Article Text

Abstract
Insulin resistance and diabetes are inextricably linked to chronic hepatitis C. Our understanding of this relationship continues to improve. This review focuses on the molecular mechanisms relating insulin resistance to hepatitis C with a subsequent overview of the consequences of hepatitis C-associated insulin resistance and diabetes, as well as perspectives for future management.
- Diabetes mellitus
- hepatitis
- C
Statistics from Altmetric.com
Introduction
Hepatitis C remains a major health concern throughout the world. As the population exposed to hepatitis C ages, the morbidity related to this disease is also increasing. Insulin resistance (IR) and diabetes are becoming more prevalent as a result of rising obesity trends and sedentary lifestyle. Our understanding of the relationship between these two disease processes continues to grow. This review focuses on the biological function of insulin and the subsequent development of IR, specifically as it relates to hepatitis C. Molecular mechanisms for direct hepatitis C involvement in insulin signalling defects are discussed. Subsequently, the consequences of IR in the setting of chronic viral infection are detailed, to include fibrosis progression and decreased response to pegylated interferon (peg-IFN_ and ribavirin (RBV) treatment. Improving IR via insulin-sensitising treatment and/or weight loss has been the goal of several recent clinical trials. The data surrounding these trials are summarised and perspectives for future management of hepatitis C and IR are provided.
Biological function of insulin
Insulin is the most potent physiological anabolic agent known, promoting the storage and synthesis of lipids, protein and carbohydrates, and inhibiting their breakdown and release into the circulation.1 Insulin is produced by the pancreatic β-cells, mainly in response to postprandial hyperglycaemia. During fasting, insulin falls and this, along with increasing levels of glucagon, epinephrine and other counter regulatory hormones, stimulates glucose production and lipolysis.
The tissues that remove glucose from the circulation and impact glucose use the most are skeletal muscle (SM), liver and adipose tissue (figure 1). The liver plays a central role in the regulation of whole-body glucose, fatty acid and amino acid metabolism. It is the main source of endogenous glucose production and amino acid metabolism; it is a a major site of fatty acid disposal (esterification and oxidation); and it is the primary site of insulin degradation. SM plays a crucial role in maintaining systemic glucose metabolism, accounting for 85% of whole-body insulin-stimulated glucose uptake.2 Insulin is also a critical regulator of most aspects of adipocyte biology.3 Insulin promotes lipogenesis through enhanced glucose transport, lipoprotein-derived fatty acid uptake, and fatty acid and triglyceride synthesis via transcriptional regulation, as well as by inhibiting lipolysis by repressing genes involved in fatty acid oxidation, which result in increased adipocyte triglyceride (TG) stores. The predominant transcription factors that mediate these changes include sterol regulatory element-binding protein-1c and adipocyte determination and differentiation-dependent factor 1.3

Summary of the biological function of insulin. Insulin is produced by the pancreatic β-cells, mainly in response to postprandial hyperglycaemia. The tissues that remove glucose from the circulation and impact glucose use the most are skeletal muscle, liver and adipose tissue. Insulin promotes lipogenesis and inhibit lipolysis.
Insulin receptor/signalling and IR
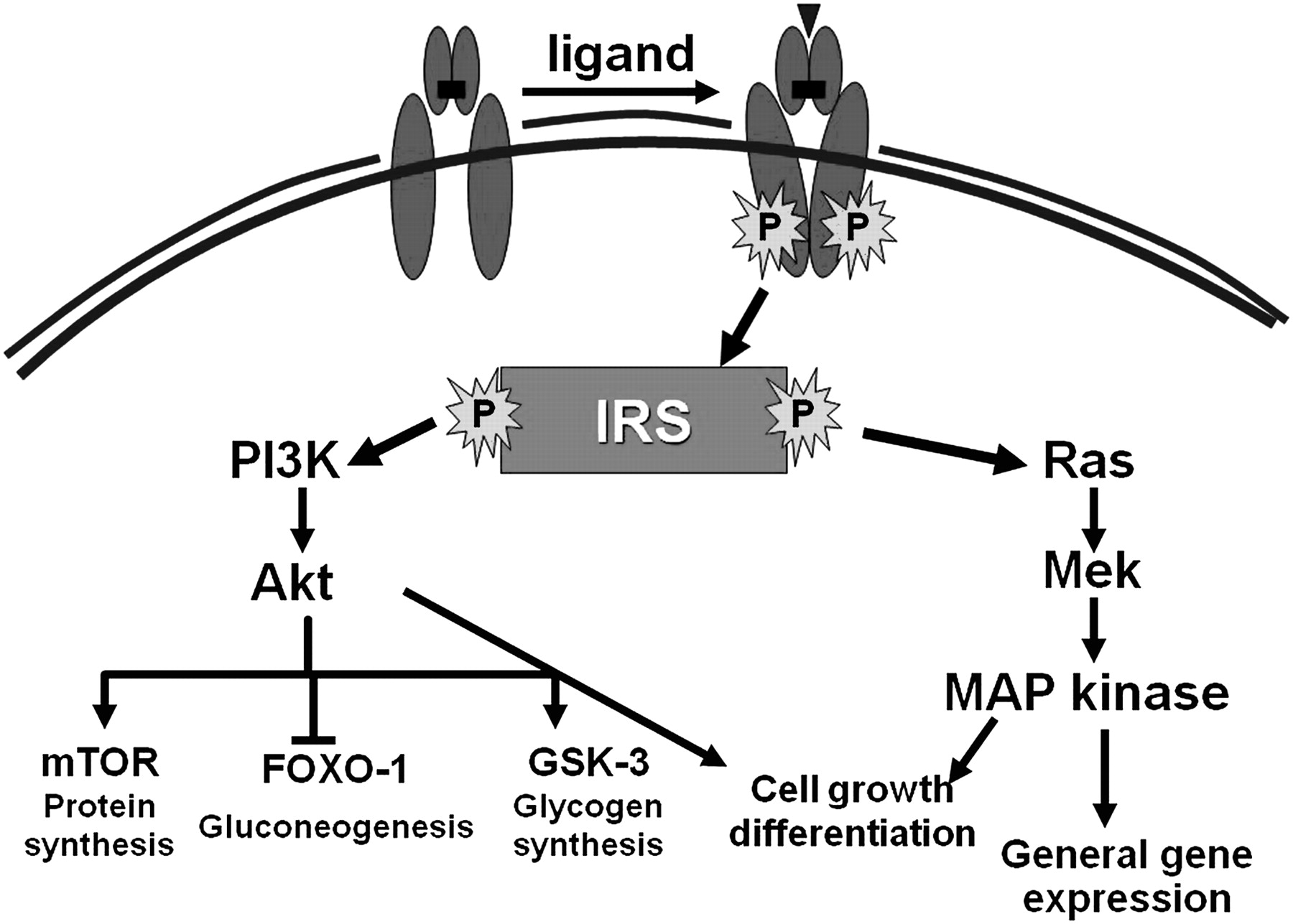
The insulin receptor is a heterotetrameric bifunctional complex, consisting of two extracellular α subunits that bind insulin and two transmembrane β subunits with tyrosine kinase activity. Insulin binding to the α subunits activates the intrinsic kinase activity located in the β subunits and subsequently initiates a cascade of phosphorylation events that leads to different biological functions. Unlike other receptor tyrosine kinases, most functions of the insulin receptor require accessory molecules known as insulin receptor substrates (IRSs) (1–4) to engage multiple downstream signalling pathways.4 5 Insulin binding results in autophosphorylation of the receptor and tyrosine phosphorylation of intracellular IRS proteins, mainly IRS-1 and IRS-2. These actions are manifested via insulin's action on a complex network of intracellular pathways in hepatocytes, adipocytes and muscle cells upon binding to its cellular receptor. Two major cellular signalling pathways, phosphoinositide-3 kinase (PI3K)/Akt and the Ras/mitogen-activated protein kinase (MAPK) pathways, can be activated. Mammalian target of rapamycin (mTOR) is another signalling pathway that is present in at least two different complexes. Activation of the mTOR branch downstream of the PI3K/Akt pathway has emerged as the critical event in rendering IRS-1 and IRS-2 unresponsive to insulin/insulin-like growth factor-I (IGF-I), and in cell growth and proliferation.6 7
These different cascades regulate diverse cellular processes, such as gene expression, protein synthesis and vesicle trafficking, which result in the regulation of glucose, lipid and protein metabolism, cell growth and differentiation (figure 2).1 8 One of the main results of these processes, is the final translocation of glucose transporter 4 (GLUT-4) from its intracellular pool to the cell membrane, facilitating glucose transport along the concentration gradient into the cytoplasm.1
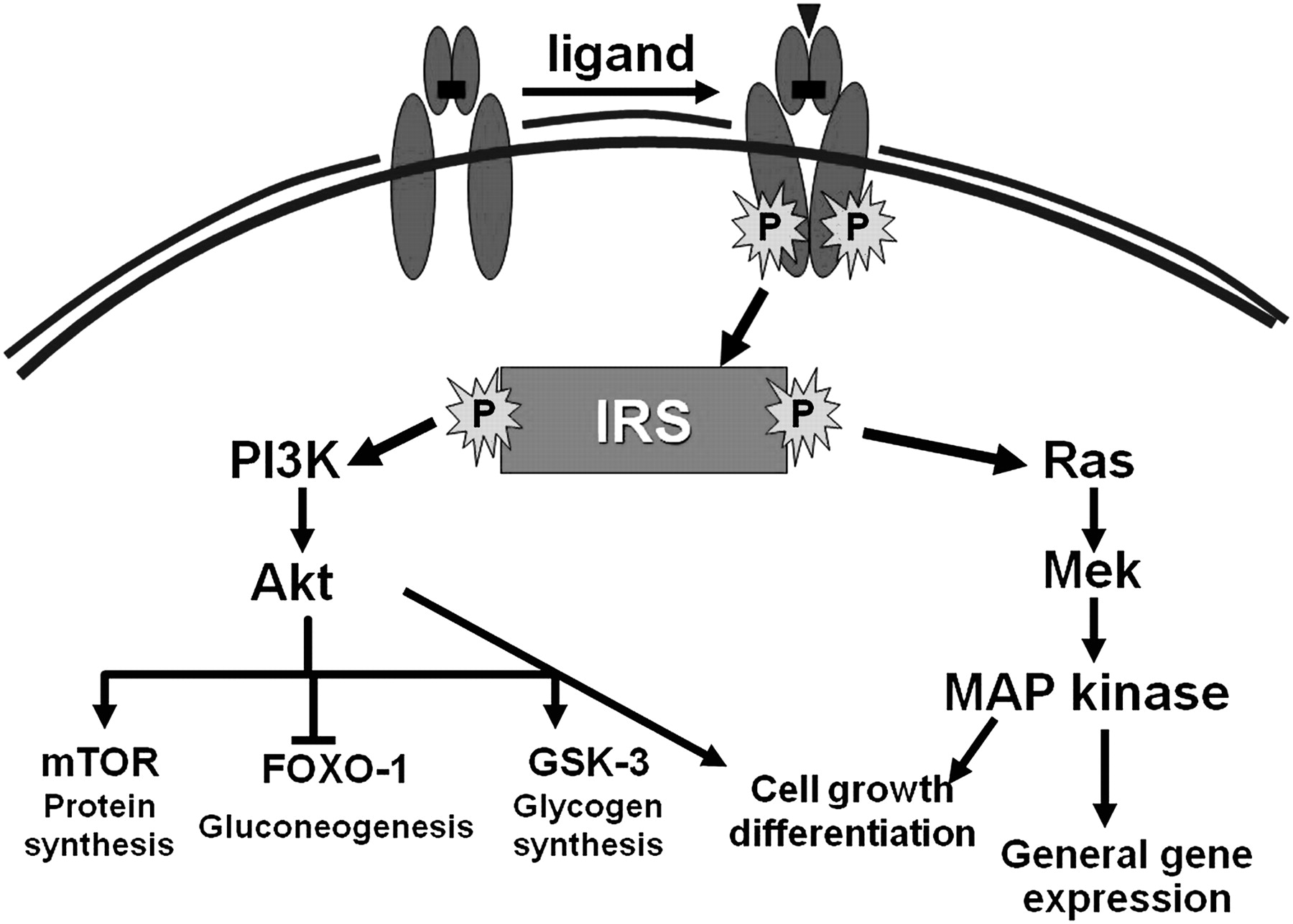
Insulin signalling pathways: insulin binds to its tyrosine kinase receptor, which requires accessory molecules known as insulin receptor substrates (IRSs) (1–4) to engage multiple downstream signalling pathways. These actions are manifested via insulin's action on a complex network of intracellular pathways. Two major cellular signalling pathways, phosphoinositide-3 kinase (PI3K)/Akt and the Ras/mitogen-activated protein kinase (MAPK) pathways, can be activated. Mammalian target of rapamycin (mTOR) is another signalling pathway that is present in at least two different complexes (see text for further explanation).
Insulin not only specifically activates its receptor, but it can also transactivate the IGF-I receptor, which is similar to the insulin receptor, a member of the receptor tyrosine kinase family of growth factor receptors.9 When insulin levels increase (as in the postprandial surge in insulin-resistant subjects or after insulin injection), insulin binds and activates the related IGF-I receptor which has a more potent mitogenic and transforming activity. Moreover, insulin decreases IGF-I-binding proteins (IGF-BP1). This results in increased free IGF-I, the biologically active form of the growth factor, the mechanism of which has been implicated in the pathogenesis of several malignancies.10 11
Insulin resistance
Insulin-mediated glucose disposal rates vary in the population by over sixfold.12 Some of this variation is because of adiposity and fitness, and some is the result of genetic origin. IR occurs when there is a decrease in the responsiveness of a target cell or a whole organism to the insulin concentration to which it is exposed, so that higher insulin concentrations are needed to achieve normal glucose metabolism.13 As blood glucose levels rise, pancreatic β-cells are stimulated to produce more insulin, leading to hyperinsulinaemia. At steady state, basal hyperinsulinaemia generates and sustains IR, irrespective of where the pathology started. Hyperinsulinaemia, IR and impairment of glucose-stimulated insulin release are intertwined biologically (figure 3). A single process could generate all three simultaneously. IR plays a fundamental role in the pathogenesis of type 2 diabetes mellitus (T2DM).

Venn diagram for dysregulated glucose metabolism. Hyperinsulinaemia, insulin resistance and impairment of glucose-stimulated insulin release are intertwined biologically. A single process could generate all three simultaneously.
Several mechanisms are involved in the pathogenesis of IR. Prereceptor, receptor and postreceptor defects have been proposed as possible mechanisms—that is, defects in insulin binding, IRS proteins, intracellular signalling or GLUT-4. From a pathophysiological point of view, IR appears to be the end result of a complex interaction between genetic predisposition and environmental factors.
Genetics
IR tends to cluster in families. The effect of genetics on insulin sensitivity as assessed by the minimal model technique is ∼30–40%.14 Candidate genes of interest that affect both liver and fat metabolism include several genes that regulate insulin action at the target organ level. Categories include genes regulating insulin receptor function (PC-1),15 intracellular insulin signalling (IRSs)16 and nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ). PC-1 is a class II transmembrane glycoprotein that inhibits tyrosine kinase activity. The K121Q polymorphism of the PC-1 gene has been correlated with insulin sensitivity independent of the degree of obesity.15
IRS-1-associated PI3K activity may be impaired by Gly972Arg substitution in the IRS-1 gene.17 A study showed that as compared with weight-matched controls, carriers of the Gly972Arg substitution were more likely to be insulin resistant.16
Though rare, mutations in the gene for PPARγ have yielded significant information. Loss-of-function mutations result in lipodystrophy, and gain-of-function mutations result in increased body fat mass.18 In humans, a Pro12Ala substitution (substitution of proline by alanine) has been detected in the PPARγ gene. This reduces the activity of PPARγ by 20–30%.19 20 Additionally, epigenic regulation of insulin signalling pathways is just beginning to be understood and may impact on the development of IR.
In brief, neither genetic factors nor environmental influence (visceral adiposity, high-fat diet) alone can explain the occurrence of IR. Complex synergistic interaction of both mechanisms results in impaired insulin signalling.
Sites of IR
IR is typically manifested as both decreased insulin-mediated glucose uptake at the level of adipose and SM tissue (peripheral IR), and as an impaired suppression of hepatic glucose output (hepatic IR). Although IR may develop simultaneously in the liver and in the periphery (SM and adipose tissue), the degree of IR can be different. In fact, IRS-1 seems to have its major role in SM. The most likely mechanism of IR within the muscle cell is specific alterations in the insulin signal transduction pathway.21
In adipose tissue, IRS-1 and IRS-2 play different roles; IRS-1 promotes brown adipocyte differentiation and IRS-2 is primarily involved with white adipose tissue lipolysis.22 In hepatocytes, the IRS-1 and IRS-2 proteins function in a complementary fashion. Both are involved in the activation of PI3K, but IRS-1 has an important function in regulating gluconeogenesis, while IRS-2 is more closely involved in lipid metabolism.23
HCV and insulin resistance
Chronic hetatitis C (CHC) can be considered not only as viral disease but also as a special type of metabolic disease. CHC interacts with lipid metabolism leading to steatosis, impairs glucose metabolism leading to IR and T2DM, and is associated with an increased risk of carotid atherosclerosis.24
The strong link between hepatitis C virus (HCV) infection and IR and T2DM was first reported by Allison et al25 who observed that diabetes was significantly more prevalent in those with hepatitis C-related cirrhosis than those with cirrhosis resulting from conditions other than CHC. Others comparing the prevalence of T2DM in a population of patients with CHC with that of a comparator group using a cross-sectional design have confirmed this association.26–33 Conversely, the prevalence of HCV infection in patients with diabetes is far higher than in the general population, ranging from 5% to 12%.34–43 The third National Health and Nutrition Examination Survey, which included 9841 subjects aged ≥20 years, showed that subjects with HCV are at threefold higher risk of developing T2DM.44 A recent large meta-analysis has re-affirmed this association.45
Further evidence for this association between HCV and IR and T2DM comes from a long-term longitudinal follow-up study assessing the incidence of diabetes in a large cohort of US subjects. Among those at high risk for T2DM, patients with CHC were 11 times more likely to develop T2DM than those without HCV infection (OR 11.58, 95% CI 1.39 to 96.6).46 T2DM was also a more frequent complication in liver and kidney transplantation among HCV+ patients compared with HCV– patients.47–54 In a recent meta-analysis of 10 studies, the pooled RR for postkidney transplantation T2DM was 2.73 (95% CI 1.94 to 3.83).55
The association between HCV infection and glucose abnormalities even at the prediabetes stages such as impaired glucose tolerance (IGT) or IR is also found.56 57 In 2003, Hui et al compared fasting serum insulin, C-peptide and HOMA (homeostatic model assessment)-IR levels between 121 patients with CHC without relevant hepatic fibrosis and 137 healthy volunteers matched by sex, body mass index (BMI) and waist-to-hip ratio. All three parameters were significantly higher in patients with CHC.56 This finding was confirmed in a recent study of 600 consecutive patients (500 with CHC and 100 controls with chronic hepatitis B) where the prevalence of T2DM was 7.6%. Among the patients with CHC without diabetes (n=462), IR was present in 32.4%. IR was less frequent in chronic hepatitis B than in matched CHC cases (5% vs 35%, respectively, p<0.001).58 Additionally, IR was associated with genotypes 1 and 4 and high serum HCV RNA levels.58 A correlation between HCV RNA levels and HOMA score has been reported by others as well.59 60
Lastly, data about amelioration of the HOMA score and decreased incidence of T2DM after completion of treatment in responder patients provide strong evidence for a causal relationship between HCV and glucose abnormalities. A study of 89 Japanese patients found that eradication of HCV led to improved HOMA scores and intrahepatic expression of IRS-1 and IRS-2.61 Similar results in HOMA scores were reported in a separate cohort of 181 genotype 4 patients.62 A longitudinal cohort study from Spain has assessed the incidence of glucose metabolism derangements after sustained virological response (SVR). Romero-Gómez et al63 evaluated the effect of SVR on the incidence of IGT and T2DM in 1059 patients with CHC treated with Peg-IFNα 2a and RBV. They concluded that SVR reduces by half the incidence of T2DM and/or IGT during a post-treatment follow-up of 27±17 months (range 9.3–67 months). Two more recent and lengthy cohort studies yielded contradictory data. Giordanino et al in a cohort of 202 patients with a longer follow-up (8.0 years, range 5–16 years) failed to show a benefit in patients obtaining an SVR, even after adjustment for several baseline risk factors of T2DM.64 In contrast, Arase et al, in a retrospective cohort study, followed 2842 HCV-positive patients for an average of 6.4 years and concluded that SVR causes a two-thirds reduction in the risk of T2DM development.65
IR is present in 30–70% of individuals with CHC.56–58 Its presence can occur early in the course of HCV infection, independent of BMI, viral load and the severity of liver disease. HCV seems to increase the risk of incident T2DM in predisposed individuals. Recent clinical data suggest that IR is genotype dependent (1 and 4), related to HCV viral load and is improved in patients with HCV clearance following antiviral treatment. Taken together, these results suggest a direct link between HCV infection and IR that is independent of BMI and visceral adiposity, and that HCV infection itself may promote IR.
Molecular mechanisms of IR in HCV
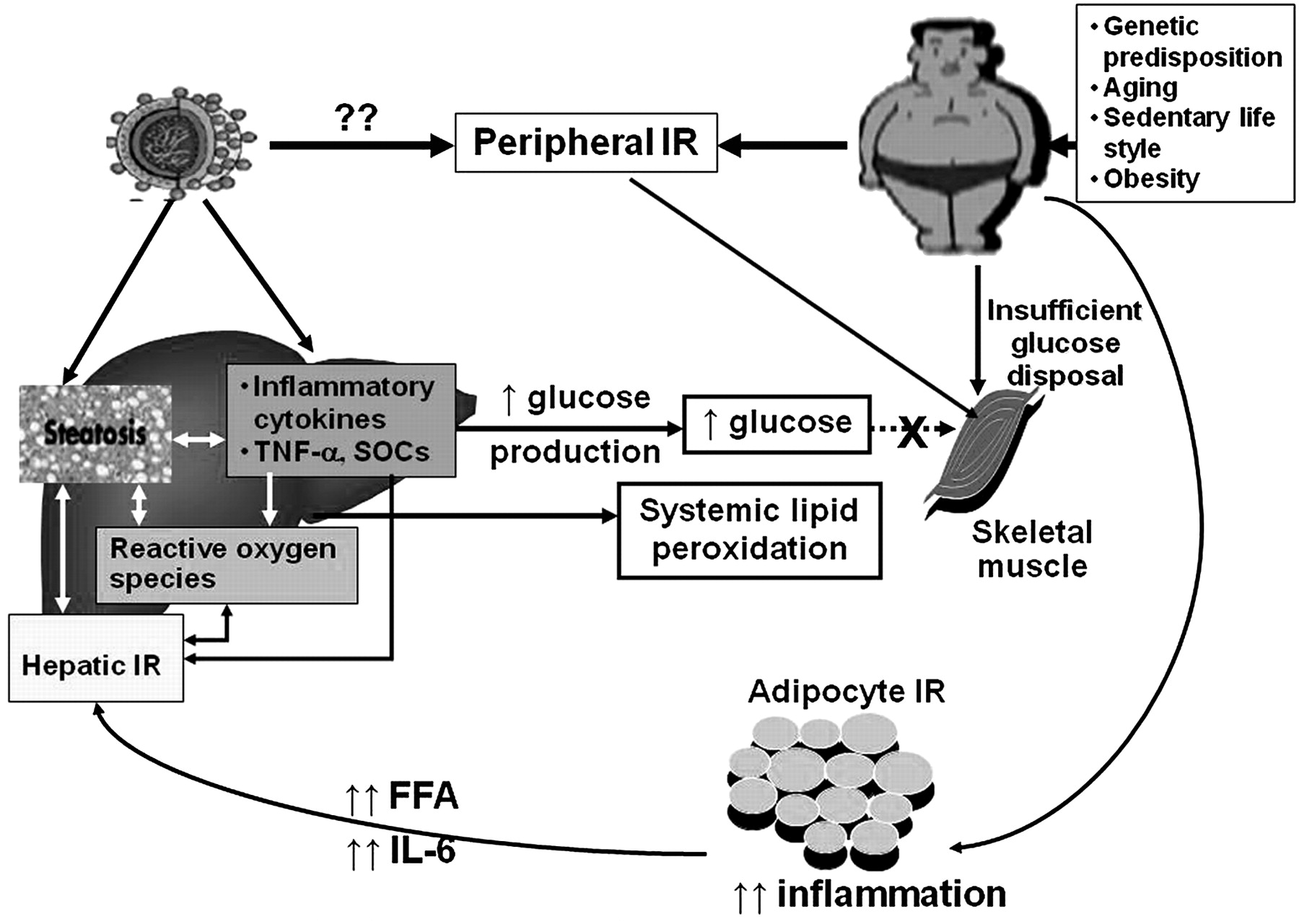
Development of IR and T2DM involves highly complex systemic mechanisms that have not yet been conclusively described. Numerous molecular pathways have been implicated. The interaction of various virus and host factors in inducing IR is the most accepted scenario (figure 4). We will discuss the potential molecular pathways by which HCV contributes to IR.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Molecular mechanisms of insulin resistance (IR) in hepatitis C virus (HCV) infection. The mechanisms are complex, with an interaction of various virus and host factors in inducing IR being the most accepted scenario. Host factors involve interplay between both environment and genetic predisposition, which interact with HCV infection in inducing IR. Hepatic IR in patients with chronic hepatis C is associated with steatosis and enhanced intrahepatic expression of inflammatory cytokines and reactive oxygen species with possible interference with insulin signalling. Steatosis promotes an increase in intrahepatic and systemic lipid oxidation and endogenous glucose production. In the peripheral tissues (muscle and adipose tissue), host factors and possibly HCV hinder glucose disposal by skeletal muscles by decreasing glucose oxidation. Both host- and virus-induced specific changes of adipocytokines are associated with an increase in both hepatic and peripheral insulin resistance (see text for further explanations). FFA, free fatty acids, SOCS, suppressor of cytokine signalling; TNF, tumour necrosis factor.
Proinflammatory cytokines and adipokines
Both adipocytes and hepatocytes lie in close proximity to immune cells, including Kupffer cells, macrophages, lymphocytes and dendritic cells, and are thus subject to the impact of immune status. This close interaction can be of importance when inflammatory states are present, such as in metabolic syndrome.66 Chronic inflammation is postulated to play a significant role in IR associated with HCV, due to increased levels of interleukin 1 (IL-1), transforming growth factor β (TGFβ), tumour necrosis factor α (TNFα) (and its soluble receptor molecules sTNFR1 and sTNFR2), IL-6, IL-8, leptin and resistin, and reduced levels of adiponectin.67
Expression of HCV core protein (genotype 1b) in transgenic mice induces hepatic IR.68 When fed a high-fat diet, these mice develop frank diabetes and hepatic steatosis that is associated with elevated circulating levels of TNFα. The IR is reversed by administering antibodies against TNFα, although the mechanism of this effect has not been clearly elucidated.68 Another study comparing proinflammatory cytokine expression in HCV+ liver tissues (chronic hepatitis, n=10) with HCV– normal liver tissues (n=6) found that intrahepatic expression of the cytokine IL-18 was significantly upregulated in CHC versus controls (p=0.02), with a concordant increase in IFNγ and TNFα expression (p=0.002 and 0.02, respectively).69 Additionally, there was an increase in intrahepatic macrophage numbers with persistent HCV infection, which is also consistent with the chronic inflammatory state seen in CHC.69
The role of TNFα-induced IR in CHC has been widely studied. TNFα has been shown to induce IR by impairing insulin signalling through serine phosphorylation of IRS-1 and IRS-2, thus downregulating (GLUT2/GLUT4) gene expression,70 preventing the uptake of glucose into hepatocytes and adipocytes and promoting a state of hyperinsulinaemia and hyperglycaemia.71
Although the hypothesis of chronic inflammation as an inducer of IR in CHC seems to be simple and acceptable, there are conflicting reports regarding the role of purely virus-induced inflammation in the development of IR in CHC. In an Italian study of 161 consecutive patients with CHC, serum TNFα levels were positively correlated with steatosis grades and HOMA-IR values, whereas serum levels of adiponectin were inversely correlated with steatosis grades, serum TNFα levels and HOMA-IR values.72 These results were independent of gender and HCV genotype. In contrast, a large Australian prospective study of 154 HCV-infected males without diabetes and 75 matched uninfected controls found higher serum levels of TNFα and IL-6 in HCV-infected patients than controls, but they did not correlate with IR. Serum levels of leptin and adiponectin were independently associated with IR (adiponectin inversely), but not with HCV infection itself. The authors concluded that virus-specific IR in CHC may be a cytokine-independent effect of the virus to modulate insulin sensitivity.73
Direct effects of HCV in modulating insulin signalling
Significant attention is presently being drawn towards the way in which HCV can induce IR directly, through specific viral effects.74 The HCV genome is composed of both structural (core, E1 and E2) and non-structural genes (NS2–NS5B), each of which has been implicated in IR development.
HCV core protein
Recent data suggest that HCV core protein reduces IRS-1 and IRS-2 protein levels and inhibits insulin signalling, although differing mechanisms have accounted for this suppression.75 76 It is not completely understood whether altered signalling results from changes in IRS expression, degradation or altered activity.68 75–79 At the molecular level, oxidative stress, dowregulation of PPAR, increasing levels of the molecule suppressor of cytokine signalling (SOCS) and proteasome activator 28γ (PA28γ), and activation of the mTOR pathway are postulated mechanisms, all of which may occur in a genotype-specific manner.79
During HCV replication, the core protein promotes an unfolded protein response that causes dysfunction of the endoplasmic reticulum (ER) and mitochondria by facilitating the uptake of calcium into the mitochondria and induces mitochondrial permeability transition.80 Following calcium accumulation, there is a stimulation of electron transport, which increases the production of reactive oxygen species (ROS).80 81 Interestingly, clearance of HCV infection has been shown to improve IR and restore the hepatic expression of IRS-1 and IRS-2.61 It is possible that the core HCV protein stimulates increased levels of SOCS-3, which leads to ubiquitination and proteasomal degradation of IRS-1 and IRS-2.75 Two clinical studies have also shown that the level of SOCS-382 and polymorphisms in the SOCS-3 gene were predictive of response to IFN treatment.83
Genotype-specific abnormalities in postreceptor insulin signalling that could help explain the clinical association of genotypes 1 and 4 with IR have not been clearly elucidated.58 It is unclear if it is based on the known differences in treatment response between these groups or due to difference in the interactions between viral proteins and host signalling pathways.
There is recent evidence that while HCV core protein from both genotypes 3a and 1b reduced IRS-1 protein levels and inhibited insulin signalling, differing mechanisms accounted for this suppression. Specifically, genotype 3a core protein appears to cause IRS-1 degradation via the downregulation of PPARγ and upregulation of the SOCS-7 protein.79 In contrast, genotype 1b core protein caused IRS-1 downregulation through a mechanism involving increased phosphorylation of IRS-1 at inhibitory serine residues (636/639), as well activation of the mTOR.79
PA28γ is an inducer of late proteasome activity that may play a role in HCV-induced IR.18 Recent work combining mice transgenic for the HCV core protein (HCVcpTg) with PA28γ (–/–) knockout mice has added to our understanding of IR induced by HCV by showing that the PA28γ-dependent pathway was required for HCV core protein-mediated suppression of IRS-1 tyrosine phosphorylation, suppression of IRS-2 expression and activation of the TNFα promoter.77 PA28γ has also been shown to play a critical role in the development of steatosis and HCC.84
Insulin regulates gene expression of key enzymes in glucose and lipid metabolism by modulating the activity of specific Forkhead box transcriptional regulators (FoxO1 and FoxA2) via the PI3K/Akt signalling pathway in the liver.85 FoxO1 and FoxA2 may have a novel role in HCV-induced IR. In a recent study, HCV core protein, either alone or together with other viral proteins from the HCV genome, impaired insulin-induced FoxO1 translocation from the nucleus to the cytoplasm and subsequently significantly reduced accumulation of FoxA2 in the nucleus.86
HCV NS5A
Nuclear factor-κB (NF-κB) and protein phosphatase 2A (PP2A) are two molecules that also may play a role in HCV-induced IR. HCV NS5A stimulates NF-κB-induced increase in the inflammatory cytokines TNFα, IL-6 and IL-8 by inducing mitochondrial ROS production and by binding to Toll-like receptor 4 (TLR-4) found on the plasma membranes of hepatocytes and B cells.87–89
PP2A is upregulated either directly by NS5A90 or due to increased ER stress.91 PP2A has been shown to mediate HCV-associated IR by dephosphorylating and thus inactivating Akt.92 Moreover PP2A has also been shown to inhibit IFN signalling, and this has been proposed as one of the potential links between IR and IFN resistance.93 NS3-induced ER and oxidative stress may also activate NF-κB and increase the risk of inflammation, IR and HCC in a similar way to NS5A.94
PPAR and HCV-induced IR
PPARs belong to the nuclear receptor superfamily and require heterodimerization with receptor X for retinoids (RXR) in order to function.95 96 The PPAR–RXR heterodimer, when bound to a ligand (including unsaturated fatty acids, eicosanoids, oxidised low-density lipoprotein and very low-density lipoprotein) changes conformation and binds to DNA at PPAR response elements, resulting in gene transcription.96 97
There are three isotypes in mammals designated PPARα (NR1C1), PPARδ (NR1C2) and PPARγ (NR1C3).98
PPARα/γ, together with their obligate partner RXR, are the three main nuclear receptors expressed in the liver and are involved in the control of lipid and glucose metabolism, inflammatory responses, and cellular differentiation and proliferation.
Expression of PPARα appears to be impaired with HCV infection.99 100 Expression of the PPARα gene in the liver was reduced by 86% compared with controls, and the expression of its target gene, CPT1A, was coordinately reduced by 85%. De Gottardi et al showed that PPARγ expression was significantly lower in genotype 3 compared with genotype 1 HCV infection. In this study, there was no significant relationship between PPAR mRNA levels and liver activity or fibrosis. In a follow-up study, treatment of genotype 3a core-expressing cells with the PPARγ agonist rosiglitazone improved insulin signalling.100 Adiponectin is another important cytokine that interacts with PPARα to regulate hepatic TG content.101 Accumulation of hepatic TG is associated with loss of adiponectin receptors in the liver and, together with reduction in circulating adiponectin, contributes to systemic IR and various other metabolic anomalies.102 As adiponectin is upregulated by PPARγ, it provides a connection between the two isotypes of PPAR and mechanisms for IR and steatosis in people with CHC. Future studies are required to define the role of adiponectin treatment clearly in humans.
Oxidative stress and IR
Oxidative stress has emerged as a key player in the development and the progression of many HCV-induced hepatic derangements, including IR and steatosis. HCV infection is characterised by increased markers of oxidative stress. Studies have indicated that HCV can directly induce oxidative stress intracellularly in hepatocytes.80 103–105 HCV core gene expression has been associated with increased ROS, decreased intracellular and/or mitochondrial glutathione content, and increased levels of oxidised thioredoxin and lipid peroxidation products.80 103–105 Contradictory data are available about the role of oxidative stress in HCV-induced IR.
A recent study found that in patients infected with HCV genotype non-3, BMI (p=0.031) and oxidative stress (measured as glutathione) (p=0.037) were independently associated with IR.106 Conversely, Vidali et al showed that in CHC genotype non-3, oxidative stress (measured as antibodies to malondialdehyde–albumin adducts) is primarily correlated with hepatic steatosis and not with IR. The authors concluded that in genotype non-3 infection oxidative stress and IR contribute to steatosis, which in turn exacerbates both IR and oxidative stress and accelerates the progression of fibrosis.107 It has been shown that oxidative stress serum markers tend to normalise in patients who achieve an SVR.108
What is the primary site of IR in CHC?
Recent trials have attempted to discriminate between the contribution of the HCV virus to ‘systemic’ and ‘hepatic’ IR and have shown that HCV infection is associated with both hepatic IR and peripheral (muscle) IR, but is predominantly peripheral.109 110 This observation is supported by decreased insulin-stimulated glucose disposal at high insulin dose clamp—that is, when endogenous glucose production is completely suppressed—whereas at low dose insulin no significant difference was noticed in insulin-stimulated hepatic glucose output between CHC subjects and controls.109 Free fatty acids tended to be higher in CHC versus controls basally, but was suppressed similarly to controls during low dose insulin. This suggests that IR is largely confined to SM and not adipose tissue.109 Although these data are derived from a highly selected group of patients with CHC with no features of the metabolic syndrome and no histological evidence of cirrhosis, the possibility of the presence of previous skeletal muscle IR independent of HCV infection cannot be completely excluded. Previous data from patients with T2DM indicate that the initiation of IR is in the periphery, with hepatic steatosis following and exacerbating the degree of IR.111 Supporting data are found in another trial that screened 400 young, healthy, lean subjects without diabetes and found that at least 12 (3%) of the screened subjects had IR.112 These young lean insulin-resistant subjects had significant IR in SM due to decreased muscle glycogen synthesis that predated hepatic IR.
The reported improvement in glucose tolerance following liver transplantation in HCV-positive patients with diabetes is not always associated with complete regression of IR.113 In fact, transplanted patients still maintained the reduced muscle glucose uptake and the decreased non-oxidative glucose disposal observed before transplantation, indicating persistence of IR in peripheral tissues, particularly in the SM.113
The possible mechanisms of HCV-induced IR in SM have not been fully elucidated. However, the potential mechanisms include viral-induced adipocytokine release or HCV viral proteins directly interfering with muscle insulin signalling or inflammatory pathways. Despite the fact that one study found no evidence of viral replication in SM,114 further studies are needed to clarify this.
Consequences of HCV-associated IR/diabetes
HCV-associated IR is involved in the development of various complications associated with HCV infection. Table 1 summarises the evidence for adverse outcomes associated with IR among patients with CHC.
Consequences of HCV-associated insulin resistance/diabetes
Hepatic fibrosis
IR is closely associated with progression of hepatic fibrosis in patients with HCV infection.56 58 115–123 Also, the presence of IR is strongly associated with more rapid progression of fibrosis after liver145 and kidney transplantation.146 Hyperinsulinaemia and hyperglycaemia may promote fibrosis through the stimulation of hepatic stellate cells, thereby increasing the production of connective tissue growth factor and the accumulation of extracellular matrix.147 Alternatively, IR-induced hepatic lipid accumulation may increase oxidative stress, resulting in progression of hepatic fibrosis.148 In these cases, IR rather than steatosis seems to predict the stage of fibrosis and its progression over time.115
Response to antiviral treatment
Increasing levels of IR are associated with reduced rates of rapid virological response (RVR)124–126 as well as SVR in patients with HCV genotype 1, 2, 3 and 4 infections treated with a combination of Peg-IFNα and RBV.127–132 However, the mechanisms of IR-induced IFN resistance are not completely understood.
Intracellular factors dysregulated by HCV and responsible for the insulin-resistant phenotype may have additional effects as they are also involved in regulating IFNα signalling. These factors include some members of the SOCS family75 79 82 83 149 and the PP2A.92 IR is known to increase hepatic lipid synthesis.150 Since the lipid droplet is an important organelle for HCV replication,151 accumulation of hepatic lipid droplets may increase HCV replication and result in poor responses to antiviral treatment.
Hepatocellular carcinoma (HCC)
IR has been recognised as an independent risk factor for the development of HCC.118 133–139 Potential pathogenic mechanisms include a direct mitogenic effect of insulin152 as well as oxidative stress and resultant steatosis that may also contribute to the development of HCC.153 154
Oesophageal varices (OVs)
IR is emerging as a risk factor for OVs in patient with cirrhosis with HCV infection.140 The pathogenic mechanism is not completely understood. IR may be associated with OVs via progression of liver fibrosis.115 Insulin modulates the endothelial synthesis of nitric oxide and endothelin155 to induce the production of TNFα and connective growth factor, and to stimulate hepatic stellate cells.147 Therefore, insulin could contribute to the pathogenesis of portal hypertension by interfering with both mechanical and dynamic mechanisms with collagen deposition, vasoconstriction and regulation of sinusoidal structure.140
Liver transplant outcome
Patients with T2DM or even IR have higher post-transplantation complication rates (either liver or non-liver related) than those without glucose metabolic disarrangement.141–145 Interestingly, the incidence of post-transplant diabetes is higher in patients with liver diseases due to CHC rather than other causes.47–49 Further data are needed to clarify the relationship between IR and post-transplant complications.
Perspectives for management
In view of the suboptimal response to the current antiviral treatments, it is imperative that thought is given to improving these potentially modifiable risk factors, especially IR and T2DM which are associated with adverse outcomes. Although increasing insulin sensitivity may be a rationale option in patients with CHC, especially those with metabolic syndrome, the ideal therapeutic modality for the prevention and management of IR and T2DM in the setting of HCV has not yet been established. Different approaches have been proposed which include both pharmacological and lifestyle interventions. However, this is rather empirical, as the mechanism by which IR leads to potential IFN resistance are not completely elucidated.
The use of insulin-sensitising agents to enhance the antiviral treatment response of Peg-IFN and RBV has been postulated to be of benefit. Potential treatments studied to date include the thiazolidinedione, pioglitazone (PIO)—a specific PPARγ agonist—and the biguanide, metformin, whose mechanism of action is specifically directed against the hepatic AMP-activated protein kinase.156
The primary data on the use of PIO are from a prospective, multicentre study aimed at investigating the efficacy and safety of a 15 mg daily dose added to once-weekly Peg-IFNα 2/RBV combination therapy in retreatment of patients with CHC who were previously non-responders to a Peg-IFNα/RBV combination. All patients had a baseline HOMA >2.157 The study was prematurely terminated as none of the first five patients enrolled in the trial had a sufficient virological response after 12 week. However, the authors surmise that their approach may have been inadequate in view of the suboptimal dose (15 mg four times a day).158
Emerging data from several recent studies using PIO in combination with Peg-IFNα/RBV have yielded conflicting results.159–162 Elgouhari et al studied PIO 30 mg/day given for 4 weeks as monotherapy and then added Peg-IFN and RBV to treatment-naive patients with CHC without diabetes. The authors showed that the triple regimen containing PIO increased the rate of virological response significantly after 4 weeks of treatment compared with Peg-IFN and RBV alone. Long-term data are keenly awaited.159 A randomised placebo-controlled study performed in the USA with PIO 30 mg/day plus Peg-IFN and RBV (ie, without preceding administration as monotherapy) clearly improved IR and steatosis, and increased the on-treatment virological response. However, SVR was not improved.160 A subsequent trial with PIO 30 mg/day plus Peg-IFN and RBV was conducted conclusively in patients infected with HCV genotype 4 and resulted in higher rates of RVR and SVR with improvement in all parameters of IR.161
In contrast to these findings, interim 12 week analysis of a large, randomised, double-blind, placebo-controlled study of CHC genotype 1 patients with IR using PIO monotherapy for 16 weeks (30 mg/day×8 weeks then 45 mg/day×8 weeks) prior to the 48 week antiviral treatment with Peg-IFN and RBV found no improvement in RVR or early virological response despite improvement in adiponectin and several glycaemic variables including plasma glucose and insulin levels, and HOMA score.162
Metformin has also been studied as potential adjuvant therapy for patients with IR, with mixed results. The TRIC-1 study,163 which involved CHC genotype 1 patients with IR who were treated with metformin plus standard of care (SOC) reduced IR significantly but afforded only a marginal, non-significant gain in the SVR rate, despite an increased RVR after 4 weeks of triple therapy. In a subset analysis, women who received metformin doubled the SVR rates compared with the placebo group (57% vs 28%, p=0.03).163
Ultimately, the use of insulin sensitisers in triple therapy raises many questions. Insulin sensitisers alone do not seem to affect viral load. While all studies to date utilising triple therapy with an insulin sensitiser and Peg-IFN and RBV have demonstrated improvement in IR, a variable virological response has been seen. This varying response suggests that there are probably many variables, including both host and viral factors, that alter responses to antiviral therapy even when insulin sensitivity is improved. Further study is needed to clarify the role of genotype, the degree of insulin sensitivity improvement, gender and genetics (such as the IL28β mutation) on viral kinetics. Although the relationships between early viral kinetics during antiviral treatment in patients with CHC and IR are not completely understood, recent data support that increasing levels of IR are associated with reduced rates of the RVR.124 126 Similar data were recently published by Nasta et al125 in HIV/HCV-co-infected patients. Moreover, in a recent study, hyperinsulinaemia reduced the 24 h virological response to Peg-IFN treatment in patients with CHC and IR.164
As an alternative to pharmacological improvement of IR, assessment of the effect of a dietary and/or lifestyle changes on IR in patients with CHC, with its impact on hindering the progression of liver fibrosis and enhancing response to antiviral treatment, is interesting and worthy of further evaluation. A 3 month study that encompassed body weight reduction and increased physical activity was associated with improvement histology and fasting insulin.165 166 Virologial response was not assessed.
In another small pilot trial, 15 patients were placed on a strict low-calorie diet for 3 months to achieve a 10% reduction in BMI before starting treatment, whereas 17 were on a free diet for the same period. All patients were offered standard combined antiviral therapy with Peg-IFN and RBV. The use of a low-calorie diet was associated with an improved virological response (60% vs 17.6%; p=0.035), although it is unclear if this represent and end of treatment response or SVR. Improvement in virological response was associated with a reduction in HOMA.167
Ultimately, there are no current clinical guidelines advocating the use of antidiabetic agents for patients with CHC and IR or diabetes mellitus. While the potential exists for improved outcomes to treatment among the various CHC genotypes, much is yet to be learned in reference to improving IR and its effect on virological response.
Major facts about insulin resistance in the setting of HCV
HCV is associated with insulin resistance and diabetes.25
The pathogenesis of insulin resistance represents a complex interplay between host and viral factors.
Insulin resistance is linked to fibrosis progression.56 58 115–132
Insulin resistance is associated with a decreased early and sustained response to current antiviral therapy.124–132
Summary
Insulin resistance in setting of HCV is still an evolving story.
Our understanding of the pathogenesis of HCV-induced insulin resistance and its deleterious effect are greatly improved.
Direct effects of HCV in modulating insulin signalling via core protein and NS5A are better understood.75 76 87–89
Insulin-sensitising agents including the thiazolidinediones and metformin have a variable response on viral kinetics and SVR.
Studies are ongoing to further address the utility of insulin sensitising agents in improving SVR or mitigating disease progression.159–163
Our improved understanding of HCV-induced insulin resistance may allow for development of better strategies to eradicate the virus or prevent disease progression in the future.
References
Footnotes
All authors contributed equally to this article.
The opinion of ascertains contained herein are the private views of the authors and are not to be construed as official or reflecting the view of the Department of the Army or the Department of Defense.
Competing interests SAH: Research support from Genentech, Merck and Rottapharm; Ad Hoc Advisory Board for Three Rivers Pharmaceuticals; Speaker's bureau for Bristol Myers Squibb.
Provenance and peer review Not commissioned; externally peer reviewed.