Article Text

Abstract
Objective The dysfunction of immune regulation plays a critical role in the pathogenesis of a number of chronic inflammatory disorders, such as IBD. A close relationship between psychological stress and intestinal inflammation has been noted; the underlying mechanism remains elusive. This study aims to elucidate a pathological pathway between psychological stress and the dysfunction of regulatory T cells (Treg), and its effect on facilitating intestinal inflammation.
Design A restraint stress model was employed to induce psychological stress in mice. The functions of Tregs were determined by assessing the immune suppressor effects in the intestine. A mouse model of intestinal inflammation was established using a low dose of trinitrobenzene sulfonic acid (TNBS) or dextran sulfate sodium (DSS) together with the challenge of chronic stress.
Results After treating mice with restraint stress, the suppressor function of intestinal Treg was compromised, although the frequency of Treg was not changed in the intestine. Further observation revealed that stress induced Tregs in the intestine to differentiate into foxhead box P3+ interleukin (IL)-17+ tumour necrosis factor (TNF)-α+ T cells. We also observed that exposure to stress-derived prolactin induced dendritic cells (DC) to produce IL-6 and IL-23 in vitro and in vivo, which played a critical role in altering Treg's phenotypes. Treating mice with chronic stress facilitated the initiation of intestinal inflammation by a low dose of TNBS or DSS, which was abolished by pretreatment with an inhibitor of prolactin, the cabergoline.
Conclusions Psychological stress-derived prolactin alters DC and Treg's properties to contribute to intestinal inflammation.
- INFLAMMATION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Psychological stress is involved in intestinal inflammation; the mechanism has not been fully understood.
-
Regulatory T cells are supposed to suppress mucosal inflammation; some papers indicate the numbers of regulatory T cells are increased in the intestine of IBD patients.
-
IL-17+ Foxp3+ T cells are reported; the origins of the cells are unclear.
What are the new findings?
-
Psychological stress did not alter the number of regulatory T cells in the intestine, but compromised their immune suppressor functions.
-
Psychological stress-derived mediator, prolactin, alters the phenotypes of regulatory T cells in the intestine.
-
Administration with inhibitors of prolactin abolished the intestinal inflammation induced by simultaneous exposure to chronic stress and a low dose of trinitrobenzene sulfonic acid or dextran sulfate sodium.
How might it impact on clinical practice in the foreseeable future?
-
Administration of prolactin antagonists may attenuate the chronic stress-induced immune deregulation and inflammation in the intestine.
Introduction
Published data indicate that psychological stress (in short, stress) breaks the established tolerance in the intestine,1 ,2 compromises intestinal epithelial barrier function3 and augments immune responses.4 One of the hallmarks of stress is the activation of the hypothalamic–pituitary–adrenal axis, and the corticotropin-releasing factor (CRF)–adrenocorticotropic hormone (ACTH)–cortisol cascade; such a physiological process represents the prototypic stress hormone system5. Some other mediators such as epinephrine and prolactin (PRL) are also involved in the process of stress.6 ,7 The relationship between stress and intestinal disorders has been noted;8 however, the underlying mechanism between stress and the pathogenesis of intestinal inflammation has not been fully elucidated yet.
The aetiology and pathogenesis of IBD are unclear. Animal model studies have made substantial progress to reveal the causative factors and the pathogenesis of IBD. The trinitrobenzene sulfonic acid (TNBS) colitis model and the dextran sulfate sodium (DSS) colitis model are the main models in the experimental studies of IBD.9 Inflammation induced by DSS mimics several features of ulcerative colitis10 while treating with TNBS mimics several features of Crohn’s colitis.11 However, the underlying mechanism of these animal models in the induction of intestinal inflammation remains to be further understood.
Dendritic cells (DC) are one of the major immune cells in the intestine, distribute immediately under the epithelial layer, recognise harmful or non-harmful stimuli and regulate lymphocyte activities.12 Upon recognising stimulation, DCs secrete cytokines, such as microbial stimuli induces DCs to release interleukin (IL)-6 and IL-23 to induce Th1 inflammation.13 ,14 The DC-derived IL-23 also facilitates Th17 cell development.15 Published data indicate that regulatory T cells (Treg) may produce IL-17 under a given environment.16 The mechanism by which DC-derived IL-6 and IL-23 dictate Tregs to differentiate into IL-17-producing cells remains to be further investigated.
Tregs, characterised by the expression of foxhead box P3 (Foxp3), are one of the major components in the immune-tolerant system of the body.17 Tregs can suppress other effector T cell activities to prevent body damage by the self-immune cells.18 The ongoing inflammation in the intestine, such as the IBD, implies the dysfunction of the immune tolerance in the intestine. Some reports indicate that the frequency of peripheral Tregs is less in IBD patients than normal controls19 while others indicate that the number of Tregs is actually increasing in the IBD intestinal mucosa.20 The paradox is explained by further studies; under given circumstances, Tregs may also produce IL-4, interferon (IFN)-γ, or IL-17, but keep the expression of Foxp3.21 ,22 However, factors altering the phenotype of CD4+ CD25+ Foxp3+ Tregs remain unclear.
Since stress can increase the expression of the proinflammatory cytokines, such as tumour necrosis factor (TNF)-α and IFN-γ,23 we hypothesise that stress is an important factor to alter the phenotypes of CD4+ CD25+ Foxp3+ Tregs. To test the hypothesis, we treated mice with chronic-restraint stress. The results showed that stress-derived PRL induced DCs to produce IL-6 and IL-23, which induced Foxp3+ Tregs to express IL-17 and became Foxp3+ IL-17+ T cells. Furthermore, the Foxp3+ IL-17+ T cells also expressed high levels of TNF-α, the latter is one of the major proinflammatory cytokines of IBD.24
Materials and methods
Mice
Male BALB/c mice and C57/B6 mice (6–8-week of age) were purchased from the Shanghai Experimental Animal Center (Shanghai, China). Animals were housed in pathogen-free animal cages under a 12-h light/dark cycle with free access to food and water. The experimental procedures were approved by the Animal Ethic Committee at Tongji University and Shenzhen University.
Restraint stress
After acclimation for 1 week, mice were individually placed in transparent plastic cylinders (3.5×10 cm) for 60 min per session, starting at 9:00, 12:00 and 15:00, respectively, for consecutive 10 days. The sham-stressed mice were left in their cages.
Flow cytometry
Cells were fixed by 2% paraformaldehyde (0.01% Triton X-100 was added to the fixative in the case of intracellular staining) for 1 h. After washing with phosphate buffered saline (PBS), cells were incubated with fluorescence-labelled antibodies (0.5–1 µg/mL). The cells were washed with PBS and resuspended in PBS-BSA (bovine serum albumin) at 106 cells/mL. The cells were analysed by a flow cytometer (BD FACSCanto II, BD Biosciences, Shanghai, China). To exclude the cell debris and dead cells, the counting threshold was set at 5000 cells.
Treg suppressor function assay
To determine the suppressor function of Tregs, we measured their ability on inhibiting the proliferation of effector T cells (Teff) (CD3+ CD4+ CD25−) T cells. The Teff cells were isolated from the spleen of naïve mice and labelled with 1.5 μM carboxyfluorescein succinimidyl ester (CFSE) and co-cultured with Tregs (Teff:Treg=5:1) in the presence of plate-bound anti-CD3 (2.5 μg/mL) and soluble anti-CD28 Ab (1 μg/mL) in 96-well plates (5×105 cells/well) in order to activate the T cells. The co-cultures were incubated for 3 days at 37°C, 5% CO2. After harvest, the CFSE-labelled T cell proliferation was analysed by flow cytometry.
Isolation DCs from the colon
Lamina propria mononuclear cells (LPMCs) were prepared as described above. The LPMCs were treated with a DC-isolation kit; the CD11c+ CD11b+ B220− DCs were isolated by magnetic activated cell sorting (MACS) following the manufacturer's instruction.
Assessment of viability of cells
The viability of DC and Treg was checked by Trypan blue exclusion assay before using for further experiments. The viability of both DC and Treg was more than 95% in all experiments.
Induction of intestinal inflammation
Following our established procedures, a colitis mouse model with a low dose of TNBS (BALB/c mice; 1.25 mg/mouse; one dose only) or a low dose of DSS (B6 mice; 15 mg/mL in drinking water) plus chronic-restraint stress was developed. In the low-dose model, TNBS was administered to the mice on day 4, and DSS was added to the drinking water starting on day 4, in the course of the 10-day stress treatment. The body weight was recorded from each mouse on days 0, 2, 4, 6, 8 and 10, respectively.
Histology and inflammatory scores
A piece of the colon was excised at sacrifice and processed for paraffin sectioning and stained with eosin and haematoxylin. The sections were observed under a light microscope. Twenty pictures were randomly taken from each sample. The sections were coded; the observers were not aware of the code to avoid the observer bias.
In addition, the degree of inflammation was scored from 0 to 3 (0, no signs of inflammation; 1, increased mucosal mononuclear cell present; 2, increased mucosal and submucosal or transmural mononuclear cell present; 3, mucosal regenerative features with crypt distortion and increased crypt proliferation or ulcers or erosions) and represented as mean±SD of 20 fields.
MPO measurements
The sera were collected from each mouse and incubated in 0.5% hexadecyl-trimethylammonium bromide (pH 5.5) and 0.026% ortho-dianisidinedihydrochloride and plus 0.018% H2O2 was added. Thirty minutes later, the myeloperoxidase (MPO) activity was measured with a spectrometer at 650 nm.
Statistics
The statistical difference between the groups was determined by ANOVA. The results were considered to be statistically significant when p<0.05.
Some experimental procedures are presented in the online supplementary materials.
Results
Restraint stress compromises Treg's suppressor function
In the first step of this study, we assessed the frequency of CD4+ CD25+ Foxp3+ Tregs among the LPMC in mice after restraint stress. The results showed that the frequency of Foxp3+ Tregs was not significantly different between the stressed mice and naïve mice (figure 1A). To test whether the Tregs from stress-treated mice still conserve the immune suppressor function, Tregs were isolated by MACS from the intestine and cultured with Teff cells in the presence of anti-CD3/CD28 for 3 days. Tregs from naïve mice effectively suppressed Teff cell proliferation while the suppressor function of Tregs from stressed mice was markedly weaker than naïve mice or sham mice (figure 1B). The results indicate that the immune suppressor functions of Foxp3+ Tregs from mice treated with stress was compromised. Whether those Foxp3+ Tregs still conserve the immune regulatory properties is questionable. To this end, we further characterised the Foxp3+ Tregs. The results showed that a portion of the gated Tregs in figure 1A were also IL-17+ (figure 1C) and TNF-α+ (figure 1D). The results indicate that psychological stress can alter Treg's properties in the intestine by converting the Foxp3+ Tregs to IL-17+ TNF-α+ Foxp3+ T cells.

Stress compromises Treg's suppressor function. Mice were treated with restraint stress. The treatment was denoted above each subpanel. (A) Colon LPMC were prepared and analysed by flow cytometry. CD4+ T cells were gated first from the LPMC, from which the frequency of CD25+ Foxp3+ T cells was determined. The dotplots and bars (A5) indicate the frequency of CD4+ CD25+ Foxp3+ T cells in LPMC. (B) By MACS, Tregs were isolated from LPMC of naïve mice (nTreg), stressed mice (sTreg) and sham-stressed mice (shTreg); Teffs were isolated from the spleen and labelled with CFSE. The histograms and bars (B5) indicate the frequency of Teff cell proliferation after activating by anti-CD3/CD28 in the culture in the absence (B1) or presence (B2-3) of Tregs (Teffs:Treg=106:2×105). The filled histogram is a CFSE staining control. (C, D) The dotplots and bars (C4, D4) indicate the frequency of Foxp3+ IL-17+ cells (C) and Foxp3+ TNF-α+ cells (D) in the gated cells of (A). Naïve, naïve mice; Stress, stressed mice; Sham, sham-stressed mice. Each group consists of nine mice. Specimens from three mice were pooled as one sample to process. The data of bar graphs are presented as mean±SD; ***, p<0.001, compared with B1, or C1, or D1. CFSE, carboxyfluorescein succinimidyl ester; LPMC, lamina propria mononuclear cells; MACS, magnetic activated cell sorting.
Stress alters the phenotypes of Tregs via modulating DC phenotypes
Published data indicate that, under a given environment, Tregs may change their phenotypes to be Foxp3+ IFN-γ+ cells,25 or Foxp3+ IL-4+ cells,21 or Foxp3+ IL-17+ cells.26 The data of figure 1 also indicate that the exposure to stress increases the frequency of Foxp3+ IL-17+ cells and Foxp3+ TNF-α+ cells in the intestine. To elucidate whether the stress-related mediators altered the phenotypes of Tregs in the intestine, Tregs were isolated from the mouse intestine and exposed to several stress-related mediators, including ACTH, CRF and PRL, respectively, in the culture for 72 h. As shown by qRT-PCR (table 1), however, little detectable changes of the mRNA expression of Foxp3, TNF-α and IL-17 were observed in the Tregs in response to the exposure to any of the stress mediators (figure 2A). The data indicate that the stress-related hormones do not directly induce Tregs to express IL-17 to become the Foxp3+ IL-17+ T cells.
Primers used in qRT-PCR

Stress modulates phenotype of Tregs via modulating IL-6 and IL-23 expression in DCs. (A) Tregs were isolated from the intestine of mice (the mice were treated with OVA (5 mg/mouse daily for 1 week to increase the amount of Tregs)). The Tregs were cultured in the presence of the stress-related mediators (denoted on the x-axis) for 72 h. The bars indicate the levels of mRNA in the Tregs. (B, C) DCs were isolated from the intestine of mice treated with or without stress. (B) The bars indicate the levels of mRNA of IL-23 in the DCs. (C) The immune blots indicate the IL-6 (C) and IL-23 (D) protein levels in the DCs. E1 and F1: The SSC and FSC plot of LPMC (E1) and splenocytes (F1). E2 and F2: The gated plots show the population of CD11c+ DCs in LPMC (E2) and splenocytes (F2). E3–E8 and F3–F8: The histograms show the frequency of IL-6+ DCs (E3–E5 and F3–F5) and IL-23+ DCs (E6–E8 and F6–F8) in the gated DCs of E2 and F2, which are summarised in (G), (H), (I) and (J), respectively. The data of bars are presented as mean±SD. ***p<0.001, compared with groups of naïve (B), or E3 (G), or E6 (H), or F3 (I), or F6 (J), respectively. The group labels on x-axis of (G, J) are the same as those in (E) and (F). Each group consists of nine mice; specimens from three mice were pooled as one sample to process. The data represent three separate experiments. DC, dendritic cells; FSC, forward scatter; LPMC, lamina propria mononuclear cells; SSC, side scatter.
We then assessed the expression of IL-17 facilitators, IL-6 and IL-23, in intestinal DCs isolated from mice treated with stress or sham stress. The results showed that the expression of IL-6 and IL-23 was detectable in the intestinal DCs of naïve mice. The exposure to restraint stress significantly (p<0.001) increased the expression of IL-6 and IL-23 in the intestinal DCs as well as spleen DCs (figure 2; p<0.001). In addition, the levels of MHC II, CD80 and CD86 were also upregulated in the DCs (see online supplementary figure S1). The results imply that the restraint stress can activate DCs and increase the expression of IL-6 and IL-23 in intestinal DCs.
To elucidate which stress-associated mediators modulated the phenotyping of DCs, we isolated DCs from the naïve mouse intestine and treated with the stress-associated mediators, including ACTH, CRF and PRL, respectively, in the culture for 2 days. As shown by qRT-PCR and western blotting, the exposure to PRL, but not CRF or ACTH, markedly increased the expression of IL-6 and IL-23 in the DCs (figure 3A–C). The results implicate that PRL can increase the expression of IL-6 and IL-23 in DCs. To confirm the results, we added recombinant PRL to the DC culture at graded concentrations for 2 days. The results showed that PRL significantly increased the expression of IL-6 and IL-23 in DCs in a PRL dose-dependent manner (figure 3D). We then measured the serum levels of PRL in the mice treated with stress or sham stress. The results showed that significant (p<0.001) increase in the serum levels of PRL was detected in the stress group on day 1, decreased on day 5 and reincreased on day 10 (p<0.001), as compared with that in sham stress group and naïve group (figure 3E). To further strengthen the results of figure 1, we treated mice with both stress and cabergoline (the PRL inhibitor) in the same procedures of figure 1. The results showed that the frequency of Tregs in the intestine was also not significantly altered in the mice treated with both stress and cabergoline. As expected, these Tregs showed strong suppressor function (see online supplementary figure S2).
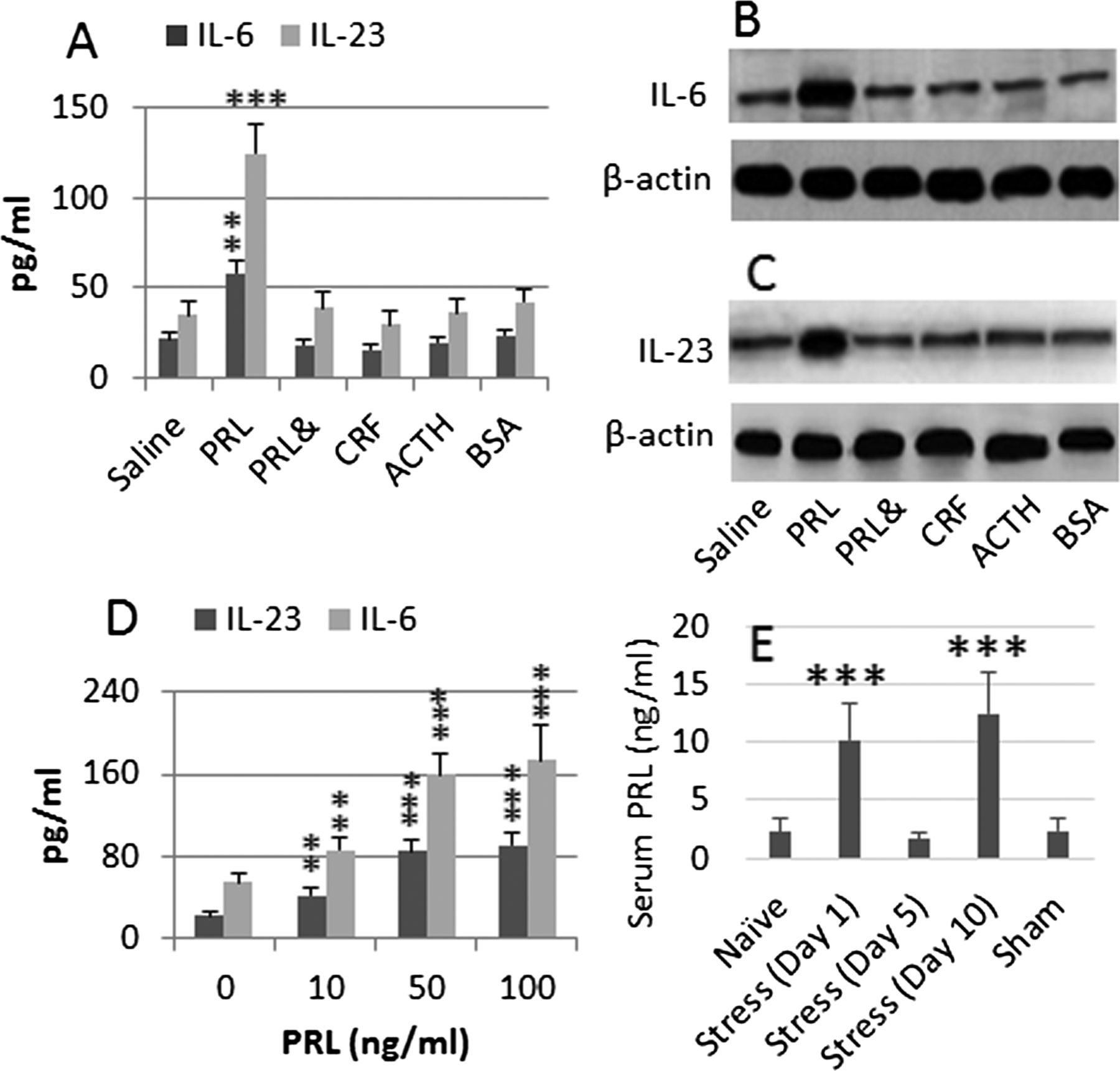
PRL modulates DC phenotypes. DCs were isolated from the naïve mouse intestine and cultured in the presence of stress-associated mediators (indicated on the x-axis; the BSA is a control protein) (A), or in the presence of PRL at graded concentrations (D) for 2 days. The culture medium was collected and analysed by ELISA. The bars indicate the levels of IL-6 and IL-23 in the culture supernatants (A, D). (B, C) The immune blots indicate the levels of IL-6 (B) and IL-23 (C) proteins in the DC extracts. (E) The bars indicate the serum levels of PRL of mice (n=9) treated with stress or sham stress. The data in bar graphs were expressed as mean±SD. **, p<0.01, ***, p<0.001, compared with naïve group (A, E), or group ‘0’ (D). The data represent three separate experiments (specimens from three mice were pooled as one sample to process). Concentration in culture: PRL=50 ng/mL. CRF=10−5 Mol. ACTH=500 ng/mL. BSA=1 µg/mL. &: The presence of NF-κB inhibitor, BAY 11-7082 (1 µM). ACTH, adrenocorticotropic hormone; BSA, bovine serum albumin; CRF, corticotropin-releasing factor; DC, dendritic cells; PRL, prolactin.
To understand the signal transduction pathway by which PRL increases IL-6 and IL-23 in the DCs, we pretreated DCs with NF-κB inhibitor, BAY 11-7082 and then exposed the cells to PRL, which abolished the increases in IL-6 and IL-23 in the culture (figure 3A–C). Treating cells with BAY 11-7082 alone did not alter the expression of IL-6 and IL-23 (data not shown). The results implicate that the stress-derived PRL increases the activities of NF-κB, the latter triggers the expression of IL-6 and IL-23 in DCs. In addition, we also repeated the experiments of figure 3A–C with bone marrow-derived dendritic cells (BMDC), which resulted in similar results (data not shown) of figure 3A–C.
Tregs were then isolated from the mouse intestine and spleen, and co-cultured with BMDCs, which were primed with or without PRL before the co-culture for 6 days. As analysed by flow cytometry, the culture with the PRL-pulsed BMDCs markedly induced the Tregs to express TNF-α and IL-17, although the expression of Foxp3 was still detectable. The presence of anti-IL-23 or anti-IL-6 antibody abolished the conversion (figure 4A–E). After culturing with BMDCs in the presence of PRL, the expression of TGF-β was almost depleted in the Tregs (figure 4F). Collectively, the results implicate that stress-derived PRL can increase the expression of IL-23 and IL-6 in DCs, which further convert Tregs to Foxp3+ IL-17+ TNF-α+ T cells.

PRL modulates phenotype of Treg. Tregs and DCs were isolated from the mouse spleen and co-cultured at a ratio of 5:1 for 6 days. The additional treatments were annotated above each panel. The cells were analysed by flow cytometry. The representative dotplots show the frequencies of Foxp3+ TNF-α+ cells (A) and Foxp3+ IL-17+ cells (B). (C) is a negative control. (D) Summarised data of (A). (E) Summarised data of (B). (F) gel a: TGF-β in Tregs; gel b: β-actin. Treg: Tregs were cultured alone. Treg/DC: Tregs and DCs were co-cultured. PRL: PRL was added to the culture medium. αIL-6 (αIL-23): Neutralising anti-IL-6 (or anti-IL-23) antibody was added to the culture medium. The data represent three separate experiments. Concentration in culture: PRL=50 ng/mL. Anti-IL-6 (or anti-IL-23)=200 ng/mL. Data of bars are presented as mean±SD. *p<0.01, compared with A1 (D), or B1 (E). DC, dendritic cells; PRL, prolactin.
PRL modulates the phenotypes of DC and Treg in the mouse intestine
We then took a further insight into the role of PRL in modulating DC and Treg phenotypes in the mouse intestine. The results showed that treating mice with PRL increased the frequencies of IL-6+ DCs, IL-23+ DCs (figure 5), IL-17+ Foxp3+ T cells and TNF-α+ Foxp3+ T cells (figure 6) in the intestine; similar results were also observed in mice treated with both stress and TNBS, but not in those treated with a low dose of TNBS alone. Furthermore, the alteration of the DC and Treg phenotypes induced by treating with both stress and TNBS was abolished by pretreatment with the PRL inhibitor, cabergoline. To elucidate whether the Foxp3+ T cells still had the immune suppressor function, we isolated CD4+ CD25+ CD127− Tregs from the mouse intestine and cultured with Teff cells (isolated from the spleen) in the presence of anti-CD3/CD28. As shown by flow cytometry, Tregs isolated from mice treated with PRL showed weaker suppressor function as compared with Tregs from naïve mice; the immune suppressor function was further weakened in Tregs from mice treated with both PRL and TNBS, or both stress and TNBS, which was abolished by pretreatment with PRL inhibitors (figure 6).
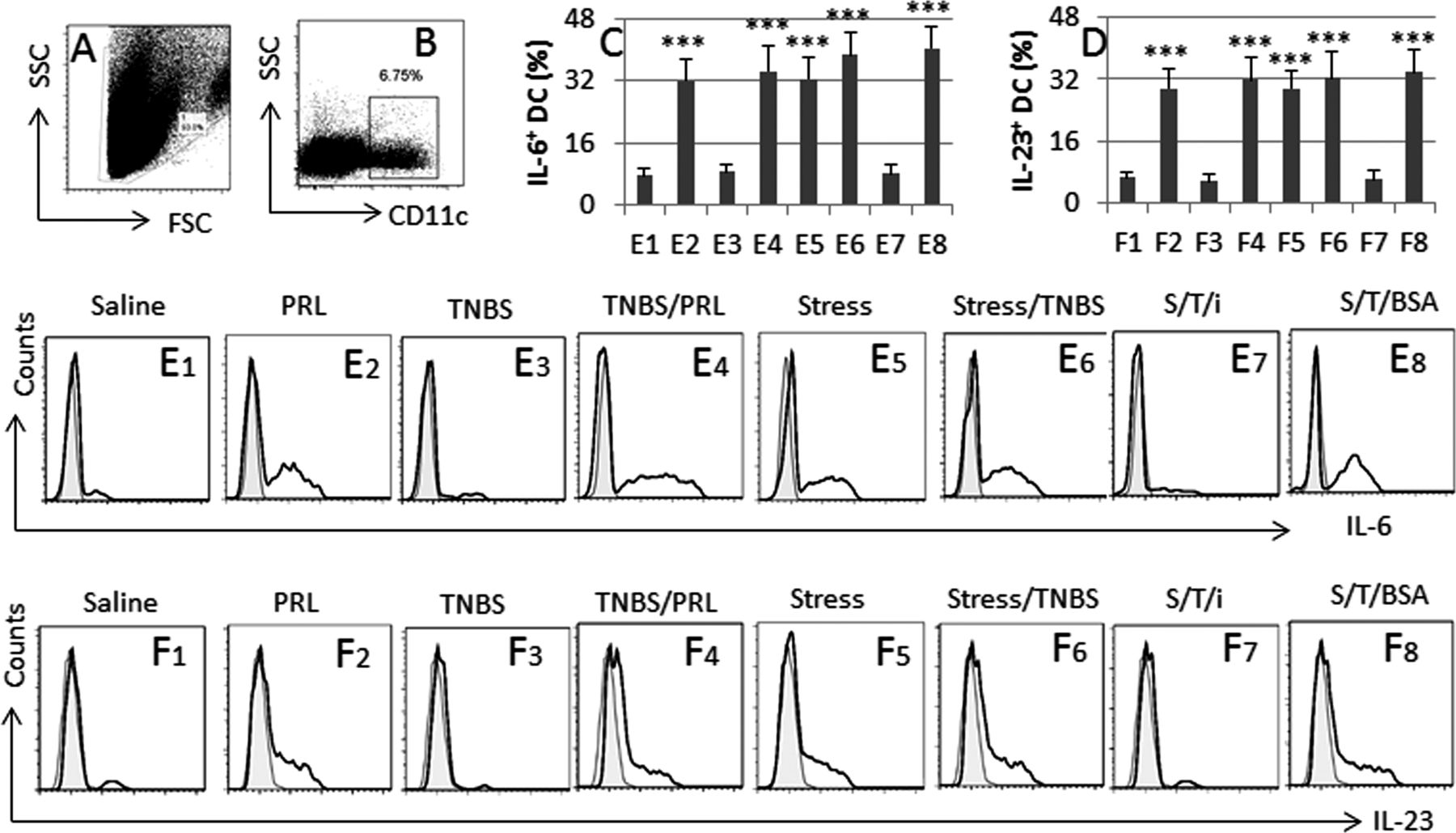
PRL modulates DC phenotyping in the intestine. BALB/c mice were grouped and treated as denoted above each subpanel of flow cytometry histogram. LPMC were prepared and analysed by flow cytometry. (A, B) The dot plots show the gating strategy of CD11c+ DCs. (C, D) The bars indicate the summarised frequencies of IL-6+ DCs and IL-23+ DCs in LPMC; the labels on the x-axis are the same as those in (E) and (F). (E, F) The histograms indicate the frequency of IL-6+ DCs and IL-23+ DCs. The filled histograms are the negative controls. The labels above each histogram indicate the treatment of mice, from which LPMC were isolated. S, stress; T, TNBS; i, PRL inhibitor (cabergolin; 500 µg/kg body weight; intraperitoneally 30 min prior to each stress session). The data of bars are presented as mean±SD; ***, p<0.001, compared with group E1 (C) or F1 (D). Each group consists of nine mice. Specimens from three mice were pooled as one sample to process. The data represent three separate experiments. DC, dendritic cells; LPMC, lamina propria mononuclear cells; PRL, prolactin; TNBS, trinitrobenzene sulfonic acid.
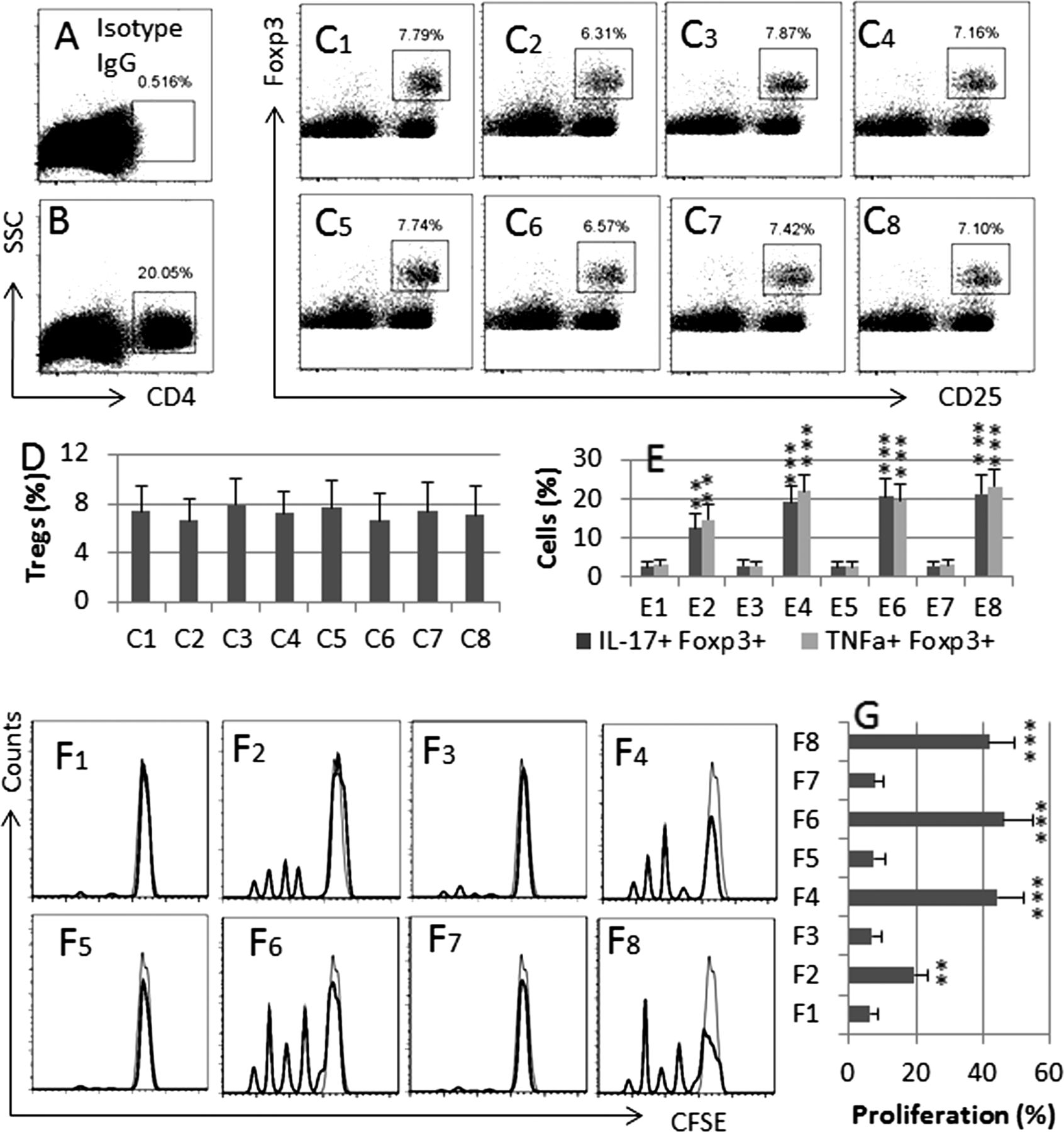
PRL modulates Treg phenotyping in the intestine. BALB/c mice were grouped and treated as described in figure 5 legends. LPMC were prepared and analysed by flow cytometry. (A) A negative staining control. (B) The dot plots indicate the frequency of CD4+ T cells in LPMC. C1–C8, the gated dot plots indicate the frequency of CD25+ Foxp3+ Tregs in the gated CD4+ T cells of (B). (D) The bars indicate the summarised data of Tregs in (C). (E) The bars indicate the summarised frequencies of IL-17+ cells and TNF-α+ cells in the gated Tregs in (C). F1–F8, the histograms indicate the Teff cell proliferation. The filled histograms are staining controls. (G) The bars indicate the summarised data of (F). Treatment of mice in C1–8, E1–8 and F1–8: 1: Saline; 2, PRL (10 µg/mouse; intraperitoneally, daily for 10 days). 3, TNBS; 4, TNBS and PRL; 5, stress; 6, stress and TNBS; 7, stress and TNBS and PRL inhibitor (cabergoline, 500 µg/kg body weight; intraperitoneally 30 min prior to each stress session); 8, stress and TNBS and BSA (a control protein). The data of bars are presented as mean±SD; **, p<0.01, ***, p<0.001, compared with saline group. Each group consists of nine mice. Specimens from three mice were pooled as one sample to process. The data represent three separate experiments. BSA, bovine serum albumin; LPMC, lamina propria mononuclear cells; PRL, prolactin; TNBS, trinitrobenzene sulfonic acid.
Psychological stress facilitates the initiation of intestinal inflammation
The data described above implicate that psychological stress-derived PRL is a facilitating factor in the initiation of intestinal inflammation. To test the hypothesis, we treated mice with a low dose of TNBS (or DSS) together with or without treating with restraint stress. The results showed that treating mice with the combinations of PRL and TNBS, or stress and TNBS, or PRL and DSS, induced apparent colitis as shown by colon histology (figure 7A–C, K); treating with PRL alone (figure 7D), TNBS alone (figure 7E), stress alone (figure 7F) and DSS alone (figure 7G) did not induce apparent colitis. Administration with a PRL inhibitor, cabergoline, prevented the combination of stress/TNBS, or the combination of stress/DSS, from inducing colitis (H, I). Mice treating with saline showed normal histology in the colon (J). Groups D–J also showed low inflammatory scores (figure 7K). In addition, the measurements of MPO levels in the intestinal extract (see online supplementary figure S4A), serum levels of IL-17/TNF-α/IFN-γ (see online supplementary figure S4B), the mouse body weight changes (see online supplementary figure S4C) and the colon length (see online supplementary figure S4D) are in parallel to the colon histology results (figure 7A–J) and the inflammatory scores (figure 7K).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PRL facilitates development of colitis. Mice were treated with low doses of TNBS (or DSS) with or without PRL (or stress). The representative colon histology images show colitis in mice treated with PRL/TNBS (A), or stress/TNBS (B), or PRL/DSS (C). The mice did not show colitis after treating with PRL alone (D), TNBS alone (E), stress alone (F) and DSS alone (G). Administration with a PRL inhibitor, cabergoline, then treated with the combination of stress/TNBS (H), or the combination of stress/DSS (I), and saline (J). (K) The bars indicate the inflammatory scores in the mouse colon (mean±SD; ***p<0.001, compared with group A). Each group consists of six mice. Image magnification: (A, C), ×50; (D, J), ×100. (P.S., two control data not shown: Treating with ethanol did not induce colitis; an irrelevant protein, bovine serum albumin, did not block the stress/TNBS-induced colitis). DSS, dextran sulfate sodium; PRL, prolactin; TNBS, trinitrobenzene sulfonic acid.
Discussion
The present study has revealed a novel phenomenon that psychological stress alters the phenotype of DC and Foxp3+ Treg in the intestine. After treating with stress, the intestinal Tregs were converted to IL-17+ Foxp3+ T cells. Conspicuously, the IL-17+ Foxp3+ T cells expressed high levels of TNF-α, one of the major proinflammatory cytokines in IBD.27 Although treating with stress alone did not induce apparent inflammation in the intestine, a novel mechanism by which psychological stress-derived PRL facilitates the induction of IBD-like inflammation in the intestine has been revealed in this study. Published studies have noted that restraint stress increases PRL in rats in the acute phase, but decreased 4 or 5 days later.28 In line with the reports, we observed that the serum PRL levels were increased on day 1–3, decreased on day 4–6 and gradually increased from day 7 to day 10.
A number of mediators are associated with stress-induced disorders in the body. For example, corticotropin-releasing hormone plays a role in causing the epithelial barrier dysfunction29 ,30; epinephrine is responsible for the stress-related cutaneous or muscle hyperalgesia.31 After testing a few major stress mediators, the present data show that PRL is responsible for the conversion of Tregs to IL-17+ Foxp3+ T cells in the intestine of mice after treatment with stress. Although PRL is not originally found in the intestine, accumulating reports indicate that PRL is involved in a number of intestinal activities, such as modulating the calcium ion movement and altering the epithelial barrier integrity.32 ,33 Our data indicate that PRL can also increase the production of IL-6 and IL-23 by DCs in the intestine. Others also found that PRL promoted the DC maturation and played a role in the pathogenesis of immune disorders, such as systemic lupus erythematosus.34 Matera et al35 indicate that PRL can increase DC maturation. Of course, the present study did not test all the stress-associated mediators; other stress-related mediators may also contribute to altering the Treg's property, which needs to be further investigated.
Several pathways of altering the phenotypes of Tregs have been reported. Feng et al25 have reported that Tregs can be converted to IFN-γ+ Foxp3+ T cells by exposing Tregs to IL-12. Voo et al26 stimulated Tregs with Phorbol myristate acetate to induce Tregs to produce IL-17. Moreover, Hansmann has also indicated that suppression of Foxp3 converts Tregs to Th2 phenotypes.21 The present data reveal a novel aspect in the alteration of Treg phenotypes; treating with stress can convert Tregs to be IL-17+ TNF-α+ Foxp3+ T cells. The underlying mechanism is also suggested by the present study that stress induces DCs to produce IL-6 and IL-23 via activating the NF-κB pathway; the IL-6 and IL-23 are involved in the induction of IL-17+ TNF-α+ Foxp3+ T cells.
The plasticity of Tregs has been observed. Under given conditions, Tregs may be converted into IL-4+ Foxp3+ T cells, or IFN-γ+ Foxp3+ T cells, or IL-17+ Foxp3+ T cells.21 ,22 ,25 ,26 The production of proinflammatory cytokines renders these cells to have the potential to contribute to the ongoing inflammation in the local tissue.21 ,22 Our results are in line with these studies, after treating with stress, the frequency of IL-17+ TNF-α+ Foxp3+ T cells was increased in the mouse intestine. Based on the fact that the total number of Foxp3+ cells is not statistically different from that of the tolerant control mice, the treatment with stress may convert Foxp3+ Tregs into the IL-17+ Foxp3+ T cells. The inference is further supported by the data of in vitro and in vivo experiments; previous studies also indicate that Foxp3+ Tregs can be converted to IL-17+ Foxp3+ T cells.26 The novel aspect of the present data is that the stress-induced IL-17+ Foxp3+ T cells barely show the immune suppressor function; furthermore, they express high levels of TNF-α, a critical proinflammatory cytokine of IBD.
A close relationship between stress and intestinal disorders has been proposed. Such a stress compromises the epithelial barrier function,36 facilitates the development of intestinal allergy,30 modulates mast cells’ function in the intestine37 and affects intestinal neural activities.3 The effect of stress on the pathogenesis of IBD has been suggested.38 The present study has expanded the knowledge in this area by showing that stress converts Tregs to IL-17+ TNF-α+ Foxp3+ T cells in the intestine. Treating mice with the stress alone was not sufficient to induce the IBD-like inflammation in the intestine. Similarly, applying a low dose of TNBS alone or DSS alone also did not induce appreciable intestinal inflammation. It seems that the event of psychological stress in this study works as a ‘trigger’ to initiate inflammation in the intestine provided other pathogenic factors exist
In summary, the present data indicate that psychological stress-derived PRL can increase the production of IL-6 and IL-23 by DCs; these cytokines modulate the plasticity of Tregs by inducing Tregs to differentiate into IL-17+ Foxp3+ T cells.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
WW, SM and ZH-P share the first authorship.
-
Correction notice This article has been corrected since it was published Online First. An additional corresponding author has been included.
-
Contributors WW, MS, H-PZ, TC, RW, CL, GY, X-RG, B-SF and ZhiL performed experiments, analysed data and reviewed manuscript. P-CY and ZhaL designed the project, supervised experiments and wrote the manuscript.
-
Funding This study was supported by the following grants: The Natural Science Foundation of SZU (no. 000004), the innovation of science and Technology Commission of Shenzhen Municipality (JCYJ20120613172559904), the Natural Science Foundation of China (nos. 30871752, 31101280, 81061120521, 81270470 and 81373176) and Shanghai Science and Technology Commission (12XD1404000).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.