Article Text

Abstract
The central nervous system interacts dynamically with the immune system to modulate inflammation through humoral and neural pathways. Recently, in animal models of sepsis, the vagus nerve (VN) has been proposed to play a crucial role in the regulation of the immune response, also referred to as the cholinergic anti-inflammatory pathway. The VN, through release of acetylcholine, dampens immune cell activation by interacting with α-7 nicotinic acetylcholine receptors. Recent evidence suggests that the vagal innervation of the gastrointestinal tract also plays a major role controlling intestinal immune activation. Indeed, VN electrical stimulation potently reduces intestinal inflammation restoring intestinal homeostasis, whereas vagotomy has the reverse effect. In this review, we will discuss the current understanding concerning the mechanisms and effects involved in the cholinergic anti-inflammatory pathway in the gastrointestinal tract. Deeper investigation on this counter-regulatory neuroimmune mechanism will provide new insights in the cross-talk between the nervous and immune system leading to the identification of new therapeutic targets to treat intestinal immune disease.
- Gut Immunology
- Enteric Nervous System
- Gastrointestinal Immune Response
- Neuroimmunology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Accumulating evidence supports the idea that an intricate communication network exists between the nervous and immune systems, and that this crosstalk could play a crucial role in the regulation of the immune response.1 The interplay between those diverse systems occurs through a complex set of neurotransmitters, cytokines and hormones that act as counter-regulatory mechanisms able to dampen inflammation and restore homeostasis.1 ,2 On a systemic level, neuroendocrine mechanisms reduce inflammation by the hypothalamic-pituitary-adrenal (HPA) axis through the anti-inflammatory effect of glucocorticoids, by the hypothalamic-pituitary-gonadal axis through sex hormones, and by the hypothalamic-pituitary-thyroid hormone axis through thyroid hormones.3 ,4 Although modulation of the immune system by the nervous system, in particular the adrenergic nervous system, has been introduced decades ago,5 interest in the role of the autonomic nervous system as a key player in immune homeostasis has recently increased exponentially. In 2000, Tracey and coworkers demonstrated that vagus nerve (VN) stimulation potently suppresses cytokine production in a rodent model of sepsis.6 This discovery has led to the introduction of the concept of the cholinergic anti-inflammatory pathway,7 a hard-wired connection between the immune and nervous systems closely interacting to regulate inflammation. It is currently supposed that inflammatory mediators activate sensory nerves and send signals concerning the state of the inflammation to the central nervous system. The latter, through efferent nerves, releases neuromediators that influence immune cells and modulates local inflammation.8 Consequently, it is now clear that the nervous system is able to regulate inflammation in peripheral tissues and to restore local immune homeostasis.
In the present review, the current knowledge and the clinical implication of the intestinal cholinergic anti-inflammatory pathway will be discussed. Readers interested in the sympathetic modulation of the immune response are referred to excellent reviews on this topic.9–11
The cholinergic anti-inflammatory pathway
While studying the anti-inflammatory effect of the inhibitor of p38 MAP kinase, CNI-1493, it became clear that this compound suppressed carrageenan-induced paw oedema at doses at least 6-logs lower when injected intracerebroventricular than required for a systemic effect.12 This potent anti-inflammatory effect was abrogated after bilateral vagotomy. Conversely, recording of the efferent VN electrical activity revealed an increase in discharge rate after infusion of CNI-1493, suggesting anti-inflammatory properties of VN activation. Similarly, electrical stimulation of the transected peripheral VN for 20 min prevented the development of an acute inflammation in response to carrageenan injection in the paw and increased survival in a model of sepsis6 by reducing cytokine (tumor necrosis factor (TNF)) production of splenic macrophages. This anti-inflammatory effect could be reproduced in vitro using isolated human macrophage cultures; the release of TNF, interleukin (IL)-1β, IL-6 and IL-18 in response to endotoxin was significantly reduced by acetylcholine (ACh) and nicotine. In a search to pharmacologically mimic the effect of VN stimulation, Wang et al identified the α7 subtype of the nicotinic acetylcholine receptor (α7nAChR) as the main receptor by which splenic macrophages are modulated.13 The anti-inflammatory effect of VN stimulation is lost in α7nAChR knock-out mice, can be blocked by specific antagonists α7nAChR, and is mimicked both in vivo and in vitro by α7nAChR agonists.13 Based on these findings, the ‘cholinergic anti-inflammatory pathway’ was introduced, whereby the VN modulates the immune response in the spleen providing an additional protective mechanism to the host (figure 1). This mechanism protects against the lethal effects of cytokines by restraining the magnitude of a potentially fatal peripheral immune response.6–8
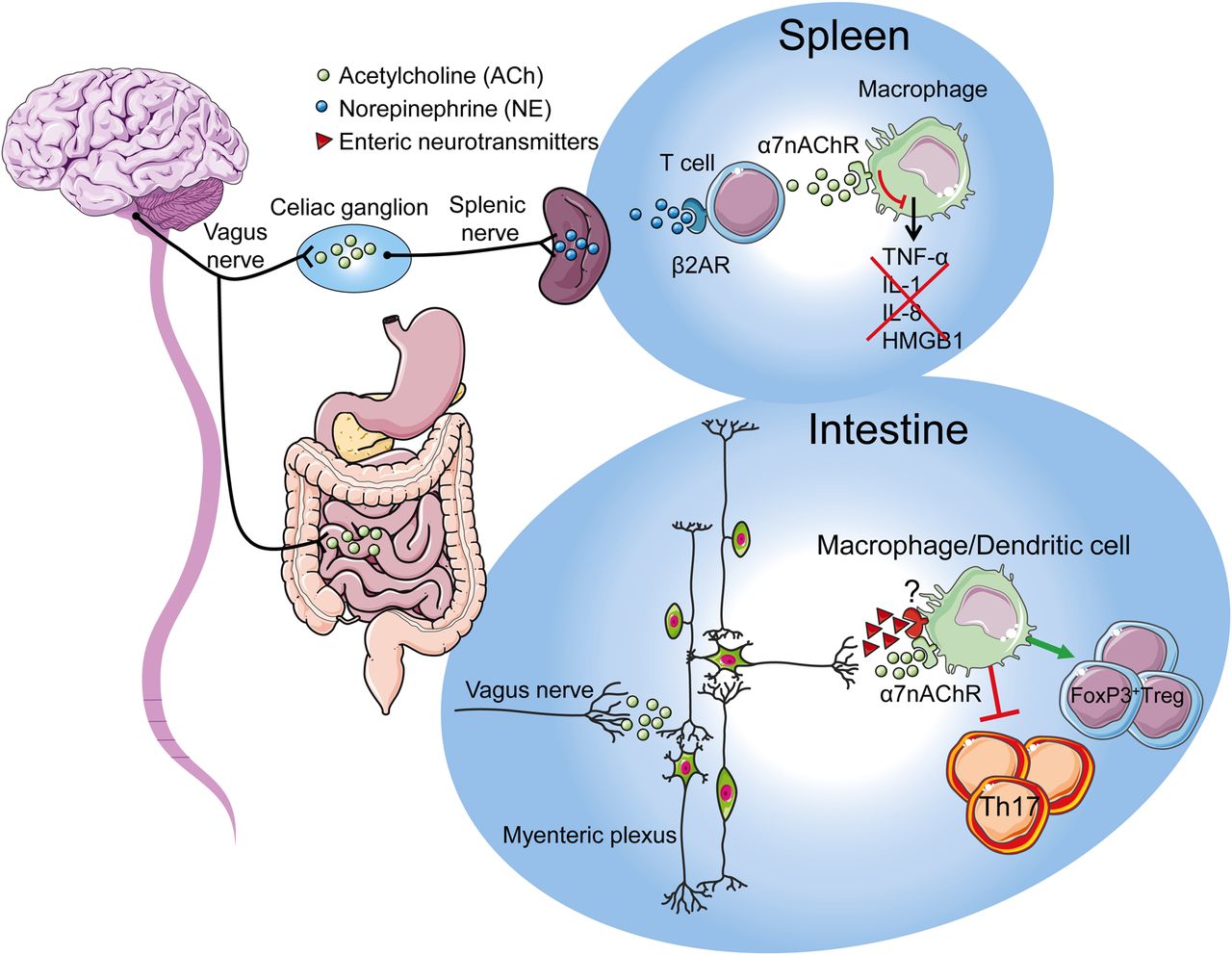
Schematic representation of the cholinergic anti-inflammatory pathway. During systemic inflammation, the central nervous system is activated via the circumventricular organs and by vagal afferents. After integration, this sensory input will trigger the efferent arm via the coeliac superior mesenteric ganglion modulating the immune response in the spleen. The activation of adrenergic neurones in the spleen leads to the release of norepinephrine that induces the release of acetylcholine by a subset of T cells. Acetylcholine interacts with α7nAChR expressed on cytokine producing macrophages reducing the release of tumour necrosis factor, IL-1, IL-18, HMGB1, and other cytokines. During intestinal inflammation the central nervous system is activated via the vagal afferents arm which will trigger activation of the vagal efferent arm directly contacting the myenteric neurones in the intestinal wall. Activation of enteric neurones induces the release of acetylcholine and possibly of other immune-modulatory neurotransmitters leading to the control of intestinal inflammation and restoring intestinal immune homeostasis.
The anti-inflammatory reflex
In view of its potent anti-inflammatory properties, the cholinergic anti-inflammatory pathway has been forwarded as an additional endogenous mechanism to regulate the immune system. In comparison with the HPA axis or the local production of anti-inflammatory cytokines, the cholinergic control seems to have several properties favouring a central role in immune homeostasis. Considering the speed of neural conductance, it is capable of providing an instantaneous modulatory input to the region of inflammation. Moreover, as the nervous system can adapt its output based on information obtained from various parts of the host, the modulatory influence of the cholinergic anti-inflammatory pathway is not only fast, but integrated with respect to the general well-being of the host. The latter, however, implies that the central nervous system receives ‘sensory’ input on the immune status of the periphery, similar to the afferent limb of a neural reflex, in order to fulfil its modulatory role.
The afferent limb of the vagal anti-inflammatory pathway
In case of systemic inflammation, cytokines and/or endotoxin are present in detectable amounts in the circulation. Animal studies have clearly shown that their presence is sensed by the brain at the level of the circumventricular organs (CVO), that is, the area postrema, the organum vasculosum of the lamina terminalis, the subfornical organ, and at the level of the area postrema. Importantly, these areas are characterised by a leaky blood-brain barrier where mediators in the blood are detected by specialised neurones. Indeed, intravenous injection of endotoxin induces signs of neural activation revealing neurones containing c-Fos in the CVO.14 As these areas project to the autonomic motor neurones in the brain stem connected to the spleen, one can postulate that systemic inflammation with circulating cytokines is detected by the brain subsequently activating the cholinergic input to the spleen, thereby modulating the immune response.
The physiological role of the neuromodulation of the spleen (immune system) has been compared with the autonomic regulation of homeostasis as demonstrated in other organs—for example, the heart.8 It can be postulated that the neural input to the spleen determines the set point for the magnitude of the innate immune response towards pathogens or tissue damage. Decreased or absent neural input will increase the set point with exaggerated proinflammatory responses and tissue damage, whereas increased input results in inadequate immune defence. As part of this concept, the ‘tone’ of the nervous input to the immune system may thus have a major impact of the host defence and development of disease.
In the case of localised peripheral inflammation, cytokine levels are absent or too low to activate the CVO. Yet, the brain is informed on the presence of inflammation through cytokine (IL-1) receptors on vagal afferents and glomus cells adjacent to the VN.15–18 Brains of mice infected with Campylobacter jejuni, for instance, revealed activation of visceral sensory nuclei in the brain stem (nucleus of the solitary tract and the lateral parabrachial nucleus) even though cytokines (TNF, IL-6, IL-1β) could not be detected in the serum.19 Similarly, we found activation of the nucleus of the solitary tract in response to intestinal inflammation evoked by manipulation of the intestine.20 Interestingly, motor neurones of the dorsal nucleus of the VN directly connected to the inflamed area were also activated compatible with the existence of a hard-wired inflammatory reflex.20 In addition, the afferent limb can also be activated by enteral feeding of a high-fat diet.21 Indeed, recent studies indicated that high-fat feeding is protective in an ischaemia-reperfusion model by activation of the cholinergic anti-inflammatory pathway, an effect mediated by activation of vagal afferents by fat-induced release of the neuropeptide cholecystokinin.22 ,23
The efferent limb of the vagal anti-inflammatory pathway
It is generally accepted that the VN densely innervates the gastrointestinal tract. The nerve fibres innervating the upper and middle gastrointestinal tract originate from two nuclei in the brain stem; the nucleus ambiguus (reaching the larynx and oesophagus),24 ,25 and the dorsal motor nucleus of the vagus (DMV) (reaching the stomach until the descending colon),26 ,27 while distal colon, rectum and the anal canal receive input from the sacral cord.28–30 By means of anterograde tracers injected into the DMV, efferent vagal nerve terminals were shown to directly synapse with postganglionic neurones located in the enteric nervous system, rather than interacting with neurones in the prevertebral ganglia.26 ,31 Using the same experimental approach, a typical rostrocaudal gradient of vagal preganglionic innervation has been reported with the highest density of innervated enteric neurones observed in the stomach followed by a subsequent decrease in the small bowel and colon.31
As the spleen is the major source of TNF and HMGB1, two key cytokines in the pathophysiology of sepsis,32–34 it represents an ideal target for neural modulation of the immune response. Initial experiments indeed indicated the spleen as target organ of the VN during sepsis.32 The anatomy of the splenic innervation, in particular the cholinergic or vagal component, is however still a matter of debate. Close interposition of immune cells with nerve terminals could be identified, but these fibres were adrenergic in nature. Although these data indicate adrenergic modulation of splenic macrophages, in vitro data strongly suggest cholinergic inhibition of splenic macrophages via α7nAChRs. By contrast with the initial hypothesis proposing direct contact between vagal nerve fibres and splenic macrophages,7 it is now clear that in the spleen the VN rather indirectly modulates the innate immune response by activating the adrenergic neurones in the prevertebral ganglia. In line with this hypothesis, only in mice with an intact and innervated spleen, VN stimulation is able to exert its anti-inflammatory effect.32 Recent studies suggest that ACh released by the VN in the celiac mesenteric ganglia activates postsynaptic α7nAChR on adrenergic neurones of the splenic nerve, leading to the release of norepinephrine in the spleen.35 There, these adrenergic nerve fibres stimulate ACh synthesis by splenic T cells interacting with α7nAChR located on adjacent macrophages.35
The intestinal immune system and the cholinergic anti-inflammatory pathway
The mucosal immune system is constantly challenged by food antigens and by the intestinal microbiota requiring a perfectly balanced equilibrium between tolerance and defense.36 Decreased tolerance to microbiota is proposed as a main pathogenetic mechanism in inflammatory bowel disease (Crohn's disease and ulcerative colitis) characterised by severe inflammation of the intestinal mucosa, whereas, decreased tolerance to food antigens may lead to food allergy. In both cases, the immune system over-reacts to innocent antigens leading to severe tissue damage, morbidity and mortality. It is becoming increasingly clear that the microenvironment in the mucosa and submucosa determines the immune response to the initial exposure of foreign antigens. Given the potent anti-inflammatory effect of the cholinergic innervation, one might assume that the cholinergic tone in the submucosal compartment may have an important impact on mucosal immune homeostasis.
Indeed, studies in vagotomised mice revealed increased susceptibility to develop colitis after exposure to the mucosal irritant dextran sulphate sodium (DSS).37–39 Notably, reduced mucosal levels of ACh in a murine model of depression were also associated with a more severe colitis in response to DSS.38 ,40 As adoptive transfer of macrophages from depressive mice induced a similar increased susceptibility to develop DSS colitis,41 macrophages were proposed to be (one of) the target cells modulated by the cholinergic tone in the submucosal microenvironment. Obviously, other immune cells (T cells, dendritic cells (DC), mast cells) residing in the mucosa/submucosa carry nicotinic receptors and may be affected by the anti-inflammatory pathway as well.
Another important but less studied population of intestinal immune cells are the macrophages located between the longitudinal and circular muscle layer at the level of the myenteric plexus. These resident macrophages play an important role in diabetic-induced gastroparesis,42 ,43 postoperative ileus (POI),44–46 LPS-induced septic ileus,47–50 and seem to represent the gatekeepers of the enteric nervous system, or the ‘little brain of the gut’. VN stimulation prevents muscular inflammation and ileus following intestinal manipulation,51 suggesting that this subpopulation of macrophages is also under cholinergic control. By determining the cholinergic tone in the enteric nervous system, the vagal innervation may modulate the intestinal microenvironment (figure 2). Hence, the set point (balance) of the gut immune system will be affected influencing not only macrophages or DCs, but theoretically any immune cell carrying cholinergic receptors, that is, T and B lymphocytes, monocytes and mast cells.52 Alternatively, mesenteric lymph nodes or the spleen may be the site of neuroimmune interaction, although splenic denervation did not abolish the anti-inflammatory effect in our model of POI.53
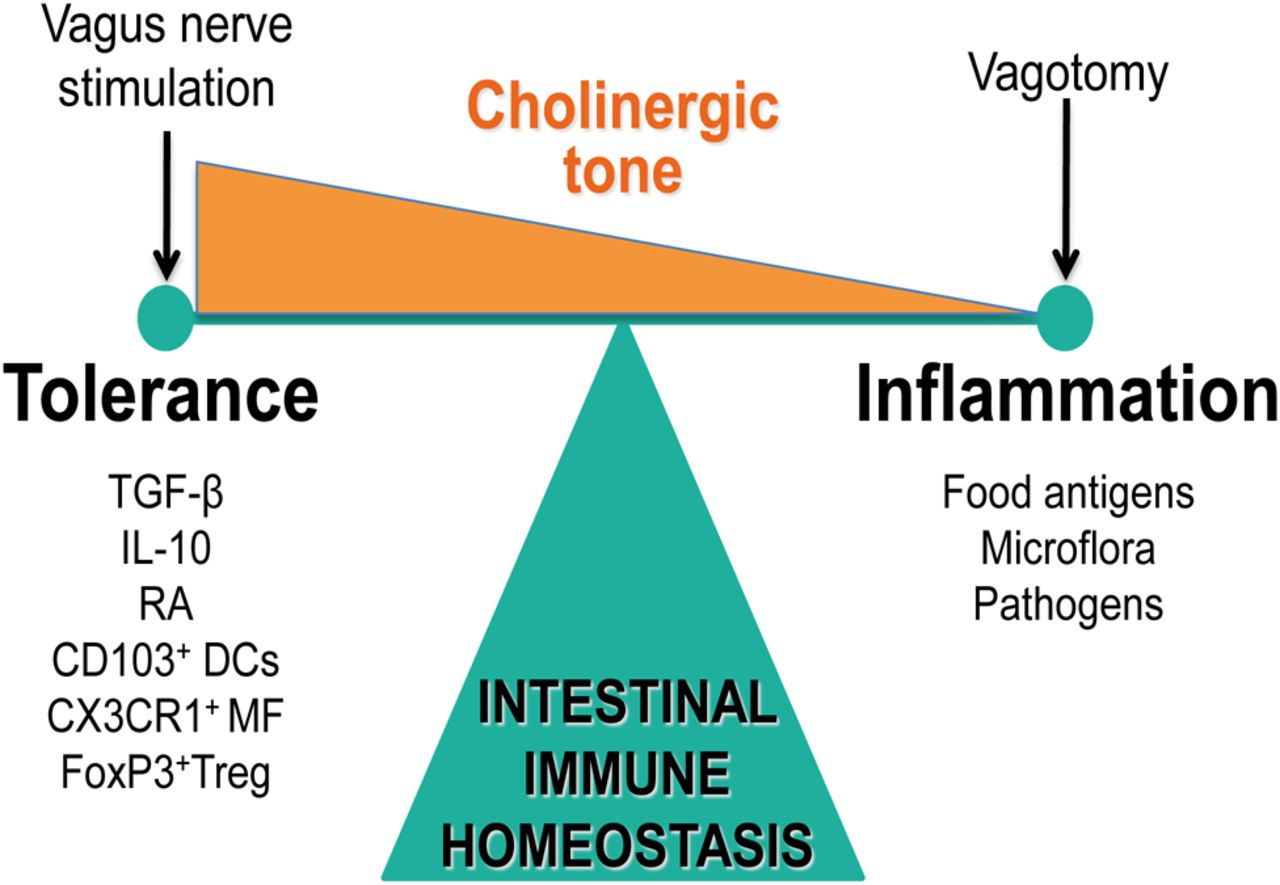
Cholinergic (vagal) tone and intestinal immune homeostasis. The cholinergic tone determines immune homeostasis either shifting the balance towards tolerance (normal to enhanced tone) or inflammation (decreased tone).
Similar to the situation in the spleen, immune cells in the gut wall will most likely be indirectly modulated by the VN as vagal efferents mainly, if not solely, synapse with enteric neurones. The latter would implicate that enteric neurones rather than vagal nerve endings interact with the intestinal immune system. The enteric nervous system forms a dense network of nerve fibres in close vicinity with intestinal immune cells, both in the submucosal (lamina propria) and muscular externa compartment of the intestine.54 This could imply that vagal signals are amplified by the enteric nervous system inducing the substantial release of ACh in the intestinal microenvironment leading to modulation of the immune response (figure 1). However, the possible release of other immune modulatory neurotransmitters by enteric neurones cannot be excluded. Indeed, several neurotransmitters, such as vasoactive intestinal peptide (VIP), glutamate and even nitric oxide have been shown to modulate immune cells.55–57
Clinical significance of the cholinergic anti-inflammatory pathway
In general, the significance of new discoveries in medicine is largely determined by its impact on clinical management. Accepting the general principle that the brain, through the VN, fulfils an important role in modulating the immune system, clearly the next important question is how this knowledge can be translated into improved clinical management of immune-mediated inflammatory disease. Preclinical models have provided a plethora of evidence supporting a beneficial effect of activation or mimicry of the cholinergic anti-inflammatory pathway in a wide variety of disorders. Most studies reported to date focus on acute innate responses triggered by ischaemia, tissue damage or bacterial products (endotoxin), that is, conditions involving sterile inflammation. Before discussing these in more detail, it is of utmost importance to stress that dampening the immune response may have detrimental effects in case of infectious diseases. Although VN stimulation and nicotine increase intestinal macrophage phagocytosis,58 reduced cytokine and chemokine production leads to impaired attraction and activation of neutrophils and T lymphocytes impairing bacterial clearance. In a model of bacterial peritonitis, α7nAChR knock-out mice efficiently cleared the infection with Escherichia coli from their peritoneal cavity and had sterile blood cultures, whereas, wild-type animals had high bacterial loads at the primary site of infection and were bacteremic.59 The knock-out animals displayed a more robust influx of neutrophils early after the infection, resulting in improved bacterial clearance, as evidenced by the much lower bacterial burden in liver, spleen, kidney and lungs. Similarly, impaired survival was also observed after nicotine treatment in septic peritonitis induced by intraperitoneal injection of E coli or fecal contamination.60 ,61 Also in a model of colitis, selective α7nAChR agonists were reported to worsen disease,62 again illustrating that dampening cytokine production is not always beneficial to the host, especially in infectious conditions.
Finally, although so far most studies focused on the innate immune system, modulation of the adaptive immune response is to be anticipated, especially as the DCs and macrophages are the key players initiating this process. One study elegantly demonstrated that antibody production against trinitrophenyl-ovalbumin was dependent on vagal innervation indeed suggesting that the adaptive immune system is under cholinergic control.14 If future studies confirm this, the therapeutic and clinical impact may be even larger than assumed so far. Preclinical evidence supporting the potential therapeutic effect of the cholinergic anti-inflammatory pathway has been recently reviewed in great detail.63 ,64 Here, we will summarise data relevant to major intestinal immune-mediated inflammatory diseases and clinical practice.
Inflammatory bowel disease
Inflammatory bowel disease (IBD) is a debilitating and chronic inflammatory disease affecting the gastrointestinal tract.65 Based on clinical presentation, endoscopic appearance and histology, two major subtypes of the disease have been identified, that is, Crohn's disease with transmural inflammation versus ulcerative colitis which involves mostly superficial inflammation confined to the mucosa. In the past decade, treatment of IBD has improved significantly with the introduction of biologicals, such as anti-TNF antibodies.66 ,67 Nevertheless, there is much room for improvement as a significant proportion of patients still requires intestinal resection for intractable disease or complications, such as abscesses, stricture formation or fistulae.
Preclinical models provide convincing evidence that the cholinergic innervation of the gut has a major impact on the intestinal immune system. Vagotomised animals develop more severe colitis (both microscopic and macroscopic) with increased levels of NF-κB and cytokines, such as IL-1β, IL-6 and TNF-α following DSS administration,37 ,39 ,40 an effect that was reversed by pretreatment with nicotine. In the same line, the acetylcholine-esterase inhibitors neostigmine or physostigmine attenuated colitis evoked by intrarectal dinitrobenzene sulphonic acid.68 As DSS colitis was more severe in α7nAChR KO mice, and the α7nAChR agonist choline chloride improved macroscopic colitis, reduces myeloperoxidase (MPO) activity, IL-6 and IL-1β levels,40 α7nAChR receptors were identified as the cholinergic receptors mediating the anti-inflammatory effect. However, also α5nAChR knockout mice subjected to experimental colitis showed significant worsening of disease parameters compared with wild-type mice,69 suggesting that the α7nAchR and also other nAChR subunits may participate in the vagus modulation of intestinal inflammation in mice.
In clinical practice, exacerbations of colitis, especially of ulcerative colitis, may coincide with episodes of depression or psychological stress. Interestingly, increased susceptibility to develop colitis was also observed in an animal model of depression (maternal separation), a phenomenon that was associated with a reduced level of ACh in the intestine and could be reversed by treatment with an antidepressant.38 ,40 The beneficial effect of the antidepressant was eliminated by vagotomy, and most interestingly normalised the intestinal ACh levels. The exact underlying mechanism is not completely understood, but increased susceptibility to colitis can be transferred to a recipient mouse by transfusing macrophages isolated from mice with depression-like behaviour.41 These data would suggest that similar to sepsis, macrophages are the target cell of the cholinergic anti-inflammatory pathway. Another study, however, provided evidence that CD4+ T lymphocytes may be involved. Transfer of CD4+CD25− T lymphocytes isolated from vagotomised animals resulted in increased susceptibility to develop DSS colitis.37 Clearly, more work is definitely needed. Nevertheless, the risk of developing colitis seems to depend on the intestinal cholinergic tone due to its impact on the intestinal immune system. Of significance to the human situation, depression and other psychological conditions, such as stress and anxiety, may trigger exacerbations by reducing this cholinergic tone.
Conversely, the differential effect of smoking on the disease course of Crohn's and ulcerative colitis patients has intrigued clinicians for decades. Interestingly, smoking has a detrimental effect on Crohn's disease increasing the risk of relapses, repeat surgeries, and the need for more aggressive immunosuppressive treatment.70–72 On the contrary, smoking seems to reduce disease severity in ulcerative colitis patients, diminishing flares and hospitalisation rates, overall leading to better disease course in smokers than in non-smokers.73–75 In addition, many ulcerative colitis patients noted disease exacerbation upon cessation of smoking, whereas symptoms relief is reported when smoking is resumed.76 Although there is no clear explanation for this difference, a recent report using two models of murine colitis mimicking Th1 or Th2 type of inflammation has shown that the divergent effect of nicotine may be explained by the upregulation of the α7nAChR by a Th2 inflammatory response (ulcerative colitis) but not by a typical Th1/Th17 inflammatory response.77 Assuming that the therapeutic response of nicotine (or smoking) is mediated by α7nAChR, these findings may explain the differential effect of smoking in Crohn's versus ulcerative colitis patients. To what extent this difference in α7nAChR expression also applies to IBD patients remains to be investigated.
Based on the beneficial effect of smoking, therapeutic trials have been initiated evaluating the effect of nicotine enemas in patients with ulcerative colitis, however, with controversial results.78–80 Most likely nicotine side effects, due to interaction with all nAChR subtypes, prevent effective dosing in patients. In view of the side effects, more ‘specific’ α7nAChR agonists have been developed. For example, choline effectively improved DSS colitis and reduced cytokine production.40 In a model of TNBS colitis, mice treated with the α7nAChR ‘preferring’ agonist, anabaseine, developed less weight loss and less severe colitis and lower levels of MPO, NF-κB and TNF.81 Taken together, these data would favour α7nAChR agonists as treatment for IBD, however, we obtained opposite results using the specific α7nAChR agonists AR-R17779 and GSK1345038A. Although both agonists reduced NF-κB transcriptional activity, IL-6 and TNF release by activated peritoneal macrophages, clinical parameters (body weight, colon weight and length), IL-6 and IL-17 levels were not augmented in vivo in two different colitis models; that is, DSS and TNBS-induced colitis.62 Most likely, the protective effect of α7nAChR activation against excessive inflammation and collateral tissue damage compromises the host defence against the increased exposure to intraluminal microbiota following mucosal damage, suggesting that care should be taken when using these compounds in patients with IBD.
Postoperative and endotoxin-induced ileus
Each abdominal surgical intervention leads to impaired motility of the entire gastrointestinal tract lasting several days with symptoms such as nausea, vomiting, intolerance to food and constipation, referred to as POI. Although some would argue that this represents a physiological response to the surgical insult and should not be regarded as a clinical problem, this iatrogenic condition is a major source of patient morbidity and a significant economic burden to healthcare.
Since more than a decade, a subtle microscopic inflammation of the intestinal muscularis, triggered by activation of resident macrophages residing between the longitudinal and circular muscle layer of the intestinal wall, has been identified as key process in the pathophysiology of POI and endotoxin-induced ileus.45 ,48 ,82 Activation of these phagocytes results in cytokine and chemokine release, followed by influx of mainly leukocytes and monocytes starting at approximately 3–4 h after surgery. As this inflammatory response has a major impact on neuromuscular function, compounds or interventions hampering this process may be instrumental in reducing POI.46
Several lines of preclinical evidence indicate that activation of the cholinergic anti-inflammatory pathway may be an efficient strategy to prevent POI (figure 3).83 VN stimulation via electrical current,51 high-fat enteral feeding84 or central application of semapimod,85 as well as peripheral α7nAChR activation via AR-R1777983 all resulted in reduction of influx of immune cells into the muscularis, dampened cytokine production and improved gastrointestinal transit. In line with the findings reported in sepsis,13 the anti-inflammatory effect of electrical VN stimulation is lost in α7nAChR-deficient mice,53 confirming the involvement of α7nAChR also in the gut. Though in contrast with other preclinical models, vagal modulation of muscularis-resident macrophages in POI results from a direct input to the gut, as selective denervation of the spleen left the modulatory effect of the VN unaffected.53 Moreover, local intestinal blockade of nicotine receptors via incubation of the intestine in a solution containing hexamethomium abolished the effect of VN stimulation51 further arguing against the spleen as site of neuromodulation, at least in this model of subtle intestinal inflammation.

{kind=link}
{kind=link}
{kind=link}
Electrical vagal nerve stimulation (EVNS) and α7-selective agonists ameliorate intestinal inflammation in a murine model of postoperative ileus. (A) Gastric retention of a semiliquid test meal in mice that had undergone laparotomy (L, empty bar) or intestinal manipulation (IM, black bar) 24 h previously after sham treatment (sham), EVNS, nicotine or the α7-selective agonist AR-R17779 (AR-R). (B) Quantitative analysis of IM-induced inflammatory cell infiltrates 24 h after indicated treatment and surgery. Reproduced with permission from The FO et al Gastroenterology, Volume 133, Issue 4, October 2007, pp. 1219–1228.83
Other GI disorders
Early loss of intestinal integrity is a common complication in polytraumatised, ischaemic or septic patients.86 Leaky gut syndrome is leading to the translocation of microbial products in the systemic circulation further aggravating the prognostic outcome.87 In several experimental settings, mimicking the loss of intestinal barrier activation of the cholinergic anti-inflammatory pathway has been reported to efficiently reduce both systemic inflammation, to limit loss of barrier function, and to reduce intestinal damage.88 ,89 Interestingly, as described above, enteral feeding with high-fat diet resulted in the activation of the cholinergic anti-inflammatory pathway reducing systemic inflammation and supporting gut barrier function in a rat model of haemorrhagic shock.21 Also in pancreatitis, the cholinergic anti-inflammatory pathway has been proven to play a crucial role also in reducing plasma hydrolases, IL-6 levels, and neutrophils infiltration.90 Interestingly, the VN has been proven also to protect the liver in a mouse model of Fas-induced hepatitis.91 Indeed, vagotomy significantly worsens the disease-increased mortality and apoptosis. On the contrary, pretreatment with nicotine, or with an α7nAChR agonist, protects mice from the detrimental effect of vagotomy supporting the idea that the vagal innervation controls hepatic inflammation.91
Human evidence
By now it is widely accepted that immune cells carry neurotransmitter receptors allowing communication between the immune and nervous systems. Human peripheral blood mononuclear cells, B and T cells, macrophages, DCs and synovial fibroblasts express a variety of cholinergic muscarinic and nicotinic receptors.92–96 From the initial in vitro work by Tracey and coworkers, we know that cytokine release by human macrophages is reduced by nicotine via the α7nAChR.13 Not only cytokine production, but also T cell skewing by DCs and B cell antibody production are affected by cholinergic input. The critical question clearly is to what extent modulation of the immune system via these receptors affects human physiology and disease.
Accepting that the VN has a major impact on the immune system through the spleen, one would anticipate increased incidence of inflammatory conditions in patients who underwent vagotomy, for instance, as treatment for peptic ulcer disease (before introduction of Helicobacter pylori eradication) or during esophagectomy. To date, no data are available supporting this. In the gastrointestinal tract, cholinergic tone, however, may be restored by compensatory increase within the enteric nervous system, possibly explaining the normalisation of the susceptibility to DSS colitis after several weeks in vagotomised mice.97 To what extent similar compensatory mechanisms are activated in other regions of the body remains to be studied. Splenectomy, on the other hand, is associated with an increased risk for potentially lethal bacterial infections, but this seems to be more related to the clearance of encapsulated bacteria in the spleen.
There is, however, increasing indirect evidence supporting an immune-modulatory role of the cholinergic anti-inflammatory pathway in humans. By using heart rate variability, several studies have evaluated the relationship between vagal tone and the activation status of the immune system (reviewed in Huston and Tracey 201198). Heart rate variability is the time difference between successive heart beats, and determined by the balance between cholinergic (parasympathetic) and sympathetic input to the heart. In healthy subjects, reduced heart rate variability indices (=low vagal tone) are independently associated with increased CRP and IL-6 levels.99–101 In the same line, low vagal tone was reported to be associated with impaired stress recovery of TNF levels, although these changes were very subtle.102 Similarly, TNF and IL-6 production by whole blood stimulated with endotoxin in healthy subjects was associated with vagal activity assessed by heart rate variability.103
In addition, in immune-mediated diseases like rheumatoid arthritis (RA), VN activity was reduced compared with healthy controls and associated with increased levels of serum HMGB1.104 Similarly, the majority of studies in patients with cardiovascular disease report that parasympathetic tone, as inferred from heart rate variability, is inversely related to inflammatory markers (IL-6 and CRP).105 ,106 Finally, increased morbidity and mortality following cardiac surgery, myocardial infarction, sepsis, RA, IBD, lupus erythematosus and sarcoidosis have been reported to be associated with decreased VN activity.98 Whereas these data suggest that reduced vagal tone increases the setpoint of the immune response with a subtle rise in proinflammatory cytokines and increased disease risk, increased vagal tone resulting from elevated intracranial pressure has been proposed as mechanism underlying the observed immune paralysis in patients with traumatic brain injury.107 Taken together, these data provide indirect evidence that basal vagal tone may represent an important determinant of the risk to develop or have progression of disease. The finding that exercise or dietary interventions, such as fish oil supplementation to enhance vagal tone may partly explain their beneficial effect on general health.108 ,109
The final evidence that the vagal anti-inflammatory pathway is indeed a breakthrough for the clinical management of immune-mediated inflammatory disease will ultimately have to come from clinical trials evaluating the different strategies discussed above. Selective α7nAChR agonists are currently evaluated as potential therapeutic drugs for the treatment of cognitive impairments in schizophrenia and Alzheimer's disease.110–114 To date, however, only a very limited number of studies have been performed to evaluate their anti-inflammatory potential in humans. Based on the observation that the selective α7nAChR agonist GTS-21 attenuated cytokine production by stimulated whole blood and human monocytes more potently than nicotine,115 ,116 its effect was tested in a human endotoxin model. However, no significant reduction in cytokine response to endotoxin injection in healthy subjects was observed between GTS-21 and placebo-treated subjects.117 Clinical studies evaluating the effect of specific α7nAChR are therefore awaited with great interest. Alternatively, more physiological activation of the cholinergic anti-inflammatory pathway using enteral feeding of a high fat solution may represent an interesting approach, for example, to prevent POI.
Finally, electrical stimulation of the VN, either in the neck or the ear, may be considered as treatment for chronic inflammatory diseases such as IBD and RA. Electrical stimulation of the VN is already an established therapeutic option used for intractable epilepsy and treatment-resistant depression. In these clinical settings, several groups have failed to detect a clear effect of vagal stimulation on serum levels of inflammatory cytokines.118–120 However, in epileptic patients, intraoperative or chronic electrical vagal nerve stimulation were able to significantly inhibit TNF-α release in whole blood samples treated with lipopolysaccharide in vitro.121 One possible explanation could be that side effects due to activation of the recurrent laryngeal nerve may hamper adequate nerve stimulation to achieve the anti-inflammatory effect of VN stimulation. Nevertheless, clinical trials in patients with RA and Crohn's disease (ClinicalTrials.gov Identifier: NCT01552941; ClinicalTrials.gov identifier: NCT01569503) will start soon, hopefully providing the first proof-of-concept study that activation of the cholinergic anti-inflammatory pathway is indeed a powerful new therapeutic tool.
Conclusion and perspective
Communication between the autonomic nervous system, in particular of the parasympathetic part, is increasingly considered to have a major impact on the immune system. The cross-talk between nerves and immune cells may be of great importance for the maintenance of immune homeostasis, thereby controlling locally inflammatory response and preventing collateral damage or disseminated disease. Nowadays, it is becoming increasingly clear that the microenvironment in the intestine largely determines the phenotype of the immune cells residing in the muscular or submucosal compartment. Mainly due to the release of mediators, such as retinoic acid, TGF-β and thymic stromal lymphopoietin by the enterocytes and/or immune cells, a unique and predominantly tolerogenic microenvironment is created in the submucosa, setting the stage for maintenance of immune homeostasis.122 Most likely, in the gut, the VN communicates with immune cells increasing the cholinergic tone, thus reducing mucosal inflammation and driving innate immune cells into a tolerogenic phenotype.
Although the mechanisms involved are gradually unravelled, the exact anatomy, and the molecular pathways involved in the cholinergic modulation of immune cells, remains a matter of debate. Overall, cholinergic activation can reduce inflammation and disease activity in various animal models of intestinal inflammation, likely via a mechanism involving activation of α7nAChR subtype, although this receptor may not be the sole receptor involved.
However, a proof-of-concept study in man is still lacking although α7nAChR agonists are available for human use. Nevertheless, the discovery of the anti-inflammatory potential of the VN is to be considered as a major breakthrough with an enormous potential for treatment. Future studies will hopefully confirm this and fulfil the high expectations, so at the end, patients will benefit with decreased morbidity and increased quality of life.
Acknowledgments
The authors would like to thank all members, past and present, of the Translational Research Center for Gastrointestinal Disorders, University of Leuven, and of the Tytgat Institute for Liver and GI research, AMC Amsterdam for their valuable contributions to the data reviewed herein, and particularly M di Giovangiulio for proofreading the manuscript and help with the figure outlines.
References
Footnotes
-
Contributors Both authors wrote part of the manuscript.
-
Funding G Matteoli is supported by a postdoctoral research fellowship from the Flemish Fund for Scientific Research (FWO), Belgium. GE Boeckxstaens is supported by grants of the Research Foundation—Flanders (FWO) (Odysseus programme, G.0905.07), the agency for Innovation by Science and Technology (IWT), Belgium, and The Netherlands Organisation for Scientific Research (NWO)(VICI), The Netherlands.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.