Article Text

Abstract
The intestine and its immune system have evolved to meet the extraordinary task of maintaining tolerance to the largest, most complex and diverse microbial commensal habitat, while meticulously attacking and containing even minute numbers of occasionally incoming pathogens. While our understanding is still far from complete, recent studies have provided exciting novel insights into the complex interplay of the many distinct intestinal immune cell types as well as the discovery of entirely new cell subsets. These studies have also revealed how proper development and function of the intestinal immune system is dependent on its specific microbiota, which appears to have evolutionarily co-evolved. Here we review key immune cells that maintain intestinal homeostasis and, conversely, describe how altered function and imbalances may lead to inflammatory bowel disease (IBD). We highlight the latest developments within this field, covering the major players in IBD including intestinal epithelial cells, macrophages, dendritic cells, adaptive immune cells, and the newly discovered innate lymphoid cells, which appear of characteristic importance for immune function at mucosal surfaces. We set these mucosal immune pathways in the functional context of IBD risk genes where such insight is available. Moreover, we frame our discussion of fundamental biological pathways that have been elucidated in model systems in the context of results from clinical trials in IBD that targeted key mediators secreted by these cells, as an attempt of ‘functional’ appraisal of these pathways in human disease.
- INFLAMMATORY BOWEL DISEASE
- MUCOSAL IMMUNOLOGY
Statistics from Altmetric.com
Introduction
The gastrointestinal tract represents perhaps the most sophisticated and complicated immune organ of the entire body.1 The intestinal epithelium, the inner single cell lining of the intestine, and evolutionarily the most ancient part of the innate immune system,2 separates the essentially sterile host from the intestinal microbiota, which is among the most intensely populated microbial habitats on earth.3 The intestinal immune system is tasked to prevent the invasion of harmful pathogens while remaining tolerant of innocuous food substances and commensal microorganisms. This carefully regulated immune balance has been shaped by several million years’ co-evolution between the host and gut microbiota and is essential for the healthy development and integrity of the intestine. By contrast, breakdown of immune homeostasis can lead to inflammatory bowel disease (IBD) and its two main forms: Crohn's disease (CD) and ulcerative colitis (UC).1
Recent years have seen a rapid and exciting expansion in our understanding of the mucosal immune system with novel insights into environmental influences of diet and the microbiota; the convergence and integration of fundamental cellular processes such as autophagy, microbial sensing and endoplasmic reticulum (ER) stress; as well as the discovery of new cell types, for example innate lymphoid cells (ILCs).
The gut is unlike the systemic immune system in several respects and much of the extensive repertoire of immune cells and their characteristics are indeed unique to the intestine. For example, the majority of intestinal effector T cells are antigen experienced and less dependent on co-stimulation, and many intestinal T cells do not express CD28.4 ,5 Such differences might have immediate clinical aspects attached to them; for example, this might help explain why abatacept (CTLA4-Ig), which blocks co-stimulation, might have failed in CD and UC, while being highly effective in mechanistically seemingly related conditions such as rheumatoid arthritis.6–8
This review aims to highlight some key recent developments, and discusses how perturbations within the many various components of the mucosal immune system can lead to inflammation. We put these fundamental biological principles in the context of IBD, whenever possible by reviewing results from clinical trials (table 1) that targeted specific molecules within these pathways since these allow functional appraisal of these biological pathways in patients. Needless to say, the essentially binary clinical outcomes measures of these trials with barely any immunological assessments usually being performed and reported, set limitations to this approach. In the same context, we also discuss the cellular mechanisms of some IBD risk genes for which functional insight has indeed been experimentally gained, but refrain from inclusion of the overwhelming majority that still awaits experimental interrogation.
Current and emerging biological inflammatory bowel disease (IBD) treatments targeting the mucosal immune cells and pathways described in this review
Mucosal immune development instructed by environment and microbiota
The human intestine covers a huge surface area of approximately 200–400 m2. Residing within the lumen are an estimated 100 trillion microbial cells.9 Whilst these commensal bacteria perform numerous metabolic functions essential to the host,3 pathogens that gain access and manage to expand within this complex ecosystem pose a constant threat of invasive disease. The mucosal immune system has evolved several layers to regulate the delicate and dynamic balance between tolerance and vigilance (figures 1A,B and 2). The development and organisation of mucosal immune cells is intimately intertwined with the microbiota, without which the immune system is immature and defective.10 An example is the requirement of a single bacterial species, segmented filamentous bacteria (SFB; also known as Candidatus arthromitus), for the differentiation of Th17 cells (see below) in the small intestinal lamina propria11 ,12; in the absence of SFB, which gets in close contact to the intestinal epithelium, mice lack Th17 cells outright, similar to mice that have been rederived germ-free.11 ,13 ,14 The full extent of the remarkable specificity between gut and microbiome for immune system development is only now emerging. Microbial transplantation between even closely related animals, that is, rat and mouse, is not an acceptable substitution in the development of the mucosal immune system.15 Colonisation of germ-free mice with a human, compared to a mouse, microbiota results in low levels of CD4 and CD8 T cells, few proliferating T cells, low numbers of dendritic cells (DCs) and low antimicrobial peptide (AMP) expression; all features characteristic for germ-free mice,15 and hence evidence for the lack of proper immune development in the absence of a species-specific indigenous flora. These observations highlight co-evolution of the host and its microbiota, and indicate that the structural composition and function of mucosal immune cells cannot be considered uncoupled from the microbiota. The gut microbial composition is most dynamic during the first years of life, when the mucosal immune system is also simultaneously still maturing and being primed for adulthood.16 We are still in the nascent stages of comprehending how these early events shape immunity and how they might later affect predisposition to disease such as IBD.

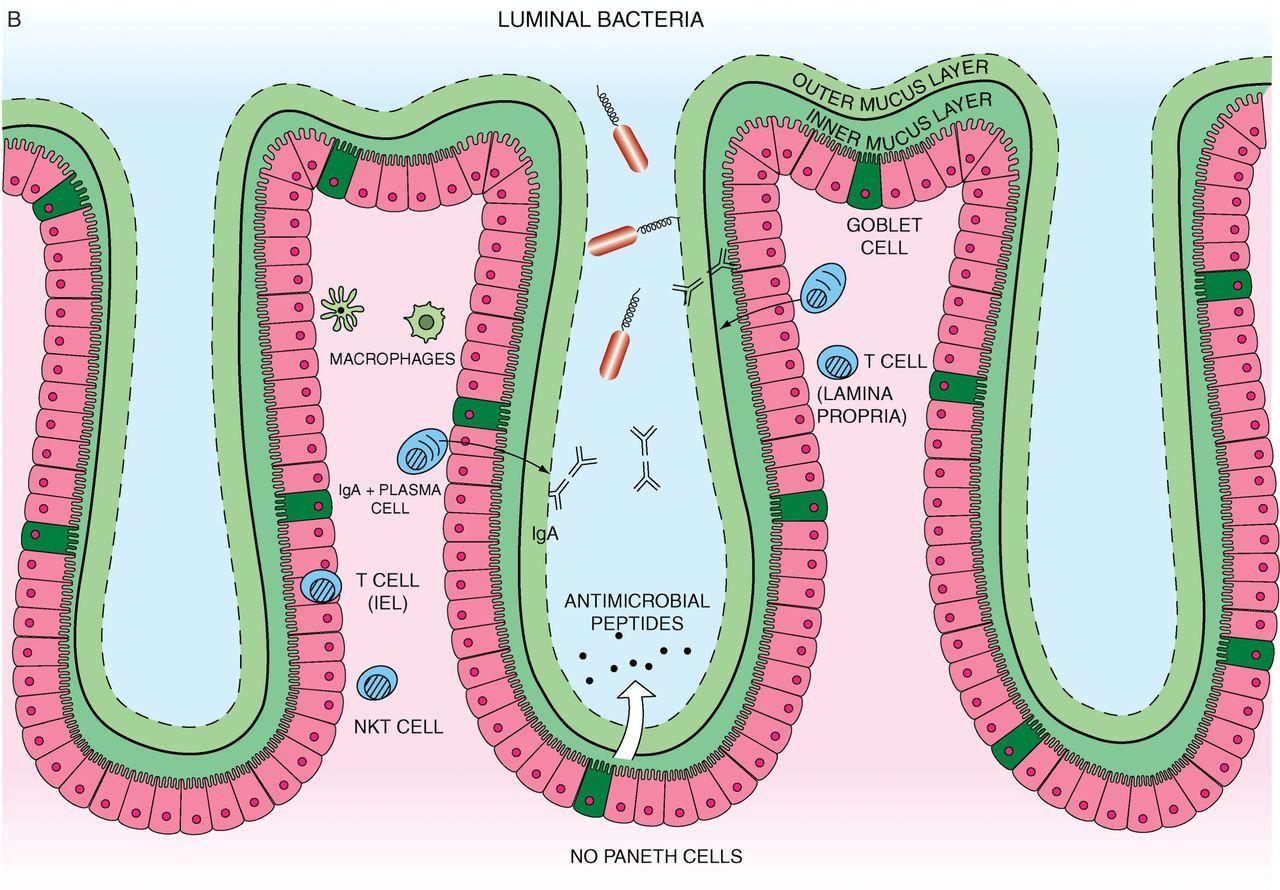
(A) Small intestine mucosal immune system landscape. The intestinal epithelial cell (IEC) layers form villi and crypt structures and are composed of different cell lineages. Goblet cells secrete mucus. Paneth cells, found only in the small intestine, can be found at the base of the crypts and are the main secretors of antimicrobial peptides. The base of the crypts also contains the IEC stem cell populations. Immune cells can be found in organised tissue such as Peyer's patches and throughout the lamina propria. They include macrophages, dendritic cells, intra-epithelial lymphocytes, lamina propria effector T cells, IgA secreting plasma cells, innate lymphoid cells and stromal cells such as fibroblasts. Antigen presenting cells in Peyer's patches or mesenteric lymph nodes interact with and activate local lymphocytes, which consequently upregulate expression of the integrin α4β7. Such cells then enter the systemic circulation but home towards the gut, in response to chemokine ligands such as CCL25. (B) Colon (large intestine) mucosal immune system. The colon has a much higher bacterial load and a markedly different immune cell composition. The colon contains only crypts, no villi. Also there are no Paneth cells, which mean that enterocytes have a much more important contribution to antimicrobial peptide production. However there is a high prevalence of goblet cells. The mucus forms dual layers, with a thick largely sterile inner layer and a thinner outer layer. There are no Peyer's patches. While the immune cell types present are similar to those found in the small intestine it is likely that there may be at least subtle differences. In particular natural killer T cells are found more frequently and have a more significant role in the colon.

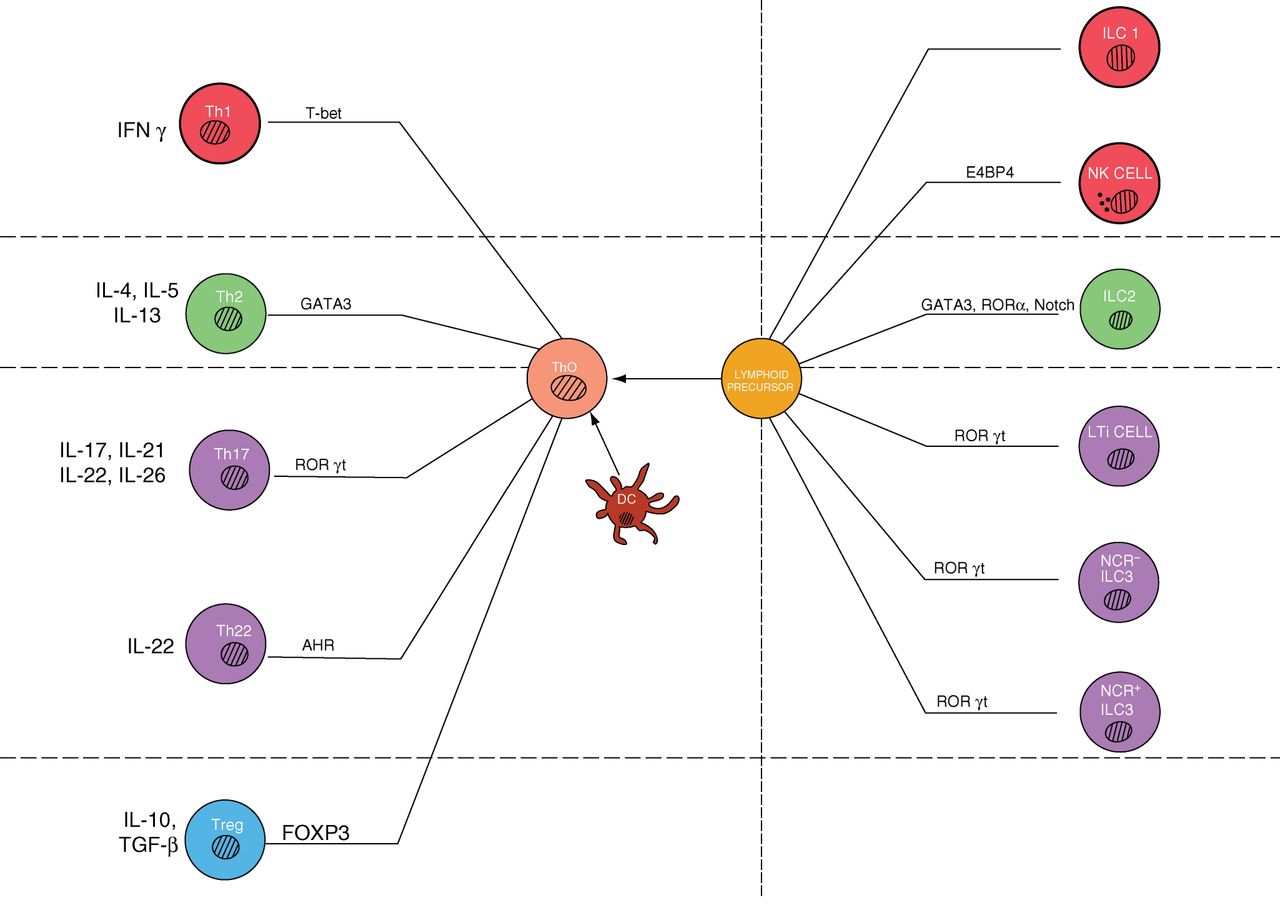
T-helper cell differentiation. Traditionally, Crohn's disease (CD) has been characterised by a Th1 and ulcerative colitis a Th2 predominant T cell infiltrate. However Th17 and Treg populations are also prevalent and have critical roles in the lamina propria. The balance of different effector cells depends on the inflammatory context and cytokines present. Dendritic cells in particular not only activate T cells, but also secrete a wide range of cytokines that can drive differentiation.
Epidemiological observations indeed suggest that early microbial exposure may be important in determining the risk of developing IBD.17 An elegant mechanism involving natural killer T (NKT) cells, the epithelium and early life exposure to the microbiota, which could contribute to this ‘hygiene hypothesis’,18 has recently been reported by Blumberg and colleagues.19 Invariant NKT (iNKT) cells, expressing an invariant TCRα chain, are activated in response to endogenous and exogenous lipids presented by the non-polymorphic MHC class I-related molecule CD1d.20 CD1d-restricted iNKT cells produce IL-13, a cytokine associated with UC,21 and have been shown to be the main effectors of IL-13-dependent oxazolone colitis, a murine model that closely resembles UC.22 Colonisation of neonatal, but not adult, germ-free mice with a conventional microbiota prevented mucosal iNKT cell accumulation and, importantly, protected against the induction of colitis.19 The accumulation of iNKT cells in the colonic lamina propria of germ-free, compared to colonised mice was due to epigenetic modification and consequent increased expression of the iNKT cell chemokine CXCL16 in the intestinal epithelium under germ-free conditions.19 Hence, age-sensitive microbial contact was able to impart adult iNKT cell tolerance, thereby determining susceptibility to experimental disease. IL-13-producing CD1d-restricted NKT cells are found in the lamina propria of human UC patients,21 and a phase II clinical trial is currently underway that aims to block IL-13 via the specific monoclonal antibody tralokinumab (ClinicalTrials.gov NCT01482884).
A further very important cellular constituent that warrants discussion in the context of microbially-instructed differentiation of the mucosal immune system is the CD4CD25 T regulatory (Treg) cell.23 Specific clusters (IV and XIVa) of the genus Clostridium, which are part of the spore-forming component of the indigenous intestinal microbiota, have been shown to promote Treg cell accumulation in the colonic mucosa.24 Remarkably, early-life exposure to Clostridium in conventionally reared mice resulted in Treg expansion and resistance to experimental colitis.24 Hence, specific ‘probiotic’ bacteria might be capable of ameliorating intestinal inflammation by augmenting Treg numbers and function.24–26 It is notable that the proportion of Clostridium clusters IV (which includes Faecalibacterium prausnitzii) and XIVa within the faecal community of patients with IBD is reduced compared to healthy controls.27 ,28
Much like the microbiota shapes the mucosal immune system, diet can also have a remarkable effect on the constitution of mucosal immune cells. A striking example comes from ingredients of cruciferous vegetables which act on the aryl hydrocarbon receptor (AhR).29 ,30 Intraepithelial lymphocytes (IELs), interspersed in the epithelial layer, have a role as a first line of defence and in wound repair.31 Remarkably, in the absence of AhR ligands, or on the receptor's genetic deletion, IELs can no longer be maintained, which results in heightened immune activation, increased susceptibility to epithelial damage in experimental colitis and a defect in controlling the proper structural composition of the intestinal microbial habitat.29 Although the characteristics of murine and human IELs differ, these insights provide a remarkable mechanistic framework of how diet affects specific immune functions. There are more examples of dietary effects including tryptophan depletion and milk-derived taurocholic acid in both altering microbial composition that drives altered immune responses and susceptibility to colitis in murine models,32 ,33 in addition to the well-known effects of the dietary constituents vitamins A and D and their effects in Treg and Th17 development.34 A final notable example of how diet affects intestinal immune function comes from studies in Ace2 −/− mice, which revealed that the dietary amino acid tryptophan has a profound effect on intestinal epithelial antimicrobial function, and deficiency resulted in increased susceptibility to experimental colitis.33
With this dietary- and microbially-instructed plasticity of the mucosal immune system in mind, we will next consider the role of intestinal epithelial cells (IECs) at the interface of microbes and the host immune system.
The intestinal epithelium and Paneth cells in IBD
The IEC layer is a polarised, columnar monolayer separating the microbiota from the subepithelial lamina propria and consists of specialised differentiated cell types: enteroabsorptive cells, goblet cells, neuroendocrine cells, Paneth cells and M cells, all deriving from a common intestinal epithelial stem cell.35 Goblet cells produce and secrete glycosylated mucins that form a mucus matrix covering the epithelium and forming the first obstacle preventing microbial invasion.36 The colon has a dual mucus layer and the inner, denser layer restricts bacterial motility and adhesion to the epithelium.37 The small intestine has a single, looser mucus layer presumably to allow the penetration of food substances, but is also substrate to glycan foraging of intestinal symbionts such as Bacteroides thetaiotamicron which contributes to ecosystem stability.38 The spatial separation of bacteria from the epithelial surface is further supported by RegIIIγ, a C-type lectin, which is essential to maintain a ∼50 µm distance of the microbiota to the small intestinal epithelial surface.39 It is interesting to note that goblet cell and mucus depletion is characteristic of UC.40 ,41 Genetic deletion of Muc2, encoding the major component of mucin, results in spontaneous colitis,42 similar to mice with point mutations in Muc2, which results in ER stress in goblet cells due to MUC2 misfolding.43 In addition to providing the intestinal mucus layer, goblet cells also serve important direct immune functions, for example by secreting resistin-like molecule β (RELMβ),44 which directs protective Th2 immunity on nematode infection,45 and by delivering small luminal antigens to a tolerogenic set of DCs.46
Paneth cells are a further IEC type that plays an important role in mucosal homeostasis, and, if functionally impaired, is thought to contribute to IBD.47 Paneth cells reside at the base of small intestinal crypts and secrete AMPs as well as inflammatory mediators.48 The products of several key genetic risk factors of human IBD affect Paneth cell function, including NOD2, the pre-eminent genetic risk factor of CD in the Caucasian population. NOD2 is expressed in Paneth cells49 and in other cell types such as DCs, macrophages and absorptive IECs.50 It is still not currently established which cell type mediates the risk conferred by NOD2 variants. NOD2 encodes a cytosolic pattern recognition receptor that is activated by N-acetyl muramyl dipeptide (MDP), derived from bacterial peptidoglycan.51 Activation involves NOD2's leucine-rich repeat domain, which leads to binding of receptor-interacting serine-threonine kinase 2 (RIPK2) and subsequent activation of nuclear factor-κB (NF-κB) via TRAF6-mediated ubiquitination of NF-κB essential modulator (figure 3).52 Patients harbouring NOD2 risk variants express decreased levels of human α-defensins 5 and 6 (HD5, HD6) in Paneth cells,53 which is mirrored in Nod2−/− mice.54 Nod2−/− Paneth cells exhibit impaired antimicrobial function, although Nod2−/− mice do not develop spontaneous intestinal inflammation, suggesting that additional factors may be required to trigger disease.54 Indeed, infection of Nod2−/− mice with a model opportunistic pathogen, Helicobacter hepaticus, caused a granulomatous type of ileocolitis, which was associated with a Th1 adaptive immune response that is also characteristic of CD.55 While there is no evidence that H hepaticus plays any role in human CD, this observation on gene–environment interaction in the murine model system is conceptually highly interesting (figure 4).

NOD2, autophagy and the unfolded protein response (UPR) in intestinal epithelial cells. The leucine rich repeat region of NOD2 binds to muramyl dipeptide (MDP) from bacteria. Activated NOD2 binds RIPK2 and downstream signalling leading to the activation of NF-κB. However, NOD2 can also directly inhibit toll-like receptor (TLR) signalling. NOD2 is also able to induce autophagy and appears to direct ATG16L1 to the bacterial isolation membrane. Autophagy and endoplasmic reticulum stress/UPR may be cross-regulated and both can be induced by TLR signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gene–environment interaction at the intestinal epithelial cell (IEC) interface. Several genetic risk factors of inflammatory bowel disease such as NOD2, ATG16L1 and XBP1 exhibit abnormalities in Paneth cells. Nod2−/− mice exhibit decreased expression of α-defensins, but do not develop spontaneous disease. However, infection with the model opportunistic pathogen (pathobiont) H hepaticus results in Th1-dominated granulomatous ileocolitis. Atg16l1HM do not develop spontaneous inflammation either, but their Paneth cells exhibit defects in their secretory apparatus on infection with murine norovirus CR6 strain. While CR6-infected mice still do not develop intestinal inflammation, they are more susceptible to colitis-inducing agent dextran sodium sulfate. Finally, Xbp1ΔIEC mice, which deplete fully mature Paneth cells and contain IECs hyper-responsive to inflammatory and microbial stimuli, develop spontaneous enteritis in their small intestine with features characteristic of human IBD. These three genetic risk factors of IBD may represent examples, established through their murine models, of the varying degree of ‘environmental’ (ie, non-host) contribution that is necessary for disease development on the background of the individual functional impact of the genetic risk variant.
A further CD risk locus that results in a Paneth cell phenotype conditional on an environmental factor is ATG16L1, whose discovery as a risk gene along with related genes identified autophagy as a mechanism involved in IBD.56 ,57 Autophagy is a cellular self-cannibalistic catabolic process that involves the formation of an autophagasome membrane around a cellular component such as a damaged organelle or intracellular bacteria, which is then marked for degradation by fusion with lysosomes.58 Patients homozygous for the CD risk-associated ATG16L1 allele (T300A) had marked structural alterations in their secretory apparatus along with transcriptional abnormalities in Paneth cells.59 Mice homozygous for a hypomorphic Atg16l1 variant had similar Paneth cell changes, but this required infection with murine norovirus (MNV CR6).59 The phenotype was not present in mice derived in an MNV-free specific pathogen-free barrier facility, or following non-persistent MNV CW3 infection.60 Again, similar to H hepaticus and NOD2, there is no evidence to imply a specific virus as trigger of IBD, nonetheless this is another notable example of gene–environment interaction (figure 4). Interestingly, the combination of hypomorphic ATG16L1 and MNV CR6 and its consequent Paneth cell abnormality did not result in spontaneous inflammation in the murine model.60 This is all the more notable as absent ATG16L1 function also results in over-activation of the inflammasome in macrophages and consequent increased IL-1β and IL-18 secretion.61 In epithelial cells, NOD2 induces autophagy and recruits ATG16L1 directly to site of bacterial entry (figure 3).62 Decreased NOD2 function resulted in impaired autophagic uptake and hence intracellular bacterial killing. Importantly NOD2 risk variant carrying CD patients were similarly unable to recruit ATG16L1.63 Remarkably the CD-associated T300A variant of ATG16L1 exhibited impaired autophagy induction on MDP NOD2 stimulation. Thus, within the diverse array of NOD2 and ATG16L1 functions, there is also a direct functional interaction that involves the autophagic process.
Autophagy also interconnects with another process that is genetically affected in IBD and that manifests in Paneth cells, which is the unfolded protein response (UPR) that arises from ER stress.64 The UPR aims to adapt the protein folding capacity of a cell to its translational output. This pathway is particularly important for highly secretory cell types such as Paneth cells. Several ER stress-related genes, such ORMDL3,65 XBP1 66 and AGR2 67 have been associated with IBD, and importantly, ER stress appears to be a general feature of the uninflamed and inflamed IBD epithelium irrespective of the presence of genetic risk variants within this pathway.43 ,66 ,68 Genetic deletion of the UPR transcription factor Xbp1 in the intestinal epithelium results in depletion of mature Paneth cells and the occurrence of Paneth cell remnants, along with spontaneous small intestinal inflammation that mimics many histological features of human IBD with crypt abscesses, neutrophil infiltration and ulcerations.66 In addition to impaired Paneth cell antimicrobial function, the XBP1-deficient epithelium also exhibited an inflammatory hyper-responsiveness which may be a critical factor in the development of enteritis.66 Similarly, Agr2−/− mice developed granulomatous ileocolitis, along with alterations in Paneth (and goblet) cell phenotype associated with unresolved ER stress.69
That these three closely connected pathways—microbial signalling (NOD2), autophagy and ER stress—are all linked with IBD and affect Paneth cell function clearly suggests that bacterial handling plays an important role, but appears not to be sufficient in its own right to instigate intestinal inflammation; this likely requires an additional inflammatory stimulus or insult that sets pathological mechanisms in motion.
Macrophages and DCs in IBD
Several types of macrophages and DCs form a further central part of the functional mucosal barrier of the intestine. Despite their importance, in both humans and mice, the exact definition of DCs and macrophages and their subtypes via surface markers remains controversial.70 ,71 One phagocytic mononuclear cell population is characterised by its expression of the chemokine receptor CX3CR1.72 ,73 Most lamina propria macrophages appear to express CX3CR1,74 and a subpopulation is located close to the epithelium and extends its processes into the intestinal lumen to sample antigens.72 Genetic deletion of the gene encoding CX3CR1 results in decreased numbers of lamina propria macrophages and increased translocation of commensal bacteria to mesenteric lymph nodes.74 Moreover, deficiency in CX3CR1 results in increased severity of experimental colitis, which can be ameliorated by either blockade of IL-17A by specific antibodies, or transfer of CX3CR1-sufficient macrophages.74
Notably, Ly6Chi monocytes, usually the precursors of these resident CX3CR1 macrophages, when recruited to the inflamed gut via the chemokine receptor CCR2, become the dominant cell type during acute colitis.75 In such an inflammatory context, they upregulate toll-like receptor (TLR)2 and NOD2, thereby becoming responsive to bacterial products and developing into proinflammatory effector cells.75 Their ablation indeed ameliorates acute experimental inflammation.75 Interestingly, adoptively grafted Ly6Chi monocytes also acquired over time a cardinal characteristic of DCs, namely migratory behaviour directed towards lymph nodes along with antigen-presenting function to prime naïve T cells.75
In light of this differentiation pathway of monocytes in the experimentally inflamed intestine, it is worthwhile to consider the characteristics of macrophages in the human intestine in health and IBD. In the healthy human gut, intestinal macrophages, which are characterised by lack of CD14 expression,76 are largely refractory to inflammatory stimulation, for example through microbial components, although they retain phagocytic and bactericidal function.77 Moreover, they express anti-inflammatory molecules, such as IL-10, and contribute to the differentiation of Treg cells, while suppressing DC-derived Th1 and Th17 immunity.78 Hence CD14− lamina propria macrophages contribute to protection from invading pathogens while at the same time restraining excess immune responses. However, in CD, another inflammatory macrophage population is present in the intestine, which is characterised by expressing both macrophage and DC markers (CD14, CD33, CD68, CD205, CD209), and which produces large amounts of pro-inflammatory cytokines, such as IL-23, tumour necrosis factor α (TNFα) and IL-6.79 Remarkably, these CD14 macrophages appear to contribute to IFNγ, rather than IL-17 production by lamina propria mononuclear cells, dependent on IL-23 and TNFα.79
Anti-TNFα antibodies in both CD and UC80 are likely targeted to TNFα originating from inflammatory macrophages (and also T cells), which might be related to induction of regulatory macrophages,81 apoptosis of activated T cells and monocytes,82 in addition to TNFα neutralisation. Similarly, it is also worth mentioning that blockade of IL-6 via anti-IL-6R antibodies has shown very promising results in an early phase clinical trial of active CD,83 further underscoring the importance of ‘inflammatory’ myeloid cells in the pathophysiology of IBD. However counterintuitively, macrophages differentiated from peripheral blood of patients with CD exhibit an impaired acute inflammatory response (including TNFα) towards E coli and TLR agonists, associated with decreased neutrophilic infiltration and clearance of killed and labelled E coli injected into the forearm.84
In contrast to primarily phagocytic macrophages, DCs are specialised antigen-presenting cells that can prime naïve T cells and induce their differentiation in inflammatory (eg, Th1, Th17) or Treg phenotypes. DCs accumulate in the mucosa of IBD patients85 and experimental models of colitis.86 Blockade of DC–T cell interaction, for example via blockade of CD40/CD40L engagement, prevents experimental T cell-mediated colitis,86 highlighting the role of DCs in originating intestinal inflammation. Similar to macrophages, several subtypes of DCs have been characterised. CD103 DCs are critical for maintaining gut homeostasis,87 which includes their capacity to promote Treg differentiation.88 The ubiquitin editing enzyme A20, encoded by the Tnfaip3 gene (located at a genetic risk locus of IBD), appears to play a critical role in preserving this homeostatic role of DCs, as genetic deletion of Tnfaip3 specifically in DCs resulted in spontaneously developing lymphocyte-dependent colitis and seronegative ankylosing arthritis.89 In contrast to this generally homeostatic role of CD103 DCs, a DC subtype that expresses E-cadherin (the receptor for CD103) has been shown to promote intestinal inflammation in experimental models.90 E-cadherin+ DCs accumulate in the inflamed colon, are characterised by expression of TLRs, and produce IL-6 and IL-23 on activation.90 Notably, adoptive transfer of this inflammatory DC subset into Rag−/− immunodeficient hosts that are reconstituted with T cells, results in increased Th17 responses, consistent with the role of the aforementioned cytokines to promote Th17 immune responses (see below).90
Inflammatory DCs also play a critical role in the Tbx21−/−Rag2−/− ulcerative colitis (TRUC) model.91 These mice spontaneously develop colitis closely reminiscent of human UC. Specifically, TNFα secreted from CD103 − DCs in TRUC mice potentiated IL-23-induced IL-17 expression in ILCs (see below),92–94 whereby neutralisation of TNFα, IL-23p19 or IL-17A ameliorated disease.91 ,92 The TRUC model is a conceptually interesting model as the host genetic defects render the intestinal microbiota colitogenic, capable of transmitting disease to genetically intact mice91 ,92; a particular proteobacterial species, Helicobacter typhlonius, was discovered to be sufficient to cause and transfer disease.92
Innate lymphoid cells
These recent insights into the mechanisms underlying the TRUC model further pointed to the importance of ILCs, newly identified members of the lymphoid lineage.95–97 ILCs play a fundamental role at the intestinal (and other) barrier surfaces by orchestrating antimicrobial immunity, tissue remodelling and inflammation. ILCs develop from a common lymphoid progenitor similar to B and T cells, the hallmarks of adaptive immunity, but lack antigen receptors generated by somatic recombination.95 Based on their cytokine expression, a nomenclature with three functional groups of ILCs has been proposed.95 All three types of ILCs have essential functions in the intestinal mucosa. Group 1 ILCs produce IFNγ and comprise NK cells and ILC1; group 2 produce Th2 cytokines such as IL-5 and IL-13 and are dependent on transcription factors GATA-binding protein-3 (GATA3) and retinoic acid receptor-related orphan receptor-α (RORα); and group 3 includes all ILC subtypes that produce IL-17 and/or IL-22 and depend on the transcription factor RORγt for their development and function.95 These three subsets broadly parallel the major T helper cell subsets with regard to their signature cytokines (eg, group 1 ILCs, Th1; group 2 ILCs, Th2; and group 3 ILCs, Th17 and Th22 cells), and may also exhibit a similar plasticity (eg, the transcription factor T-bet can induce IFNγ secretion in ILC3s).92 ,98 We will discuss some key aspects of these cells below.
Lymphoid tissue inducer (LTi) cells, assigned to group 3 ILCs, interact with stromal cells and induce lymph node organogenesis, which involves molecules such as lymphotoxin-β (LTβ). In their absence due to genetic deletion of RORγt, lymph nodes and Peyer's patches do not develop,99 and they are also required for the generation of isolated lymphoid follicles in the mucosa which arise from cryptopatches.100
The other major type of group 3 ILCs are NCR+ ILC3 cells, which were almost simultaneously discovered by three groups as a lineage marker-negative cell population in the intestinal mucosa that expresses the pan-NK marker NKp46 and secretes IL-22, and is dependent on RORγt.101–103 IL-22, which acts through the IL-22R whose expression is restricted to epithelial cells, plays a key role in ‘patrolling the border’ by orchestrating epithelial defence through induction of RegIIIβ and RegIIIγ, β-defensins, and other antimicrobial peptides, and by inducing an epithelial wound-healing response on injury caused by bacterial or fungal infection.104 In a similar manner, IL-22 protects from experimental colitis induced by chemical agents breaching the intestinal barrier.103 ,105 These newly discovered NCR+ ILC3s are the predominant source of intestinal IL-22 in mucosal homeostasis.13 ,101 ,102 In contrast to RORγt+ Th17 cells (see below), NCR+ ILC3 develop independent of the commensal flora, and in this way pre-empt colonisation by commensals.13 ,106 In fact, IL-25 expression by IECs induced by the commensal microbiota restrains IL-22-secreting NCR+ ILC3s, only to have them regain full activity on epithelial damage to instigate a protective regenerative response.13 NCR+ ILC3 cells are primarily found in the intestinal tract, located within villi, hence providing the anatomical basis for intimate cross-talk with ICSs.103 ILC3s also play a remarkable and important role in the spatial containment of commensal bacteria.107
In contrast to these primarily homeostatic roles of ILCs, a further subset of group 3 ILCs (NCR − ILC3s) has been demonstrated to promote two types of experimental intestinal inflammation which are induced in Rag2−/− mice that lack adaptive immune cells.93 Specifically, an IL-17-, IL-22- and IFNγ-secreting ILC population, which was activated by IL-23, accumulated in the intestine and was the major driver of these colitis models.93 ,108
There might be plasticity between homeostatic ILC3 and ILC1 cells. ILC1 are grouped together with classic NK cells as group 1 ILCs.95 In response to IL-12 and IL-23 derived from activated DCs and macrophages, a proportion of ILC3 can downregulate RORγt and acquire the capacity to produce IFNγ.108 The transcription factor T-bet (encoded by Tbx21) appears to play an important role for this induction of IFNγ.92 ,98 Importantly, ILCs with similar characteristics as the NCR − ILC3s observed in the murine colitis models accumulated in the colon of patients with CD, but not UC.109 A further study reported an expansion of NKp44 − NKp46+ ILC1-like cells in CD that responded to IL-23 to produce IFNγ, whereas NKp44+NKp46 − ILC3-like cells were decreased.110 Polymorphisms in the gene encoding the receptor chain that confers specificity for IL-23, IL23R, are among the strongest genetic risk factors for both CD and UC and several other immune-related disease conditions.111 ,112 IL-23 receptor binding engages JAK2 and STAT3, both also encoded at genetic risk loci of IBD,112 hence implying signalling downstream of the IL-23R as a key, genetically affected pathway in IBD. Indeed, the IBD-protective R381Q IL23R variant has been reported to result in decreased IL-17 and IL-22 production compared to individuals carrying the wild-type variant.113 While until very recently, IL-23 has been thought to contribute to IBD through enhancement of pathogenic Th17 immunity (see below), it might actually contribute to disease through induction of IFNγ-secreting ILCs.114 Indeed, the colitogenic, IFNγ-secreting ILC subtype described above may have retained its lymphoid tissue-inducing potential,108 thereby positioning it to organise the hyperplastic lymphoid clusters found in IBD.114 ,115
Finally, ILC2 secrete the signature cytokines of UC,21 namely IL-5 and IL-13.116–118 ILC2s are activated by the epithelial-derived cytokine IL-25, and play a particularly important role in anti-helminthic responses. IL-13-secreting ILC2s have been reported in the lamina propria during oxazolone colitis, an experimental colitis model dependent on CD1d-restricted iNKT cells secreting IL-13 as alluded to in the introduction.22 Further studies are needed to characterise ILC2s in human UC, and define their inter-relationship with CD1d-restricted iNKT cells.
Adaptive immune cells in IBD
Loss of tolerance of the adaptive immune system towards the commensal flora has for a long time been proposed as a major converging theme of IBD pathogenesis (figure 2). CD and UC have been associated with exaggerated T cell responses, Th1 (IFNγ) in CD, CD1d-restricted NKT cells (IL-13) in UC,21 ,22 and more recently Th17 (IL-17A, IL-17F, IL-21, IL-22, CXCL8 and G/GM-CSF) cells, primarily in CD.1 ,14 ,119 An elegant recent study demonstrated that induction of a protective, pathogen-specific T cell response is notably accompanied by loss of tolerance towards the commensal flora as an integral part of the mucosal immune response; however, these commensal-specific inflammatory effector and memory cell immune responses did not propagate intestinal inflammation in their own right.120
IL-12, a heterodimeric cytokine consisting of p40 and p35 chains, is critically involved in Th1 differentiation, and its neutralisation in experimental colitis ameliorates disease.121 Likewise, anti-IL-12p40 antibodies (ustekinumab and briakinumab) appear effective in a sub-population of CD patients resistant to anti-TNF treatment,122 ,123 indicative that TNFα and IL-12 may drive distinct biological pathways of immune pathology. Interestingly, though a trial of IFNγ neutralisation with the mAb fontolizumab in active CD has not met its primary endpoint chosen after a single dose, it is noteworthy that longer-term treatment exhibited significant improvement in clinical activity over placebo.124
However, IL-12p40 antibodies not only block IL-12, they also block the biological function of IL-23, which shares the p40 chain with IL-12. IL-23, constitutively secreted from a small population of DCs in the terminal ileum,125 induced by microbial signals, and released in large quantities from CD14 intestinal macrophages in CD patients,79 expands and activates Th17 cells (for a review, see Weaver et al 14), but also has profound effects on innate immune activation as alluded to in the previous section. As noted in the introduction, the microbiota plays an important role in the differentiation and preferential intestinal localisation of Th17 cell, which are characterised by the expression of RORγt.11 ,12 Th17 cells, and a population of IL-17A+IFNγ+ T cells accumulate in the inflamed intestine.126 ,127 Th17 cells may have evolved to combat bacterial and fungal infection via orchestration of the neutrophilic inflammatory response,14 which also explains their localisation in the intestine, both under healthy conditions and on injury. Through secretion of their effector cytokines and chemokines, Th17 cells fulfil important homeostatic functions in the intestine in addition to their role during infections; this may distinguish the intestine from other body locales, such as the brain, joints and skin, where the presence of Th17 cells is primarily associated with pathology.14 It is therefore also not surprising that Th17 effector cytokines have both protective and pathogenic properties, as revealed through the conflicting results obtained from experimental colitis.1 ,128–130 In this context it is notable that the anti-IL-17A antibody secukinumab appears to worsen active CD,131 while the same strategy is highly beneficial in other, genetically and pathophysiologically seemingly related immune diseases.112 ,132 ,133 While much focus has been placed on CD, considering the neutrophil-dominated inflammatory infiltrate in UC47 on the background of IL23R and related genes equally conferring risk for UC, Th17 cells and their effector cytokines might deserve further functional interrogation in human UC.
Finally, Treg cells have an important function in restraining effector T cell populations, but also in restraining innate inflammatory mechanisms.1 This restraining function involves IL-10 and transforming growth factor (TGF) β,134 and T cells from IBD patients appear refractory to TGFβ.135 Treg and Th17 cells are reciprocally regulated in the intestine,136 whereby under inflammatory signalling (eg, IL-6 and IL-23) Th17 cells expand at the expense of Treg cells, thereby promoting effector T cell responses and host defence.1 Loss-of-function mutations in the key Treg transcription factor FOXP3 results in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked),137 which can be accompanied by intestinal inflammation. It might be speculated that the anti-CD25 antibodies basiliximab and daclizumab, which largely failed in clinical trials in active UC,138 ,139 may not only have blocked pathogenic T cell activation as intended, but could also have depleted or functionally impaired Treg cells in the mucosa.
Intestinal homing of T lymphocytes critically involves the α4β7 integrin heterodimer, which is imprinted on T cells via all-trans retinoic acid, a vitamin A metabolite specifically synthesised by gut-associated DCs.140 The binding of α4β7 to its ligand on the endothelium is a critical step in lymphocyte extravasation, and blockade with mAbs targeting the α4 chain (Natalizumab) or the α4β7 heterodimer (Vedolizumab) is therapeutically effective in CD and UC, respectively.141 ,142
Concluding remarks
The mammalian intestine has evolved an astounding, multi-layered cellular interface that mutually interacts with the microbial flora and simultaneously provides the means to gradually respond to pathogens. A syndromic nature of IBD can be conceptualised,47 ,143 where multiple hits1 may impact on each of those layers, gradually exhausting the system's capacity to adapt, leading up to a tipping point, whereupon a self-sustained pathogenic process is set in motion. These hits may involve genetic risk, infectious or toxic injury, pathobionts/opportunistic pathogens, antibiotics and dysbiosis, and exogenous (yet unknown) agents in variable combinations; monogenic disease (eg, IL10RA mutations144) and—perhaps—bona fide yet-unknown infections may represent the extreme ends of the spectrum of this complex, polygenic disease.143 Despite this heterogeneity at the level of originators and perpetuators of disease, the inflammatory mechanisms that are engaged manifest as the two clinically quite distinct and characteristic phenotypes of CD and UC, posing the important question on what drives this distinction and which further biological-mechanistic subtypes of CD and UC may exist. Considering this dichotomy, it is notable that only a tiny fraction of the currently known genetic risk loci of IBD are exclusive for either CD or UC, while the overwhelming majority of genetic risk is shared between both, and, in fact, with other immune-related diseases (eg, 82% of psoriasis risk loci overlap with IBD).112 The differential contribution of smoking as an environmental factor is important, but obviously in no way sufficient to explain the biological differences between CD and UC either; this dichotomy may also be reflected in the variance in responsiveness towards established and novel therapeutics between the two forms of IBD (eg, 5-ASA; tofacitinib145). Further, almost diametrically opposing outcomes of clinical intervention in IBD compared to genetically seemingly similar non-IBD, immune-related diseases (eg, secukinumab in CD and psoriasis131 ,146) highlight the uniqueness of the intestinal immune system (and its immunopathology) as it distinctively caters to the requirements of intense host–microbial engagement. This also reminds us on the quintessential importance of disease-specific studies to reveal underlying patho-mechanisms and therapeutic opportunities.
Summary
-
The intestinal immune system has co-evolved with the microbiota, which is required for its normal development and function.
-
The mucosal immune system exhibits substantial plasticity affected by various factors, including the diet.
-
Perturbations in intestinal epithelial cells, in particular Paneth cells, are related to important genetic risk factors of inflammatory bowel disease (IBD) and can originate intestinal inflammation.
-
Intestinal macrophages have an important homeostatic and inflammation-restraining role, although a particular subtype can potently promote inflammation in IBD.
-
Dendritic cells can also both restrain and originate intestinal inflammation by activating T cells and secreting cytokine mediators.
-
Innate lymphoid cells (ILCs) resemble lymphocytes, but lack the key characteristics of adaptive immune cells, namely an antigen-specific receptor.
-
ILCs interact closely with IECs and fulfil an important role in early defence including wound healing, but can also differentiate into potent inflammatory effectors.
-
Antigen-specific T helper and NKT cells, homeostatically restrained by Treg cells, contribute to intestinal inflammation and their influx can be therapeutically targeted.
Acknowledgments
Work in the Kaser lab is supported by the European Research Council under the European Community's Seventh Framework Programme (FP7/2007–2013)/ERC Grant agreement no. 260961 (AK), and the National Institute for Health Research Cambridge Biomedical Research Centre (AK). ZC is supported by the Cambridge Institute for Medical Research (CIMR) Wellcome Trust PhD Programme for Clinicians.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.