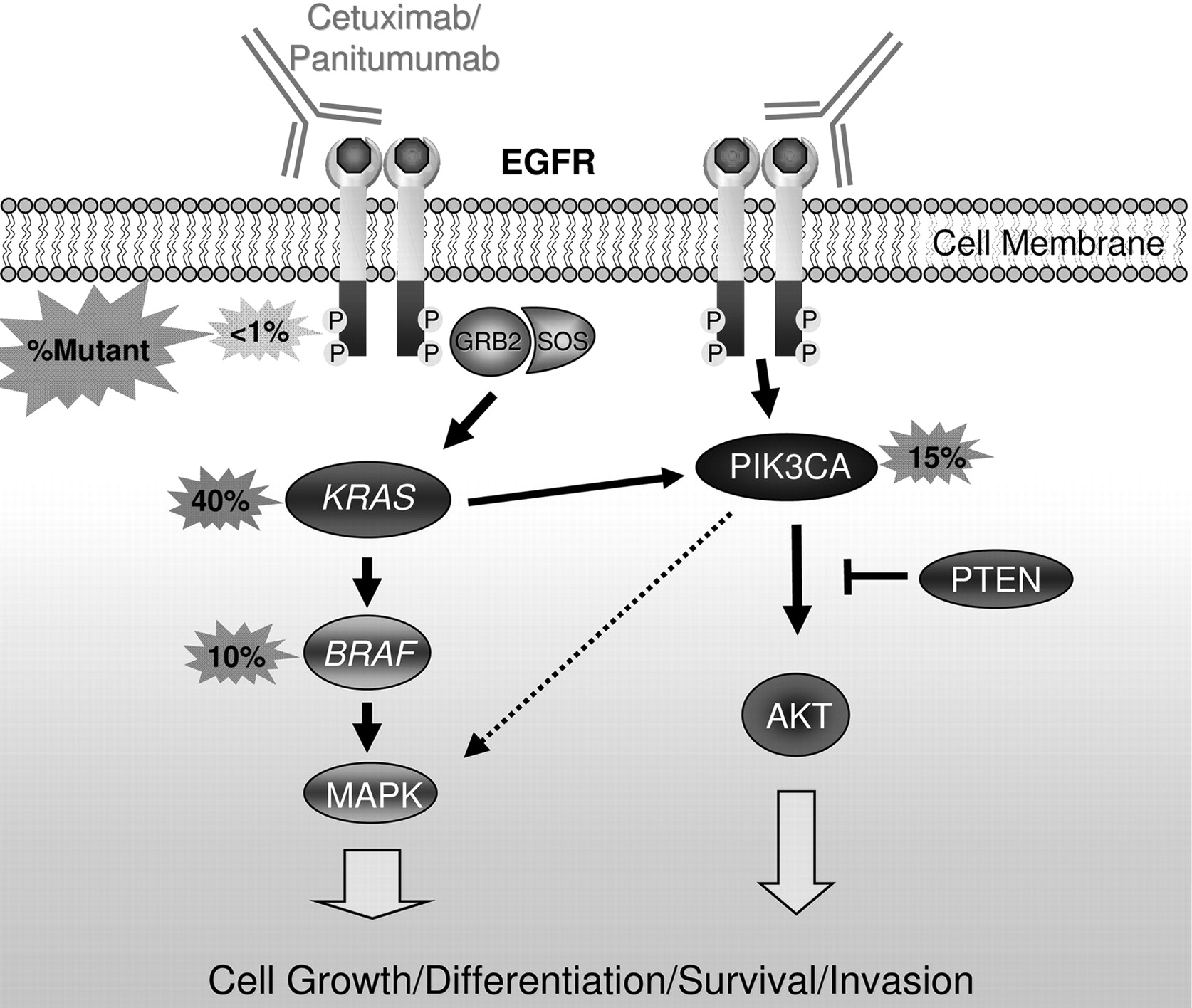
KRAS, a member of the RAS family of proto-oncogenes, is the most frequently mutated gene in all of human cancer and arguably the most clinically important oncogene in colorectal cancer. The KRAS protein is a downstream effector of EGFR that signals through BRAF to activate the MAPK pathway and promote cell growth and survival (figure 3). Mutations in KRAS codons 12 or 13 occur in ∼40% of colorectal cancers and lead to constitutive signalling by impairing the ability of GTPase-activating proteins to hydrolyse KRAS-bound GTP.53 KRAS mutations occur after APC mutations in the adenoma–carcinoma progression sequence, but are still a relatively early event in tumourigenesis (figure 1).6 Acquired KRAS mutations are maintained throughout carcinogenesis, as evidenced by the nearly perfect concordance of KRAS mutation status in primary and metastatic colorectal cancer.54 55 This fact is critical to the utility of KRAS mutational analysis on archived primary tumour specimens in patients with metastatic disease and usually eliminates the need for additional biopsy tissue.
{kind=link}
{kind=link}
{kind=link}
Mediators of epidermal growth factor receptor (EGFR) signalling and anti-EGFR antibodies. EGFR forms a homodimer after ligand activation, which results in phosphorylation/activation of the intracellular kinase domain and a cascade of downstream signalling including activation of the Ras/Raf/MAPK and phosphatidylinositol 3-kinase (PI3K) pathways that are associated with cell growth, differentiation, survival and invasion. Monoclonal antibodies used to treat patients with metastatic colorectal cancer including cetuximab and panitumumab bind to the extracellular portion of EGFR and inhibit signalling in some patients. Activating mutations in KRAS occur in ∼40% of colorectal cancers and are thought to confer resistance to these drugs by bypassing the need for upstream EGFR signals. Activating mutations in BRAF—the direct downstream effector of KRAS—occur in ∼10% of colorectal cancers and also probably confer resistance to anti-EGFR monoclonal antibodies. Emerging evidence supports an additional role for oncogenic aberrations in the PI3K pathway in cetuximab and panitumumab resistance.
The BRAF gene, mutated in ∼10–15% of colorectal cancers, encodes a protein kinase that is the direct downstream effector of KRAS in the Ras/Raf/MAPK signalling pathway. The majority of BRAF mutations are a single base change resulting in the substitution of glutamic acid for valine at codon 600 (V600E; sometimes referred to as ‘V599E’).5 KRAS and BRAF mutations are mutually exclusive, supporting the hypothesis that an activating mutation in either gene is sufficient to promote tumourigenesis via increased MAPK signalling.56 As discussed above, BRAF mutations are much more frequent in MSI tumours (∼50%) compared with MSS tumours (∼5%) and are very tightly linked to CIMP cancers and the serrated neoplasia pathway.56 57 Emerging evidence supports a role for BRAF as a genetic marker for prediction, prognosis and risk stratification.
Alterations in EGFR ligands and the EGFR gene itself are also observed in a subset of colorectal cancers. There are some data to support that upregulation of the EGFR ligands epigregulin and amphiregulin are associated with an anti-EGFR drug response.58 59
The PI3K pathway
Mutations in PI3K pathway genes are observed in up to 40% of colorectal cancer and are nearly mutually exclusive of one another.60 The most frequent mutations of the PI3K pathway occur in the p110α catalytic subunit PIK3CA, which are reported in up to 32% of colorectal cancers and may promote the transition from adenoma to carcinoma (figure 1).61 Mutations are also observed in PTEN, a tumour-suppressor gene that negatively regulates PI3K signalling in as many as 30% of MSI tumours and 9% of CIN tumours.62 The PI3K pathway is modulated by EGFR signalling in part via KRAS activation, and there is a plausible role for both PIK3CA and PTEN mutations as predictive markers of anti-EGFR treatment (figure 3).63 64 Currently, there is not sufficient evidence from clinical studies to support the use of PI3K pathway mutations as predictive or prognostic biomarkers.
Risk stratification and early detection
One use of molecular markers in the management of colorectal cancer is in risk stratification for identifying individuals at high risk for developing colorectal cancer and for the early detection of colon adenomas and early-stage colorectal cancers. With regard to risk stratification, the most robust molecular markers to date are germline mutations in genes that cause the hereditary colon cancer syndromes (eg, APC mutations and FAP, BMPR1A and juvenile polyposis, etc.) and MSI tumour status, which is an indicator of the possibility of Lynch syndrome. The use of MSI tumour testing in the diagnosis of Lynch syndrome will be discussed below in the context of MSI testing being a risk stratification marker because of its association with Lynch syndrome.
Lynch syndrome/HNPCC syndrome
Identifying individuals with Lynch syndrome (also known as HNPCC) dramatically alters their clinical management, and can lead to effective colorectal cancer prevention programmes for these individuals and their family members. However, currently the definitive molecular diagnosis of Lynch syndrome requires expensive germline DNA mutation analysis of multiple DNA MMR genes (MLH1, MSH2, MSH6 and PMS2). To facilitate the most cost-effective strategies for identifying patients at high risk for Lynch syndrome who are candidates for genetic testing, the evaluation of molecular features of colorectal cancers that have occurred in these individuals or other family members can be used to predict the likelihood of identifying a germline mutation in one of the MMR genes. It is now common practice for molecular diagnostics laboratories to offer a stepwise series of molecular tests that are used to identify colorectal cancers that probably arose in the setting of Lynch syndrome. These tests are based on the molecular pathology of colorectal cancer (figure 2).65 A common approach is initially to test the tumours for loss of MMR gene products (MLH1, MSH2, MSH6 and PMS2) by immuohistochemistry (IHC) and for MSI by PCR as the first-tier screening test (see, for example, http://www.mayomedicallaboratories.com/test-catalog/Clinical+and+Interpretive/17073), although there is support for the use of IHC alone as a first-line test.66 Tumours that display MSI and loss of MLH1 protein expression by IHC are then subjected to reflex testing for BRAF V600E mutation status and MLH1 promoter hypermethylation to help distinguish sporadic MSI tumours (∼50% BRAF-mutant and 99% MLH1-methylated) from Lynch syndrome MSI tumours (BRAF-WT, infrequent MLH1 methylation) (figure 2, table 3).30 67 68 This strategy is most effective in excluding individuals who are unlikely to have an MMR gene mutation from undergoing germline mutation testing. It is notable that in those tumours that have MSI and loss of MSH2, MSH6 or PMS2 the likelihood of having a germline mutation is extremely high. Also of interest, it is now recognised that a strategy that relies on clinical criteria alone for the diagnosis of individuals at risk for Lynch syndrome underdiagnoses this syndrome.69 In light of the substantial effect of a missed diagnosis on an individual's likelihood of developing cancer in the future, a strategy that employs universal testing of all colorectal cancer is being advocated by some experts in this area.70 It remains to be determined if this strategy is cost-effective and if the benefits outweigh the risks.
Biomarkers used in the diagnosis of Lynch syndrome (HNPCC)
Molecular markers and early detection of colorectal cancer
Colonoscopy is the most accurate test currently for colorectal cancer screening; however, it is expensive and associated with procedure-related complications and poor patient compliance. In contrast, another commonly used colorectal cancer screening test, faecal occult blood testing (FOBT) is inexpensive and simple to perform, but has a relatively low sensitivity and specificity.71 Advances in our understanding of the molecular pathology of colorectal cancer have led to the identification of promising early detection molecular markers for use in non-invasive colorectal cancer screening assays.72 73 Stool-based methylated VIMENTIN (mVim) is a clinically validated marker for early colorectal cancer detection that is now commercially available in the USA (table 1).74 The test relies on the fact that a majority of colorectal cancers (53–84%) carry an aberrantly methylated vimentin (VIM) gene. A PCR-based assay that simultaneously measured mVim and DNA integrity reported a sensitivity of 83% and a specificity of 82%, with approximately equal sensitivity in patients with stage I–III colorectal cancer .75 At this time, methods are under development to enhance the performance of stool- and plasma-based methylation assays for clinical purposes.76 The use of molecular assays, such as the faecal methylated VIM assay, in the clinical care of patients is an area that is likely to undergo rapid advances in the near future.
Genetic markers and prognosis
Genomic instability and prognosis: MSI versus CIN
Meta-analyses across a diverse range of patients have firmly established that MSI colorectal cancer have a better prognosis and that CIN tumours have an unfavourable prognosis (table 4).18 25 The combined HR for MSI colorectal cancers for overall survival (OS) was estimated to be 0.65 (95% CI 0.59 to 0.71) with only one of 32 included studies reporting an HR >1.0.25 Conversely, the overall HR associated with CIN colorectal cancer was determined to be 1.45 (95% CI 1.27 to 1.45) based on 63 eligible studies and >10 000 patients.18 Despite the clear association of MSI and CIN with prognosis, these markers have not yet been adopted into routine clinical decision making. It is most likely that MSI testing will be adopted into clinical practice before CIN testing because of the availability of a reliable assay for assessing MSI status.
Prognostic biomarkers in colorectal cancer
18qLOH and prognosis
Patients with colorectal cancer with 18qLOH appear to have a worse prognosis compared with patients with tumours that do not carry 18qLOH. A meta-analysis of 17 independent studies that was limited by evidence of publication bias found an overall HR of 2.00 (95% CI 1.49 to 2.69) for 18qLOH across all patients, and an HR of 1.69 (95% CI 1.13 to 2.54) in the adjuvant setting.22 Candidate genes in the 18q region, including DCC and SMAD4, have been studied individually for prognostic roles, with inconsistent results.77 The independent prognostic contribution of 18q deletion in colorectal cancer has been called into question due to the tight association between 18qLOH and CIN, and the inverse association of 18qLOH and MSI.16 This assertion is supported by a recent study that found no difference in prognosis attributable to 18qLOH in a prospectively collected cohort of 555 non-MSI tumours stage I–IV.78 Thus, it is unclear at this time if 18qLOH represents an independent prognostic marker, or is merely a surrogate marker for CIN/MSS colorectal cancers.
Mediators of EGFR and prognosis
Several recent studies have assessed the prognostic significance of KRAS, BRAF and PIK3CA mutations in colorectal cancer.24 79–83 Mutant KRAS was not independently associated with differences in relapse-free survival or OS in stage II or III colorectal cancer, but mutant BRAF was prognostic for OS in this group of patients.24 79 In contrast, mutant KRAS and BRAF have been reported as markers of poor prognosis in advanced colorectal cancer. In the largest study that has addressed the prognostic role of KRAS mutations in advanced colorectal cancer to date, patients with mutant KRAS cancers had a worse OS (HR 1.40; 95% CI 1.20 to 1.65) but a similar progression-free survival (PFS) compared with patients with tumours bearing wild-type (WT) KRAS.81 The potential prognostic value of KRAS mutations is of particular interest in advanced colorectal cancers because the KRAS mutational status of tumours is now being routinely collected in this setting in order to assess for eligibility for treatment with cetuximab or panitumumab. At this time, the use of KRAS mutation status for prognosis in colorectal cancer is still premature but appears to have significant potential to be adopted into clinical use in the near future.
Predictive biomarkers
Although the treatment of colorectal cancer still primarily relies on the surgical resection of the primary tumour to achieve a cure, considerable progress in the medical treatment of stage III and IV colorectal cancer has occurred over the last 15 years. The adjuvant therapy of stage III colorectal cancer has become more effective as the standard regimen has advanced from 5-fluorouracil (5-FU) and leucovorin to 5-FU and oxaliplatin or irinotecan.84 Furthermore, the treatment of patients with stage IV colorectal cancer has expanded to include targeted treatments (cetuximab, panitumumab, bevacizumab; see table 2) in addition to 5-FU, oxaliplatin and irinotecan. With the identification of multiple effective agents for the treatment of colorectal cancer has come a need for predictive markers for selecting optimal treatment regimens for patients. This is particularly applicable to colorectal cancer because of the heterogeneity in response among colon cancers and because of the toxicity and cost of the medical treatments. The potential of genetic and epigenetic alterations to be effective predictive molecular markers has received considerable attention lately and has led to the use of some of these markers in the routine care of patients with colorectal cancer (table 5).
Colorectal cancer biomarkers as predictors for drug selection
The advent of cancer therapeutics that target specific molecules and pathways highlights the potential for underlying genetic and epigenetic lesions in colorectal cancer to guide personalised treatment decisions. A clear demonstration of the potential of mutant genes to direct treatment is that of mutant KRAS and treatment with cetuximab. Only ∼15% of patients with metastatic colorectal cancer respond to monoclonal antibody (mAb) therapies targeting the EGFR, which prompted intense research into resistance mechanisms that could be secondary to alterations in the EGFR gene and/or mutations in downstream effectors. These studies have produced one well-validated and exceedingly robust predictive marker (mutant KRAS) and several more promising biomarkers that require further validation (mutant BRAF, PIK3CA and PTEN).85 Research efforts are also focused on identifying molecular features of colorectal cancer that predict response to adjuvant chemotherapy with cytoxic agents: 5-FU, irinotecan and oxaliplatin.16 In this section we will discuss genetic features of colorectal cancer that have been evaluated for a role in guiding treatment selection. We have focused primarily on acquired tumour mutations as predictive markers, but it is important to note that inherited (germline) polymorphisms also influence the effects of chemotherapy on cancers and the risk for drug toxicity, particularly in the case of 5-FU and irinotecan (reviewed in Walther et al16).
Predictors of response to anti-EGFR mAb treatments
EGFR-targeted monoclonal antibodies cetuximab (Erbitux), and the fully humanised mAb panitumumab (Vectibix), have proven to be effective in patients with metastatic colorectal cancer both as single agents and in combination with traditional chemotherapy.86–88 However, while these treatments improve both PFS and OS, they are effective in only a minority of patients with metastatic colorectal cancer.85 These drugs are generally well tolerated, but are still associated with treatment-related morbidity, including skin rash, diarrhoea and nausea, and are also expensive. To better target anti-EGFR mAb treatment to patients most likely to benefit, KRAS mutation status and additional molecular markers of cetuximab and panitumumab resistance have been extensively evaluated.5
KRAS is an accurate predictive biomarker
Results of four large phase III randomised trials have established unequivocally that patients with metastatic colorectal cancer with KRAS mutations in codon 12 or 13 do not benefit from cetuximab or panitumumab treatment.4 89–91 Prior to the publication of these pivotal trials, the link between KRAS mutation status and anti-EGFR mAb response was already firmly supported by several smaller studies,92–94 but the data were not sufficient to warrant routine clinical testing. The recently published randomised trials have established the use of KRAS mutational analysis as a predictive marker for anti-EGFR mAb resistance in patients with metastatic colorectal cancer in most of the relevant clinical settings. These settings include the use of cetuximab or pantumimab in combination with conventional cytotoxic chemotherapy (eg, 5-FU, FOLFOX or FOLFIRI) as first-line treatment of metastatic disease,90 95 96 and as monotherapy in relapsed/refractory patients.4 89 91
A second relevant question related to anti-EGFR mAb treatment is whether mutant KRAS predicts an adverse outcome in the setting of these treatments. The reported HRs were almost exactly 1.0 in a total of 348 KRAS-mutant chemotherapy-resistant or refractory cancers treated with either panitumumab89 or cetuximab4 as monotherapy, confirming lack of benefit, but also suggesting no harm from anti-EGFR mAb treatment related to PFS or OS in this population. In contrast, the reported HRs were usually >1.0 in studies of cetuximab or panitumumab as first-line treatment in combination with FOLFOX4 (fluorouracil, leucovorin and oxaliplatin) or FOLFIRI (fluorouracil, leucovorin and irinotecan) chemotherapy.85 The results of the OPUS trial (Oxaliplatin and Cetuximab in First-Line Treatment of Metastatic Colorectal Cancer) in particular suggest it may be harmful to add anti-EGFR mAb treatments to 5-FU, leukovorin and oxaliplatin in patients with KRAS-mutant metastatic colorectal cancer.90
Based on the evidence from large trials, European and US practice guidelines either recommend or require KRAS mutational analysis on colorectal cancer tumour tissue prior to the initiation of cetuximab or panitumumab treatment.1–3 The European health authority confines use of panitumumab monotherapy, and cetuximab as mono- or combination therapy, to patients with metastatic colorectal cancer who are found to carry non-mutated WT KRAS in the primary tumours.1 The American Society for Clinical Oncology recently published a provisional opinion stating that ‘All patients with metastatic colorectal carcinoma who are candidates for anti-EGFR antibody therapy should have their tumors tested for KRAS [codon 12 and 13] mutations…and [KRAS-mutant] patients should not receive anti-EGFR antibody therapy’.3 Similarly, the National Comprehensive Cancer Network (NCCN) guidelines require evidence of WT KRAS prior to cetuximab or panitumumab treatment in all metastatic colorectal cancer settings.2
Despite the nearly perfect negative predictive value of mutant KRAS, it is still only a minority (∼30%) of KRAS codon 12/13 WT patients who respond to anti-EGFR mAb treatment.85 This has led to research into additional biomarkers that might predict lack of benefit in those individuals with tumours that have WT KRAS. There is evidence that rare KRAS mutations in codons 61 or 146 (∼2% of colorectal cancer) behave similarly to codon 12/13 mutations,97 but incorporating these mutations into routine clinical practice will require analysis of a larger group of patients. Other promising markers of anti-EGFR mAb resistance are BRAF V600E mutations, PIK3CA mutations and loss of PTEN protein expression.5
BRAF: another predictor of the anti-EGFR mAb response?
The biological rationale for BRAF V600E mutations as an additional biomarker of anti-EGFR mAb resistance is strong: (1) BRAF is the immediate downstream effector of KRAS in the Ras/Raf/MAPK signalling pathway (figure 3); and (2) BRAF V600E activating mutations are 100% mutually exclusive of KRAS mutations in colorectal cancer, implying that activation of either protein is sufficient for colon tumorigenesis. Existing limited data support BRAF V600E mutations as a negative predictor of response to anti-EGFR mAb treatment, leading to the evolving use of BRAF mutation testing in KRAS-WT patients prior to treatment as a means to stratify patients further into responders and non-responders. A retrospective analysis showed that 0/11 tumours with mutant BRAF responded to cetuximab or panitumumab compared with 22/68 (32%) of BRAF-WT/KRAS-WT patients.98 Similar results were observed for patients treated with cetuximab plus irinotecan. None of the patients with tumours with mutant BRAF (n=13) responded compared with 24/74 (32%) patients with tumours with BRAF-WT/KRAS-WT.97 These finding were supported by work presented at the 2009 American Association of Cancer Research and American Society of Clinical Oncology annual meetings,85 although not all studies have found as robust a relationship between BRAF V600E mutation status and anti-EGFR antibody response.82 99 BRAF mutations also appear to be associated with worse prognosis independent of treatment, which can confound the assessment of its role as a predictive marker for response to EGFR-directed treatments.82 99 Despite the currently limited data, and lack of complete consensus, it is likely that BRAF mutation status has a role in anti-EGFR mAb treatment decisions and soon will be adopted into the planning for treatment with cetuximab and panitumumab.
PI3K pathway activation and anti-EGFR mAb resistance
Molecular lesions in the PI3K pathway, which in colorectal cancer are primarily mutations in PIK3CA and loss of PTEN protein expression, have been proposed as additional anti-EGFR mAb resistance markers because the PI3K pathway is also stimulated by EGFR.85 However, the relationship of oncogenic alterations in PI3K signalling and cetuximab or panitumumab response is much less clear than that of KRAS and BRAF mutations. In several small studies published to date, PIK3CA mutations or PTEN loss have been associated with lack of response to cetuximab.64 100–102 Both PIK3CA mutations and PTEN loss may co-exist with KRAS or BRAF mutations, which weakens the biological rationale of the activation of this pathway as an absolute predictor of anti-EGFR mAb therapeutic response. Nonetheless, the balance of evidence points towards a probable predictive role for molecular events that activate the PI3K pathway for being negative predictive markers for EGFR monoclonal antibody-based treatment. In fact, there are modest data demonstrating that when PIK3CA mutations and PTEN loss of expression are combined with KRAS and BRAF mutational analysis, up to 70% of patients unlikely to respond to cetuximab or panitumumab may be identified.85 102 This observation has led to the idea that colon cancer may be able to be classified like breast cancers (eg, triple-negative breast cancers), and these cancers have been termed ‘quadruple-negative’ for patients who do not have alterations in any of these four biomarkers.85 102 However, at this time, further studies are needed to determine if mutant PIK3CA or PTEN loss should be incorporated into clinical practice.
EGFR mutations and amplification
The most obvious candidate biomarker for resistance to antibodies which target EGFR is the EGFR gene itself. Early studies that focused on EGFR overexpression assessed by immunohistochemistry did not show a consistent relationship with treatment response, in part because of lack of standardisation of the assay, which were based on either immunostaining, fluorescent in situ hybridisation (FISH) or quantitative RT–PCR, and interobserver variability inherent in the technique.103 EGFR gene amplification is more promising for being a predictive biomarker, but has also been fraught with technical challenges that limit the interpretation of existing data, such as dilution of tumour DNA with WT DNA in PCR-based assays, and lack of consistent tissue processing and scoring systems in FISH assays.5 Activating mutations in the EGFR catalytic domain are seen frequently in lung cancer and are associated with sensitivity to anti-EGFR tyrosine kinase inhibitors, but these mutations are quite rare in colorectal cancer.5 Thus, EGFR does not appear likely to be a clinically useful predictive marker for anti-EGFR monoclonal antibody therapy. Furthermore, although preliminary studies have shown that the EGFR ligands amphiregulin and epiregulin are overexpressed in colorectal cancer and may predict response to cetuximab, lack of standardisation of the assays and studies that reproducibly demonstrate the same effect have prevented amphiregulin and epiregulin expression levels from being used as clinical biomarkers for directing treatment with EGFR mAbs.58
Predictive molecular markers for response to 5-FU, irinotecan and oxaliplatin
Currently, the tumour biomarkers that demonstrate the greatest promise for guiding adjuvant chemotherapy with conventional drugs in patients with colorectal cancer include MSI and 18qLOH.
MSI
5-FU-based regimens have been shown to be ineffective for, or even detrimental to patients with MSI tumours.28 104 Evidence that a functioning MMR system is required for the cytotoxic effect of fluorouracil provides a plausible biological rationale for 5-FU resistance in MSI tumours.17 27 However, the finding of 5-FU resistance in MSI colorectal cancer is not uniform, and may vary with tumour stage.105 106 An ongoing phase III randomised trial of patients with completely resected stage II colorectal cancer (NCT00217737) will prospectively assess the role of MSI in predicting response to adjuvant chemotherapy in localised cancers.16
MSI tumours appear to be more responsive to irinotecan-based adjuvant chemotherapy.26 Recently published results from a large randomised trial of stage III colorectal cancer demonstrated improved outcomes (both PFS and OS) in MSI patients treated with an irinotecan-containing regimen that included 5-FU compared with 5-FU/luekovorin alone.107 In light of the prior results of the CALGB 98303 study showing no benefit of adding irinotecan to 5-FU as adjuvant therapy in unselected patients with stage III colorectal cancer, the finding that MSI is a predictive biomarker for irinotecan suggests that MSI could be useful for adjusting adjuvant therapy for patients with colorectal cancer.108 Replication of these results in independent studies is required to validate MSI status as an inclusion criterion for irinotecan-based adjuvant chemotherapy. Currently, neither the European Group on Tumour Markers nor the American Society of Clinical Oncology have recommendations on the use of MSI for guiding treatments in patients with stage II or stage III colorectal cancer.
An important issue to consider with regards to MSI is that the majority of colorectal cancers that have MSI are sporadic colorectal cancers that have inactivated the MLH1 gene through aberrant promoter methylation. The majority of these sporadic MSI tumours can be classified as CIMP cancers as well. It is not known whether the associations seen between 5-FU and irinotecan effects in sporadic MSI tumours also apply to MSI tumours that arise in the setting of Lynch syndrome.
Loss of 18q
Loss of 18q has been associated with an adverse response to 5-FU-based adjuvant chemotherapy.52 109 There is some evidence that this effect is due to loss of the SMAD4 gene located in the 18q21 deleted region, although this remains to be determined with more definitive studies.51 52 A number of ongoing clinical trials are assessing the predictive value of 18qLOH and MSI status for the treatment of colon cancer. These include an ECOG trial of patients with stage II colorectal cancer being treated with 5-FU, oxaliplatin and bevacizumab (NCT00217737), a trial in patients being treated with olaparib for metastatic disease (NCT00912743), as well as a retrospective analysis assessing MSI and 18qLOH in patients with colorectal cancer (stage II or III) treated with 5-FU or 5-FU and irinotecan (CLB-9581 or CLB-89803).
Topoisomerase 1 (Topo1)
In a large randomised trial that compared 5-FU alone with 5-FU + irinotecan and 5-FU + oxaliplatin in advanced colorectal cancer, higher expression of Topo1 measured by immunohistochemistry was significantly correlated with responsiveness to irinotecan.110 Conversely, cancers with low Topo1 expression (602/1269; 47%) did not appear to benefit from the addition of irinotecan (HR 0.98; 95% CI 0.78 to 1.22). Irinotecan is a Topo1 inhibitor; thus the level of Topo1 expression has a clear biological rationale as a biomarker for predicting irinotecan response. Replication of these initial results in multiple independent studies is required before Topo1 should be considered for use as a predictive marker.
Polymorphisms and their role as molecular markers for colorectal cancer
We emphasise again that germline polymorphisms that alter pharmacokinetics and pharmacodynamics of adjuvant chemotherapy are also potential biomarkers for guiding treatment selection. For example, alterations in thymidylate synthetase and dehydropyrimidine dehydrogenase have been extensively studied in relation to 5-FU response, and look promising. However, very few of these polymorphisms have been thoroughly validated and so the majority are not ready to be used clinically.111 112 One exception to this generalisation is a homozygous polymorphism that reduces the activity of UDP-glucuronosyltransferase (UGT1A1, an enzyme that detoxifies irinotecan), which is associated with a dose-related increased incidence of irinotecan toxicity.113 114 This has led to a commercial UGT1A1 genotyping test that was approved by the Food and Drug Administration in 2005 to help guide irinotecan dosing.
Conclusions and future directions
More than three decades of investigations into the molecular mechanisms of colorectal cancer carcinogenesis is finally culminating in biomarkers that are sufficiently validated for routine clinical use. KRAS-mutational analysis to guide anti-EGFR treatment stands as one of the first successes in the era of personalised medicine. MSI and BRAF mutations already have a clear role in triaging molecular genetic testing in Lynch syndrome, and these markers are poised to take on a much greater role in prognostication and prediction of therapeutic responses for sporadic colorectal cancers.
The use of assays for mutant KRAS, mutant BRAF and MSI demonstrates how the molecular testing of colorectal cancer tissue can reduce medical costs and improve patient outcomes by targeting therapies to the appropriate patient population. Thus, it is anticipated that the use of molecular genetic markers in clinical decision making is likely to expand as more markers are identified and validated. For example, studies are in progress assessing the efficacy of the multikinase/BRAF inhibitor sorafinib, and specific inhibitors of PI3K signalling in the treatment of colorectal cancer.5 There is evidence that sorafinib restores sensitivity to anti-EGFR mAb treatment in BRAF-mutant cell lines, which has prompted an ongoing phase II National Cancer Institute-sponsored clinical trial of sorafinib plus cetuximab in patients with metastatic colorectal cancer (NCT00343772).98 If these initial findings are validated, the indications for mutational analysis of BRAF and KRAS would expand. Furthermore, patients with colorectal cancer with tumours carrying mutant BRAF might also benefit from newer selective BRAF inhibitors such as PLX-4032 combined with anti-EGFR mAb treatment. PIK3CA mutations or PTEN loss are likely to become clinically relevant for the treatment of patients with colorectal cancer as specific PI3K pathway inhibitors (such as XL147, BGT226, GDC0941, XL765 and NVP-BEZ325) move into phase II clinical trials.115 The expanding repertoire of drugs designed to inhibit specific oncogenes and oncogenic signalling pathways again highlights that molecular mechanisms of colorectal cancer will increasingly play a role in the clinical care of patients with colorectal cancer. The use of molecular markers for risk stratification and early detection of colorectal cancer is also showing promise, and will be part of the era of molecular medicine that is rapidly emerging.
Acknowledgments
We thank Jonathan Tait for his help with reviewing the manuscript.
References
Footnotes
Funding This work was supported by a Burroughs Welcome Fund Clinical Scientist in Translational Research Award, NCI RO1CA115513, and P30 CA015704 (to WMG).
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.