Article Text

Abstract
Background The gut microbiota has profound effects on host physiology but local host–microbial interactions in the gut are only poorly characterised and are likely to vary from the sparsely colonised duodenum to the densely colonised colon. Microorganisms are recognised by pattern recognition receptors such as Toll-like receptors, which signal through the adaptor molecule MyD88.
Methods To identify host responses induced by gut microbiota along the length of the gut and whether these required MyD88, transcriptional profiles of duodenum, jejunum, ileum and colon were compared from germ-free and conventionally raised wild-type and Myd88−/− mice. The gut microbial ecology was assessed by 454-based pyrosequencing and viruses were analysed by PCR.
Results The gut microbiota modulated the expression of a large set of genes in the small intestine and fewer genes in the colon but surprisingly few microbiota-regulated genes required MyD88 signalling. However, MyD88 was essential for microbiota-induced colonic expression of the antimicrobial genes Reg3β and Reg3γ in the epithelium, and Myd88 deficiency was associated with both a shift in bacterial diversity and a greater proportion of segmented filamentous bacteria in the small intestine. In addition, conventionally raised Myd88−/− mice had increased expression of antiviral genes in the colon, which correlated with norovirus infection in the colonic epithelium.
Conclusion This study provides a detailed description of tissue-specific host transcriptional responses to the normal gut microbiota along the length of the gut and demonstrates that the absence of MyD88 alters gut microbial ecology.
- Bacterial overgrowth
- cardiovascular disease
- gut microbiota
- Helicobacter pylori
- host response
- intestinal epithelium
- intestinal tract
- lipid metabolism
- mucosal immunity
- obesity
- Toll-like receptors
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Bacterial overgrowth
- cardiovascular disease
- gut microbiota
- Helicobacter pylori
- host response
- intestinal epithelium
- intestinal tract
- lipid metabolism
- mucosal immunity
- obesity
- Toll-like receptors
Significance of this study
What is already known about the subject?
The gut harbours a vast ensemble of bacteria, the gut microbiota, encoding 150-fold more genes than our own genome.
The gut microbiota is altered in inflammatory bowel disease and obesity.
Germ-free mice are protected against inflammatory bowel disease and diet-induced obesity.
Toll-like receptors recognise microorganisms and signal via MyD88.
What are the new findings?
Provides an extensive survey of host responses to the normal gut microbiota along the length of the gut.
Analysis of the gut microbial ecology along the length of the gut.
Demonstrates that Myd88-deficient mice harbour norovirus in the colonic epithelium.
How might it impact on clinical practice in the foreseeable future?
Understanding the fundamental factors underlying host–microbial interactions in the mammalian gut is essential for future studies directed at targeting the gut microbiota in order to improve health. We also provide a web-accessible database, http://microbiota.wall.gu.se, to investigate whether specific genes are regulated by the gut microbiota and/or MyD88. This resource will facilitate the identification of microbially regulated genes for researchers interested in all aspects of gastroenterology.
The human gut is home to trillions of bacteria (gut microbiota) that have co-evolved with us and established a finely tuned symbiosis.1 Their combined genomes (metagenome), which contain 150-fold more genes compared with our own genome, provide us with functions that we did not have to evolve ourselves.2 ,3 Recent data suggest that if this symbiosis is disrupted we are exposed to the increased risk of developing common diseases such as inflammatory bowel disease and obesity.4 Germ-free mice provide a powerful tool to gain mechanistic insights into host–microbial interactions and their effect on host physiology. Colonisation of germ-free mice is associated with profound morphological changes in the small intestine such as shortening and widening of the villus,5 increased vascularisation,6 recruitment of lymphocytes7 and activation of the innate and adaptive immune systems.8
The innate immune system recognises bacteria and other infectious agents by pattern recognition receptors such as intracellular nucleotide-binding oligomerization domain-like receptors and Toll-like receptors (TLR).9 All TLR (except TLR3) as well as interleukin (IL) 1 and IL-18 signal via MyD88-dependent pathways, which activate nuclear factor kappa B-driven pro-inflammatory signalling. Accordingly, MYD88 deficiency in humans is associated with increased susceptibility to pyogenic bacterial infections and Myd88-deficient mice have enhanced susceptibility and morbidity to most viral and bacterial experimental infections.10
The microbial ecology of the gut is governed by several factors, such as age, diet and host genotype, and recent findings suggest an important role for the innate immune system.11 In particular, TLR5 has been shown to have a significant impact in shaping the colonic gut microbiota, whereas TLR2 and TLR4 appear to play a minor role.12 ,13 Furthermore, in a mouse model of non-obese diabetes, Myd88 deficiency is associated with altered microbial community composition that confers protection against developing the disease.14
The function and architecture of the gut differs along its length: for example, nutrient absorption is most prevalent in the duodenum and jejunum, whereas the ileum is more immunologically active.15 The colon is more of a fermentative reactor producing short chain fatty acids and is the major site of water reabsorption.15 However, little is known about how the host responds to gut microbiota along the length of the gut. The concentration of bacteria increases along the length of the gut, from 104 cells/g in the duodenum to 1012 cells/g in the colon,15 and most published studies of gut microbial ecology have focused on the colon and feces, with less emphasis on the less populated and more inaccessible small intestine. The aim of our study was to identify the influence of innate immunity on microbiota-induced host responses and microbial composition along the length of the gut; we used Myd88 deficiency as a model for the loss of innate immune signalling.
Materials and methods
Mice
Germ-free C57Bl6/J and Swiss Webster male mice (three to five per cage) were maintained in flexible film isolators under a 12-h light cycle and freely fed an autoclaved chow diet (Labdiet, St Louis, Missouri, USA). The Myd88−/− were backcrossed at least eight generations to C57Bl6/J and the last two crossings were performed using mice from our colony. These two lines were thereafter separated by a maximum of two generations. Germ-free and conventionally raised mice were separated by a maximum of three generations. Conventionally raised mice were fed the same autoclaved diet upon weaning and all mice were used at 12 weeks of age.
Mice were killed by cervical dislocation and the small intestines, cecum and colon were removed. The small intestine was divided into eight equal-sized segments and the colon into three. For RNA analyses, we used the first (duodenum), fifth (jejunum), eighth (ileum) segments and the proximal piece of the colon. For analysis of the gut ecology, all segments were analysed. Epithelial cells were isolated as described previously.16 Animal protocols were approved by the Research Animal Ethics Committee in Gothenburg.
RNA isolation
RNA was isolated from the gut tissues and epithelial cells immediately after cell harvest using the RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA concentration and quality were evaluated by spectrophotometric analysis (ND-1000; NanoDrop Technologies, Wilmington, DE, USA) and capillary electrophoresis on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Microarray processing and statistical analysis
RNA labelling, microarray hybridisation and scanning were performed at the Uppsala array platform core facility at Uppsala University using MoGene 1.0 ST chips (Affymetrix, Santa Clara, CA, USA), according to the manufacturer's instructions. Normalisation and probe set summarisation were performed on each tissue separately using the expression console software (Affymetrix). CEL files and normalised data were deposited into the NCBI GEO repository, accession number GSE17438. Downstream analyses were performed in Matlab using proprietary scripts as and functions from the Bioinformatics toolbox. Genes were annotated against the ENSEMBL17 set of genes using the MoGene 1.0 ST probe set mapping provided using BioMart.18 Probe sets that were mapped to more than one gene were excluded while redundant probe sets were averaged. This resulted in a final dataset of 22 073 unique genes, each associated with one or more unambiguously mapped probe sets. Principal components analysis and hierarchical clustering (average linkage with Pearson's correlation coefficient) was performed on the processed dataset. Differential expression was evaluated in each tissue by calculating two-way analysis of variance (ANOVA) p values, including a test for interaction, using colonisation status (germ-free or conventionally raised) and genotype (Myd88−/− or wild type) as independent variables. This test was applied to each individual gene, and was followed by adjustment for multiple comparisons using the q value method proposed by Benjamini and Hochberg.19 Gene ontology (GO) statistics, based on ENSEMBL annotated terms and their ancestors in the GO hierarchy, were calculated using Fisher's exact test. To account for multiple testing, we used permutation simulations to determine that an enrichment p value threshold of 10−4 resulted in a low fraction of false positives (<5%). Genes without annotated GO terms were excluded from the analysis.
cDNA synthesis and quantitative PCR
An aliquot of 0.5 μg of total RNA was reverse transcribed (high capacity cDNA reverse transcription kit; Applied Biosystems, Foster City, California, USA) and SYBR green-based quantitative reverse transcription (qRT)–PCR was performed as described previously.16 Primers are listed in supplementary table S1, available online only.
Bacterial community composition analysis
Intestines were removed, flash frozen in liquid nitrogen and divided into 12 segments. The small intestine was divided equally into eight segments, the ninth segment corresponded to the cecum, and the large intestine was sectioned into three equal pieces. Whole community DNA was isolated from the segments using the tissue and cells DNA isolation kit (MoBio, Carlsbad, CA, USA). DNA concentration and quality was evaluated by Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Carlsbad, CA, USA) and a plate reader.
PCR amplification of 16S ribosomal RNA genes was carried out using barcoded primers 27F-338R for the V1–V3 region of the 16S rRNA gene.20 Three independent 50 μl PCR reactions were performed for each sample, each consisting of 2.5 U EasyA high fidelity enzyme, 1× buffer, 2 μM of each primer and 10–100 ng of the DNA template. Reaction conditions consisted of one cycle of 2 min at 95°C, 30 cycles of 30 s at 95°C, 45 s at 57°C, 60 s at 72°C and a final cycle of 2 min at 75°C on a thermocycler (Eppendorf, Hamburg, Germany). The PCR amplification products from each reaction were pooled and purified using Ampure magnetic purification beads (Agencourt Bioscience, Brea, CA, USA) and sequenced at the Cornell University Life Sciences Core Laboratories Centre (using Roche/454 FLX genome sequencer; Roche, Basel, Switzerland).
Sequences were analysed using the quantitative insights into microbial ecology (QIIME) software package.21 Sequences were removed from further analysis if their length was outside the range of 200–1000 nt, or if they contained ambigious bases, primer mismatches, homopolymer runs greater than six nucleotides, or uncorrectable barcodes. The remaining sequences were denoised22 and assigned to operational taxonomic units using UCLUST,23 with a 97% pairwise identity threshold, and classified taxonomically using Greengenes.24 Changes in bacterial abundance were compared using a two-tailed Student's t test with a Bonferroni correction for multiple comparisons. For tree-based analyses, single representative sequences for each operational taxonomic unit were aligned using PyNAST, and the phylogenetic tree used in the UniFrac25 analysis was built using FastTree.26 Sequences were deposited into MG-RAST, accession number MGP151.
Results and discussion
Gene expression profiling of germ-free and conventionally raised wild-type and Myd88−/− mice
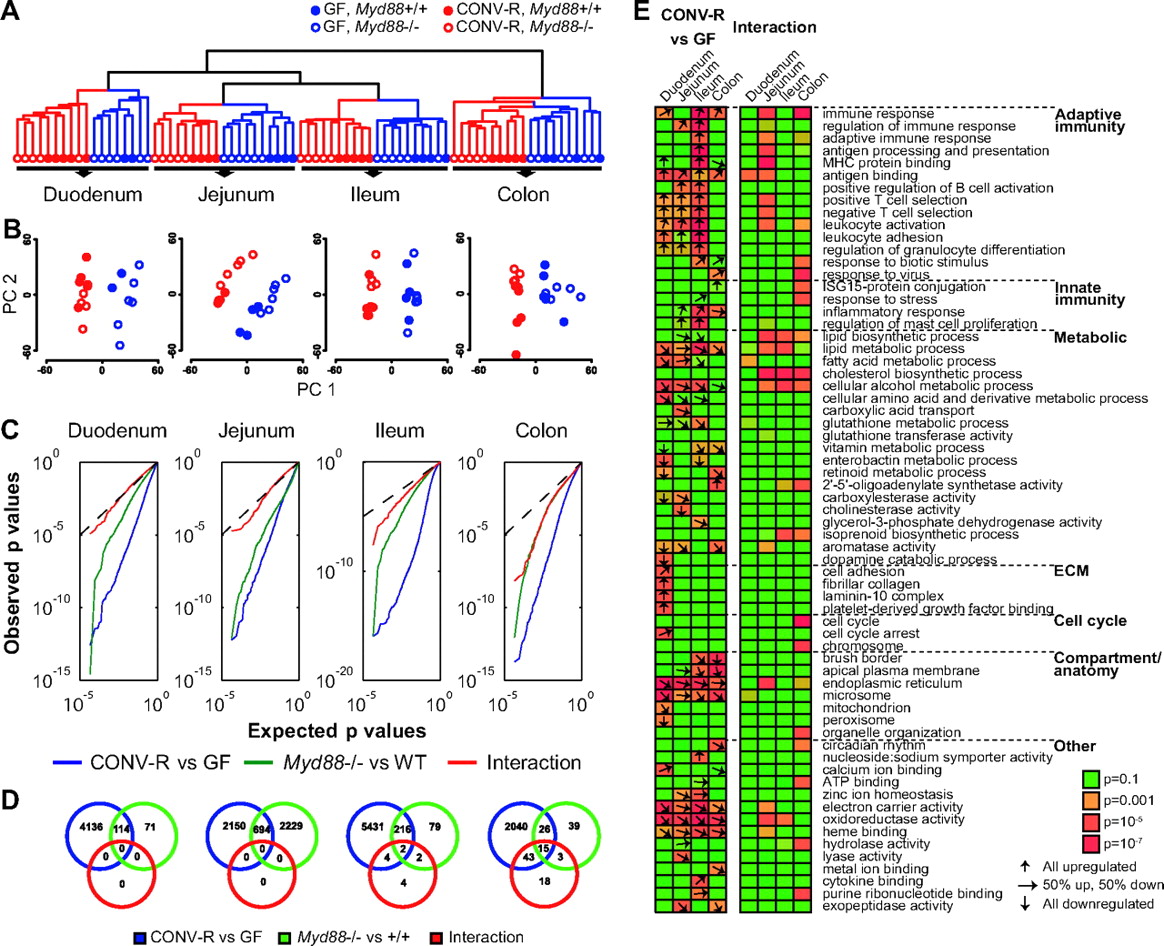
We performed 77 microarray hybridisations to obtain global transcriptional profiles from duodenum, jejunum, ileum and colon from germ-free and conventionally raised wild-type and Myd88−/− mice. Hierarchical clustering revealed distinct clusters representing each of the four tissues and further subclustering based on colonisation status and genotype (wild type or Myd88−/−; figure 1A). Clustering by genotype was most clearly seen for colonised animals. A similar pattern was revealed by principal component analysis (figure 1B). We next applied a two-way ANOVA test to score for differential expression depending on colonisation status or genotype, as well as interaction between these factors to identify cases in which MyD88 modulates the microbial response. We observed dramatic transcriptional responses to the microbiota in all segments of the gut (figure 1C). In the small intestine, between 2844 and 5653 genes were regulated by the microbiota (5% false discovery rate; blue circles in figure 1D; a list of all microbiota-regulated genes is provided in supplementary table S2, available online only). Fewer genes were regulated by the microbiota in the colon compared with other segments (2124 genes; figure 1D). The limited microbial regulation in the colon may be explained by the presence of a thick continuous mucosal layer, which shields the epithelium from direct bacterial exposure.27

Transcriptional profiling of intestinal tissue samples from germ-free (GF) and conventionally raised (CONV-R) mice. (A) Hierachical clustering dendrogram of whole-transcriptome expression profiles obtained using DNA microarrays. (B) Principal component analysis of transcription profiles performed separately on each tissue. PC1, principal component 1; PC2, principal component 2. (C) All genes were evaluated by two-way analysis of variance for differential expression in germ-free versus conventionally raised mice (blue), Myd88−/− versus wild-type (green), as well as interaction between these two factors to reveal microbial responses that were modulated by Myd88 genotype (red). Quantile–quantile plots illustrate expected versus observed distributions of p values obtained in these tests. Dotted lines show the expected distribution under the null hypothesis (no genes regulated). Strong deviations from this line indicate large effects on the transcriptome. (D) Venn diagram showing the number of differentially expressed genes (at 5% false discovery rate) between germ-free and conventionally raised mice in blue, Myd88−/− and wild-type mice in green, and genes that are regulated by the microbiota in a MyD88-dependent fashion (interaction) in red. (E) Enriched gene categories among regulated genes. The heat map indicates the level of statistical enrichment (Fisher's exact test) for select gene ontology (GO) categories among the 500 most significantly regulated genes. Representative GO terms were selected from the complete list of significant (p<10−4, see Methods section) categories (see supplementary table S2, available online only). The arrows indicate, in cases in which the enrichment is p<0.01, the proportion of genes (among top 500) in each category that are up or downregulated. Arrows are not shown for the interaction test as these per definition respond differentially depending on genotype. n=4–6 mice per group, but note that due to poor RNA quality of two duodenal samples from wild-type germ-free mice we only included two samples in the analyses.
Given the importance of TLR as pattern recognition receptors for bacteria, we anticipated a major role for MyD88 in regulating the transcriptional responses to the gut microbiota. We observed a significant impact of MyD88 on host transcriptional profiles in the jejunum but not in other segments of the gut (figure 1D, green circle); however, these responses were independent of the gut microbiota (figure 1D, red circle). Although few microbiota-induced changes in gene expression required MyD88, such interaction effects became increasingly larger in the distal segments; 79 genes were significant in the interaction test in the colon at 5% false discovery rate (red circles in figure 1D).
Genes involved in immune responses and metabolism are predominantly regulated by the gut microbiota in the small intestine
To identify major regulated pathways and processes, we evaluated the 500 most significant microbiota-regulated genes in each tissue for enrichment of functional gene categories. Several GO categories were statistically linked to microbial status in all tissues (figure 1E, see supplementary table S3, available online only). Similar to previous studies28–30 we observed significant microbial induction of genes related to immunity. The majority of these genes were associated with adaptive immunity, and are probably the result of lymphocyte migration to the mucosa and differentiation in response to the gut microbiota.7 ,28 ,31 In addition, we evaluated transcriptional responses to the gut microbiota that depended on MyD88 (500 most significant genes in the interaction test). Immune categories were only weakly enriched in this set, although a stronger association was observed in the jejunum (figure 1E). Interestingly, these categories were also identified to be affected by MyD88 under germ-free conditions in the jejunum and ileum.
Metabolism-related genes including lipid and fatty acid metabolism-related categories, as well as genes related to cellular compartments involved in nutrient absorption and metabolism, were regulated in response to the microbiota throughout the gut (figure 1E). Interestingly, genes related to cholesterol biosynthesis were enriched among the top genes that responded differentially to the microbiota depending on genotype in the jejunum, ileum and colon. Furthermore, genes associated with energy-yielding compartments, mitochondrion and peroxisome, were found to be among those downregulated in the duodenum of conventionally raised compared with germ-free mice.
Most microbially regulated genes involved in immune responses and barrier function are not dependent on Myd88
Expression levels of genes encoding immunoglobulins were altered in all segments of the gut, and this regulation was independent of MyD88 (see supplementary table S2, available online only). In agreement, we observed increased chemokine gene expression in gut tissues from conventionally raised compared with germ-free mice (table 1). By isolating primary epithelial cells from the ileum in a separate set of animals, we found that these genes were regulated by the gut microbiota in the epithelium (see supplementary figure S1A, available online only). An increase in chemokine levels would be expected to result in lymphocyte infiltration to the mucosa.7
Selected microbially regulated genes
We also observed microbiota-induced upregulation of genes associated with barrier function (table 1). Genes encoding small proline-rich protein 1A and 1B, which play important roles as components of the cell envelope, and are cross-bridging proteins,32 were significantly upregulated in the colon of conventionally raised mice (table 1). Expression of MUC2, the major mucin component of the colonic mucus layer, and expression of MUC4 and MUC13 were significantly elevated in all segments of the small intestine and colon (table 1). These responses were evident in epithelial cells (see supplementary figure S1B, available online only), but were unaffected by Myd88 deficiency (table 1).
The inability to signal through MyD88 is associated with increased susceptibility to viral infections in conventionally raised mice
Antiviral genes were induced by the microbiota in the colons of Myd88−/− mice but not in wild-type counterparts (figure 2). These results were corroborated by qRT–PCR (see supplementary figure S2, available online only) and could be localised to the epithelial layer (see supplementary figure S3, available online only). In contrast, we did not observe any increases in antiviral gene expression in the small intestine of Myd88−/− mice (see supplementary table S2, available online only).

Relative expression levels of antiviral genes in the colon of germ-free (GF) and conventionally raised (CONV-R) wild-type (WT) and Myd88−/− mice. Data are from the microarray experiment and were corroborated by quantitated reverse transcription PCR in supplementary figure S2 available online only. n=4–6 per group. *p<0.05, **p<0.01, ***p<0.001; analysis of variance.
At first these results may be counterintuitive as MyD88-independent pathways are mainly invoked in the regulation of antiviral response genes. However, Myd88-deficient mice exhibit increased mortality in several models of experimental viral infections.10 To gauge whether the elevated expression of antiviral genes in the colonic epithelium of conventionally raised Myd88-deficient mice could be attributed to a viral infection, we performed a PCR-based screen of 15 selected viruses. This analysis revealed that our Myd88-deficient mice were infected by murine norovirus (table 2). Murine norovirus is common in animal facilities around the world and does not cause symptoms in immune-competent mice.33
PCR-based screening for mouse viruses in isolated colonic intestinal epithelial cells from conventionally raised mice
Our data thus suggest that Myd88-deficient mice are susceptible to naturally occurring viral infections and that the presence of norovirus in the colonic epithelium of Myd88-deficient mice is the most likely cause of antiviral gene expression in these mice.
Microbial regulation of REG3β and REG3γ requires MyD88 in colon but not ileum
Two recently identified antimicrobial peptides, REG3β and REG3γ, which are mainly produced by the Paneth cells but also by the absorptive epithelium,34 ,35 were among the most significantly upregulated genes by the presence of a microbiota in both the small intestine and the colon (table 1). Interestingly, microbially induced expression of REG3β and REG3γ was dependent on MyD88 in the colon but not in the small intestine, (table 1), demonstrating distinct signalling pathways in these tissues. We also observed microbiota-induced expression of genes involved in the production of reactive oxygen (ROS) and nitrogen species (NOX1, NOS2 and DUOX2 and the maturation factor DUOXA2), which are also potent antimicrobial agents,36 ,37 with the most pronounced effect in the ileum (table 1; validated by qRT–PCR in supplementary table S4, available online only). However, in contrast to REG3β and REG3γ, regulation of genes related to ROS production was independent of MyD88. qRT–PCR analysis of isolated epithelial cells from ilea and colons of germ-free and conventionally raised mice revealed that most of these genes were microbially regulated in the epithelium (see supplementary figure S4, available online only). Interestingly, we also noted a similar gene expression profile in Swiss Webster mice (see supplementary table S5, available online only), which are considered to have a TH2-biased immune system.
MyD88 deficiency is associated with altered bacterial community composition
We also examined bacterial diversity and community structure along the length of the gut in conventionally raised wild-type (n=7) and Myd88-deficient (n=4) mice using 454-based pyrosequencing of 16S rRNA gene (see supplementary figure S5, available online only). Myd88-deficient mice had a significant increase in the relative abundance of segmented filamentous bacteria (SFB), especially in the jejunum and ileum (figure 3A), indicating that SFB are particularly affected by the loss of MyD88. SFB are tightly associated with the epithelium, especially in the small intestine,38 and play a fundamental role in the maturation of intestinal T-cell responses and in the induction of gut inflammation, antimicrobial defence as well as mucosal IgA production.28 ,39 ,40 Therefore, it is possible to speculate that localisation of SFB close to the epithelium may make them more sensitive to antimicrobial molecules that are expressed by the epithelium in a MyD88-dependent fashion compared with other bacteria located further away from the surface of the epithelium.

{kind=link}
{kind=link}
{kind=link}
Effect of host genotype on the microbiota composition along the length of the gut from conventionally raised wild-type and Myd88−/− mice. (A) Relative abundance of segmented filamentous bacteria (SFB) in the two genotypes (Myd88+/+ or −/−) along the length of the gut. ***p<0.001; two-tailed Student's t test with Bonferroni correction for multiple comparisons. (B) Principal coordinates analysis of unweighted UniFrac distances between bacterial communities determined from 16S rRNA genes. Percentage variation explained by each principal coordinate (PC) is indicated on the axis. Symbols repesent individual intestinal segments obtained from replicate mice: duodenum, first three segments; jejunum, segments 4–5; ileum, segments 6–8; cecum, 9; large intestine, 10–12. n=4–7 mice per group. Low sequence counts were obtained for some samples, which were removed from further analysis.
To assess how the overall diversity of the microbiota was affected by Myd88 deficiency irrespective of the relative abundances of taxa, we compared samples using the unweighted UniFrac distance metric, which assumes that similar communities have similar evolutionary histories that can be represented as shared branches in a joint phylogenetic tree.25 This analysis revealed that the two genotypes harbour different bacterial communities (evidenced as clustering of samples by genotype in each of the different segments of the intestine, figure 3B). In addition, duodenum, jejunum and ileum-associated microbial communities show significantly higher levels of inter-mouse variation in bacterial diversity in Myd88-deficient mice compared with wild-type mice (see supplementary figure S6, available online only). In contrast, the bacterial communities in the cecum and colon clustered tightly (low mouse–mouse variation in microbial diversity, supplementary figure S6, available online only) and very clearly segregated by host genotype (figure 3B). Generally, difference in mouse to mouse variation along the length of the gut may reflect the biomass difference between gut segments. However, the greater mouse to mouse variation in Myd88-deficient animals compared with wild-type mice may signify a loss of control over microbial diversity.
Earlier studies have shown that the host genotype can affect the microbial ecology of the gut: for instance, α-defensin deficiency has recently been shown to modify the composition of gut microbiota significantly in mice.41 We therefore propose that the MyD88-dependent increases in REG3β and REG3γ gene expression in the colon could contribute to the genotype-specific clustering of bacterial communities in the colon, but further experiments are required to prove this hypothesis.
Conclusion and prospectus
In summary, we have extensively analysed the influence of the microbiota on gene expression and microbial ecology along the length of the gut, in both wild-type and Myd88-deficient animals. Furthermore, we identified limited but selective roles of MydD88 in increasing susceptibility to norovirus infections and modifying microbial ecology. Despite relatively small effects on mediating microbial-induced host gene expression changes, MyD88 affects host microbial ecology in the gut.
To make our gene expression data easily available to the scientific community, we constructed a searchable database (http://microbiota.wall.gu.se). The database enables interactive queries on gene name and detailed gene views with graphical illustrations of measured levels in different tissues and conditions, as well as statistical results and additional information about tissue expression profiles.42 Data on host microbial responses in tissues (liver, epididymal and subcutaneous fat), that were not analysed in this report, are provided through the web interface.
Acknowledgments
The authors would like to thank Carina Arvidsson, Anna Hallén and Gunnel Östergren-Lundén for superb technical assistance, Rosie Perkins for editing the manuscript, Aymé Spor for statistical support, and Jeffrey Gordon for supplying germ-free Myd88−/− mice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 3 - Online table 3
- Data supplement 2 - Online table 2
Footnotes
Funding This work was supported by the Swedish Research Council (K2007-65X-20421-01-04), Swedish Foundation for Strategic Research, EU-funded ETHERPATHS projects (FP7-KBBE-222639, http://www.etherpaths.org) and TORNADO (FP7-KBBE-222720, http://www.fp7tornado.eu/), Åke Wiberg, Torsten and Ragnar Söderberg, Chalmers, Knut and Alice Wallenberg, and Novo Nordisk Foundations, and a LUA-ALF grant from Västra Götalandsregionen, as well as a Beckman young investigator award to REL.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement CEL files and normalised array data are available at NCBI GEO repository, accession number GSE17438. The expression data are also freely available through http://microbiota.wall.gu.se/. The 454 data set will be available at MGRAST—accession number MGP151.