Article Text

Abstract
Background Besifovir (LB80380) is an acyclic nucleotide phosphonate effective in hepatitis B virus (HBV) DNA suppression for both treatment-naive and lamivudine-resistant chronic hepatitis B (CHB) patients in preliminary studies.
Design We aimed to compare the safety and antiviral activity of two doses of besifovir (90 mg and 150 mg daily) with entecavir 0.5 mg daily in CHB patients. 114 patients were randomised to receive besifovir 90 mg daily (n=36), besifovir 150 mg daily (n=39) or entecavir 0.5 mg daily (n=39). HBV DNA and liver biochemistry, including serum L-carnitine levels, were monitored.
Results At week 48, in the intention-to-treat population, the proportion of patients achieving undetectable HBV DNA (<20 IU/mL) were 63.6%, 62.9% and 58.3%, respectively (p>0.05). The serum mean log10 HBV DNA changes from baseline for the HBeAg-positive patients were −5.84, −5.91 and −6.18, respectively; and for the HBeAg-negative patients were −4.65, −4.55 and −4.67, respectively (p>0.05). There were no differences in the proportions of patients achieving normalisation of alanine aminotransferase (91.7%, 76.9%, 89.7%, respectively) and HBeAg seroconversion (11.11%, 15%, 9.52%, respectively) among all three groups. None of the patients had resistant mutations or increase in serum creatinine of >0.5 mg/dL from baseline. 64 (94.1%) patients on besifovir had lowering of serum L-carnitine (not tested in entecavir patients). L-carnitine levels returned to normal with carnitine supplement.
Conclusions At 48 weeks, 90 mg and 150 mg daily of besifovir were non-inferior to entecavir 0.5 mg daily in treatment-naive CHB patients. The only significant side effect of besifovir was L-carnitine depletion, requiring carnitine supplementation.
- HEPATITIS B
- LIVER
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Besifovir has potent viral suppression of hepatitis B virus (HBV) replication in early studies.
-
Besifovir is effective for treating wild type and drug-resistant HBV.
-
Entecavir is one of the most potent antiviral agents for chronic hepatitis B (CHB) infection.
What are the new findings?
-
Treatment of besifovir for 48 weeks is associated with more than 5 and 4.5 logs HBV DNA reduction for HBeAg-positive and negative hepatitis B patients, respectively.
-
Compared with entecavir, treatment of besifovir for 48 weeks achieved equal potency of HBV suppression.
-
No serious adverse event were observed in both groups of patients treated with besifovir and entecavir.
How might it impact on clinical practice in the foreseeable future?
-
With the high potency of besifovir for treatment-naive and lamivudine-resistant patients, and its absence of renal toxicity up to 48 weeks of treatment, besifovir (taken together with carnitine supplement) is a potential alternative agent for the treatment of CHB.
Introduction
Despite the success of hepatitis B virus (HBV) vaccination in preventing new infection in endemic areas,1 ,2 chronic hepatitis B (CHB) still poses a heavy burden globally. There are approximately 350 million carriers in the world, with an estimated 60 000 persons dying annually of hepatitis B-related cirrhosis and hepatocellular carcinoma.3
Long-term nucleoside/nucleotide analogue therapy has been shown to reverse cirrhosis of the liver4 and to decrease the risk of hepatocellular carcinoma.5 ,6 The major problem with the L-nucleosides, lamivudine and telbivudine, and the less potent nucleotide, adefovir, is the development of resistant HBV mutations.7 ,8 Currently, the recommended first-line agents include entecavir and tenofovir.
Besifovir (previously known by the designation of LB80380) is a novel and potent acyclic nucleotide phosphonate with similar chemical structure to adefovir and tenofovir. It is rapidly converted in the liver and intestine into LB80331, and then further metabolised to LB80317. LB80317, the active metabolite, is a nucleotide analogue of guanosine monophosphate which inhibits HBV replication after phosphorylation into the diphosphate, and then triphosphate forms.
To date, a phase Ib/IIa dose-finding study in treatment-naive hepatitis B e antigen (HBeAg)-positive CHB patients9 and a phase IIa study in lamivudine-resistant HBeAg-positive patients10 have been performed. In the former study, it was shown that viral suppression reached the optimal level with besifovir given in dosages above 60 mg per day. In the latter study, besifovir was found to be effective at reducing viral load in lamivudine-resistant patients. In both studies, besifovir was safe and well tolerated up to the dose of 240 mg daily.
In the current study, we report the results of viral suppression and safety of two doses of besifovir, with entecavir as a comparator in treatment-naive subjects for 48 weeks.
Materials and methods
Study objectives
The primary objective of the study was to select the optimal dose of besifovir (by analyses at weeks 24 and 48) as well as to show non-inferiority of besifovir to entecavir in terms of HBV DNA reduction. The other objectives were to investigate the safety and tolerability of the two doses of besifovir, and to document the occurrence of resistant viral mutations, if any, at week 48.
Study design
The study was a Phase IIb open, multicentred randomised study to compare the safety and antiviral activity of two doses of besifovir (90 mg and 150 mg daily) with entecavir 0.5 mg daily in patients with CHB for 48 weeks. After 48 weeks of treatment, all patients were rolled over to another study in which they were given the same medications and followed-up regularly every 12 weeks till week 96. The doses of 90 mg and 150 mg daily of besifovir were chosen based on the findings of our previous dose-escalating study showing the maximal HBV DNA reduction of 3.92 and 4.16 logs at week 12 in patients receiving these two doses, respectively.10 Patients receiving besifovir 240 mg daily did not achieve more potent HBV DNA reduction (4.0 logs).
The patients were randomised through interactive web-based response to receive either besifovir 90 mg daily (Group 1), or besifovir 150 mg daily (Group 2) or entecavir 0.5 mg daily in the ratio of 1 : 1 : 1. For the two besifovir groups, the dosage of besifovir was blinded for the patients and the investigators, with each patient receiving two tablets of either one 90 mg tablet of besifovir and one placebo tablet, or one 90 mg tablet and one 60 mg tablet of besifovir. Entecavir was provided in the licensed tablet form. Patient compliance was checked by counting of pills during each visit.
Patients were between the ages of 18 and 65 years. They must be documented to be positive for HBsAg for >6 months before screening. For patients who were positive for the hepatitis B e antigen (HBeAg), their HBV DNA levels were higher than 20 000 IU/mL (approximately 105 copies/mL) for at least 6 months, and for patients negative for HBeAg, their HBV DNA levels were higher than 2 000 IU/mL (approximately 104 copies/mL) for at least 6 months. They were not treated with antiviral therapy including interferon for more than 12 weeks within 6 months before screening. Their serum alanine aminotransferase (ALT) levels were between 1.2 and 10 times the upper limit of normal (ULN). They all had compensated liver disease. Written informed consent was obtained from all participants. Female patients of child-bearing potential, and male patients with partners of child-bearing potential were willing to practice contraception by two birth-control methods.
Patients were excluded if they had coinfection with hepatitis C or D or HIV; decompensated liver disease; evidence of hepatocellular carcinoma; history of alcohol or illicit drug use; creatinine clearance of less than 50 mL/min as calculated by the Cockcroft–Gault formula; abnormal peripheral blood counts; serum amylase or lipase of >2×ULN; and serious medical illnesses.
After randomisation, patients were seen biweekly for the first 4 weeks, then every 4 weeks until week 24, and thereafter at 8-weekly intervals. At each visit, patients were assessed for adverse events (AE) and compliance. Blood was taken for liver and renal function tests, blood counts and serum amylase, lipase, creatine phosphokinase, calcium, magnesium, phosphate and glucose. The phosphorus toxicity was graded as follows: grade I, 2.51 mg/dL—<lower limit of normal; grade II, 2.01–2.50 mg/dL; grade III, 1.01–2.00 mg/dL; grade IV, <1.00 mg/dL. Serum HBV DNA levels were also measured using the COBAS Taqman HBV assay (Roche Diagnostics, Branchburg, New Jersey, USA). The lower limit of detection was 20 IU/mL. HBV serology was tested at baseline, week 24 and week 48. HBV genotype by direct sequencing, using a Big dye terminator cycle sequencing kit (Biosystems, USA) were performed at baseline for all patients, and at week 48 for patients with HBV DNA levels ≥2000 IU/mL (approximately 104 copies/mL). The surveillance of antiviral resistance was performed by direct sequencing of the whole reverse transcriptase (RT) region. The sensitivity of detection was estimated to be 15–20% of the mutants existing in the total viral population.
For the besifovir treatment groups but not for the entecavir group, 12-lead electrocardiogram and serum L-carnitine levels were also performed from week 2 onwards, every 4 weeks till week 24, and then every 8 weeks till week 48. L-carnitine loss is associated with the presence of the pivalic moiety which is absent from the entecavir molecule. It is therefore deemed unethical to take blood from patients receiving entecavir to check for L-carnitine. L-carnitine supplements were given at 660 mg daily whenever the levels fell below the reference value. The study was approved by the institutional review boards in all the study centres in Hong Kong and Korea. It was conducted in accordance with the current International Conference on Harmonisation and Good Clinical Practice Guidelines, and the 1964 principles of the Declaration of Helsinski and its subsequent revisions. The trial was registered under ClincalTrials.gov (NCT01026610). All authors had access to the study data and had reviewed and approved the final manuscript.
Statistical analysis
The sample size of 117 subjects (29 per group, and an additional 25% for possible patient attrition) (39 subjects for each group of total three groups) was calculated to give a power of 90% to show that besifovir was non-inferior to entecavir, using data based on the phase III pivotal trials of entecavir and the comparative trials of entecavir versus adefovir. A total of 114 patients were finally enrolled.
Baseline characteristics, efficacy data (log10 reduction in HBV DNA from baseline and other secondary efficacy variables) and AEs were tested for any differences among groups. For continuous variables, comparisons between two groups were performed using two sample t test/Wilcoxon's rank sum test. Comparisons among three groups were performed using Analysis of Variance (ANOVA)/Kruskal–Wallis test. χ2 and Fisher's exact test were used for categorical variables. Subgroup analysis for efficacy endpoints was performed after stratification into HBeAg-positive subgroups.
For serum total/free L-carnitine in the two besifovir groups, the frequency and percentage of subjects out of the reference range before and during treatment was calculated. The differences between pretreatment and post-treatment levels were analysed using McNemar's test.
In the analysis of efficacy variables, missing HBV DNA and ALT values at weeks 24 and 48 were imputed using both the ‘last observation carried forward’ and the ‘baseline observation carried forward’ methods. For the results of undetectable HBV DNA (<20 IU/mL), the values were regarded numerically as 20 IU/mL.
Results
Baseline characteristics
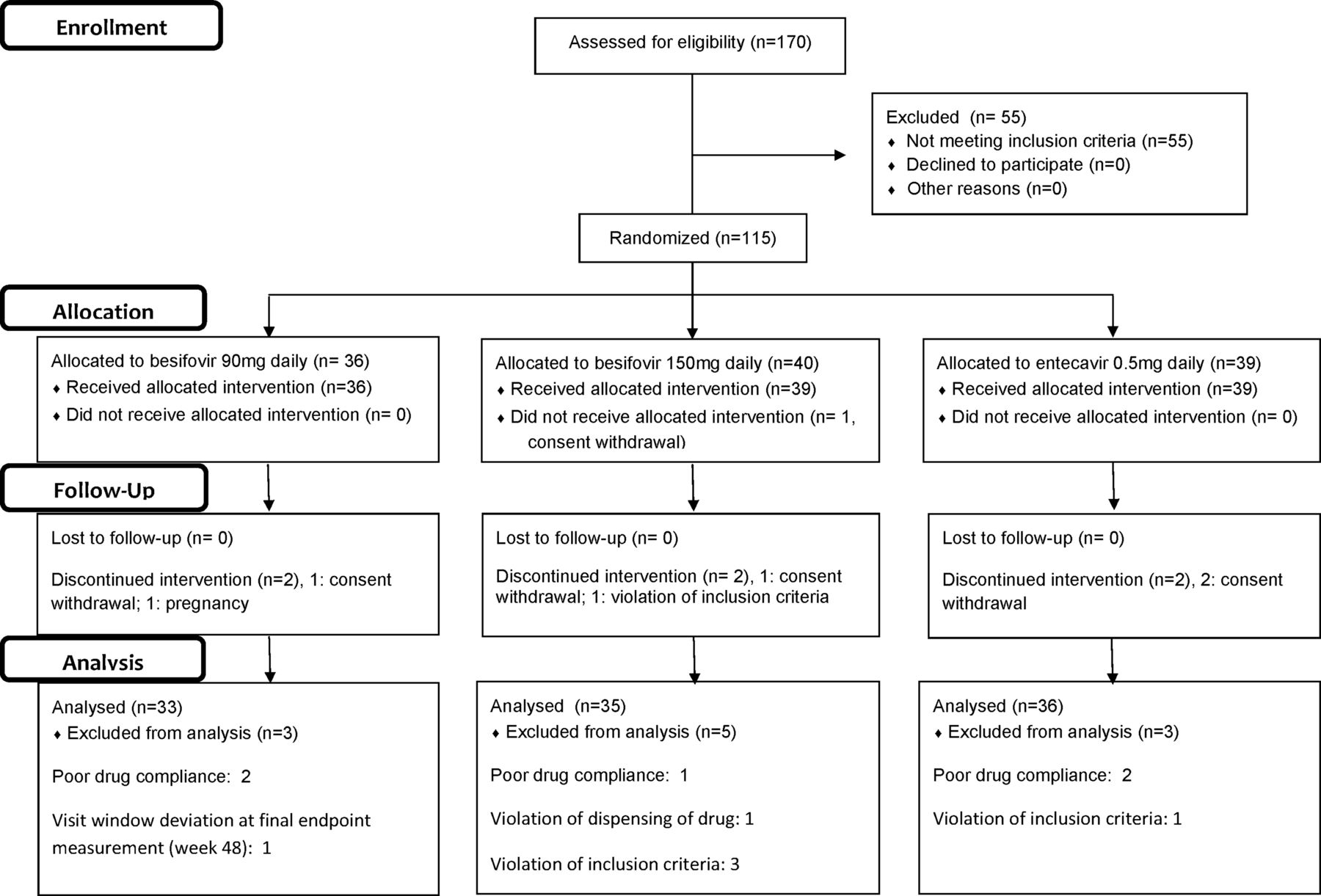
Ten investigational sites (nine in Korea and one in Hong Kong) screened a total of 170 Asian patients. One hundred and fourteen patients were randomised: 36 to besifovir 90 mg daily (Group 1), 39 to besifovir 150 mg daily (Group 2) and 39 to entecavir 0.5 mg daily (Group 3). The details of the patient disposition are described in figure 1. Three patients from Group 1, five from Group 2 and three from Group 3 were excluded from the per-protocol (PP) analysis at week 48, because of consent withdrawal, protocol violation or treatment compliance <80%. The baseline characteristics of the intention-to-treat (ITT) patients are shown in table 1. There were no differences between the three groups of patients except for the gender ratio and mean creatinine clearance. None of the patients had any clinical evidence of cirrhosis. None of the recruited patients had been treated with oral nucleoside/nucleoside analogues, or interferon in the past.
Baseline characteristics of intention-to-treat patients receiving besifovir 90 mg, besifovir 150 mg and entecavir 0.5 mg daily

Patient disposition of the whole study population.
Serum HBV DNA reduction
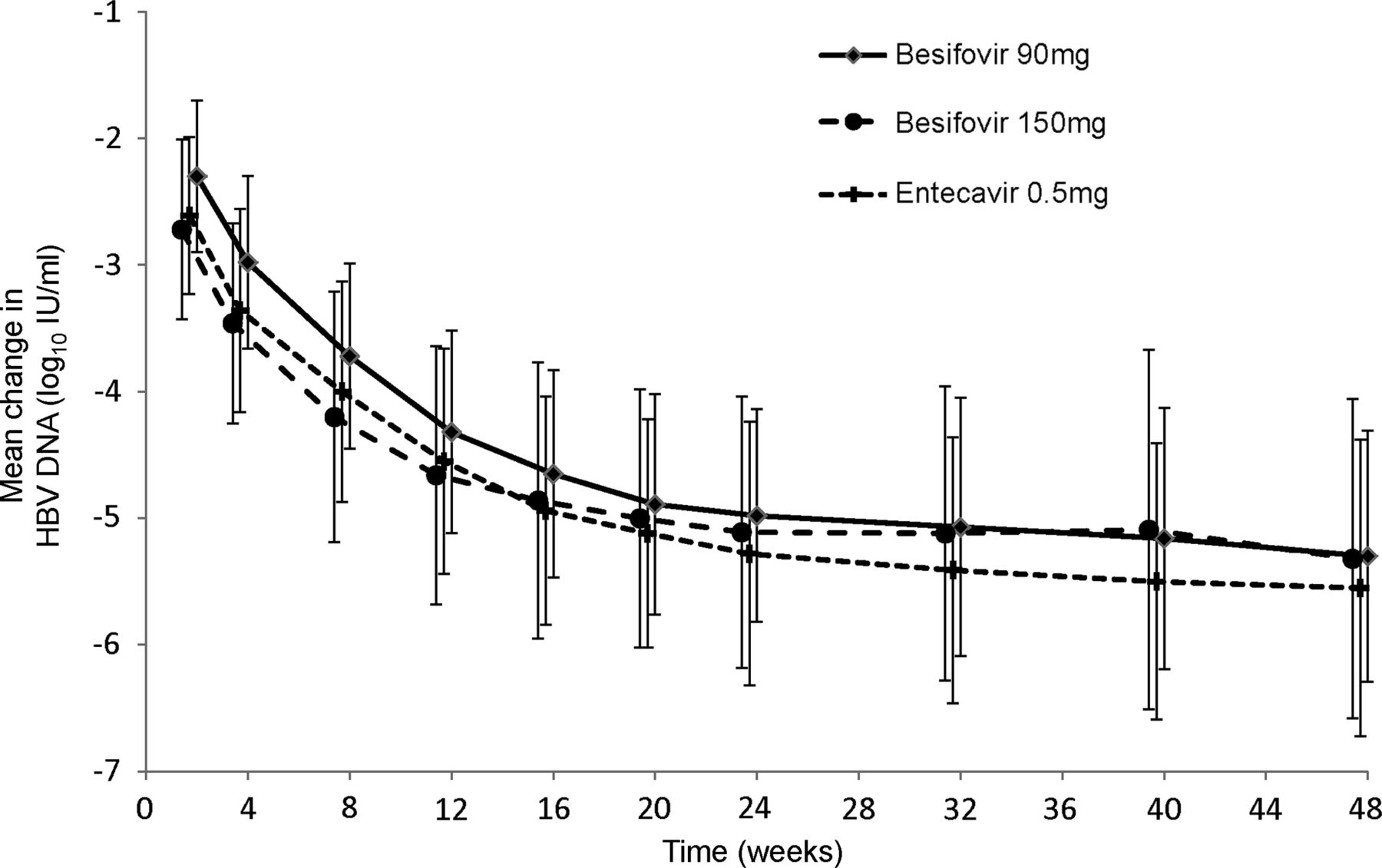
The rate of reduction of the mean log10 HBV DNA levels in the three groups of patients is shown in figure 2. The baseline, week 24 and week 48 log10 HBV DNA levels, as well as the reduction in levels at these two time points of both ITT and PP analyses are shown in table 2. The serum mean log10 HBV DNA changes from baseline at week 48 for 90 mg besifovir, 150 mg besifovir, and 0.5 mg entecavir for the HBeAg-positive patients were −5.84, −5.91 and −6.18, respectively; and HBeAg-negative patients were −4.65, −4.55 and −4.67, respectively. There were no significant differences between the three groups of patients in the reduction of HBV DNA at any time point in both the HBeAg-positive and HBeAg-negative patients. Both doses of besifovir were shown to be non-inferior to entecavir as the lower limit of two-sided 90% CI (−0.69 for 90 mg and −0.71 for 150 mg) was larger than the predefined non-inferior margin (−2).
The decrease in log10 HBV DNA in the three groups of patients at weeks 24 and 48 (per-protocol and intention-to-treat analyses)

Reduction of mean log10 serum hepatitis B virus DNA level from baseline to week 48 in the three groups of patients.
The proportions of patients with HBV DNA levels below 20 IU/mL (approximately 116 copies/mL), the lower limit of detection by the COBAS Taqman assay) at each time point till week 48 are shown in figure 3 (all p=NS). The median HBV DNA levels for patients with detectable HBV DNA receiving besifovir 90 mg, besifovir 150 mg and entecavir 0.5 mg at week 24 were 2.4 (range 1.4–3.6) versus 2.0 (range 1.5–4.0) versus 2.1 (1.5–4.1) logs IU/mL and at week 48, were 2.2 (range 1.5–3.2) versus 1.7 (range 1.3–4.7) versus 1.6 (1.3–4.1) logs IU/mL, respectively (all p=NS). The proportions of patients with HBV DNA levels below 60 IU/mL (approximately 300 copies/mL) at week 48 were 69.7% versus 82.9% versus 80.6%, respectively (p=NS).
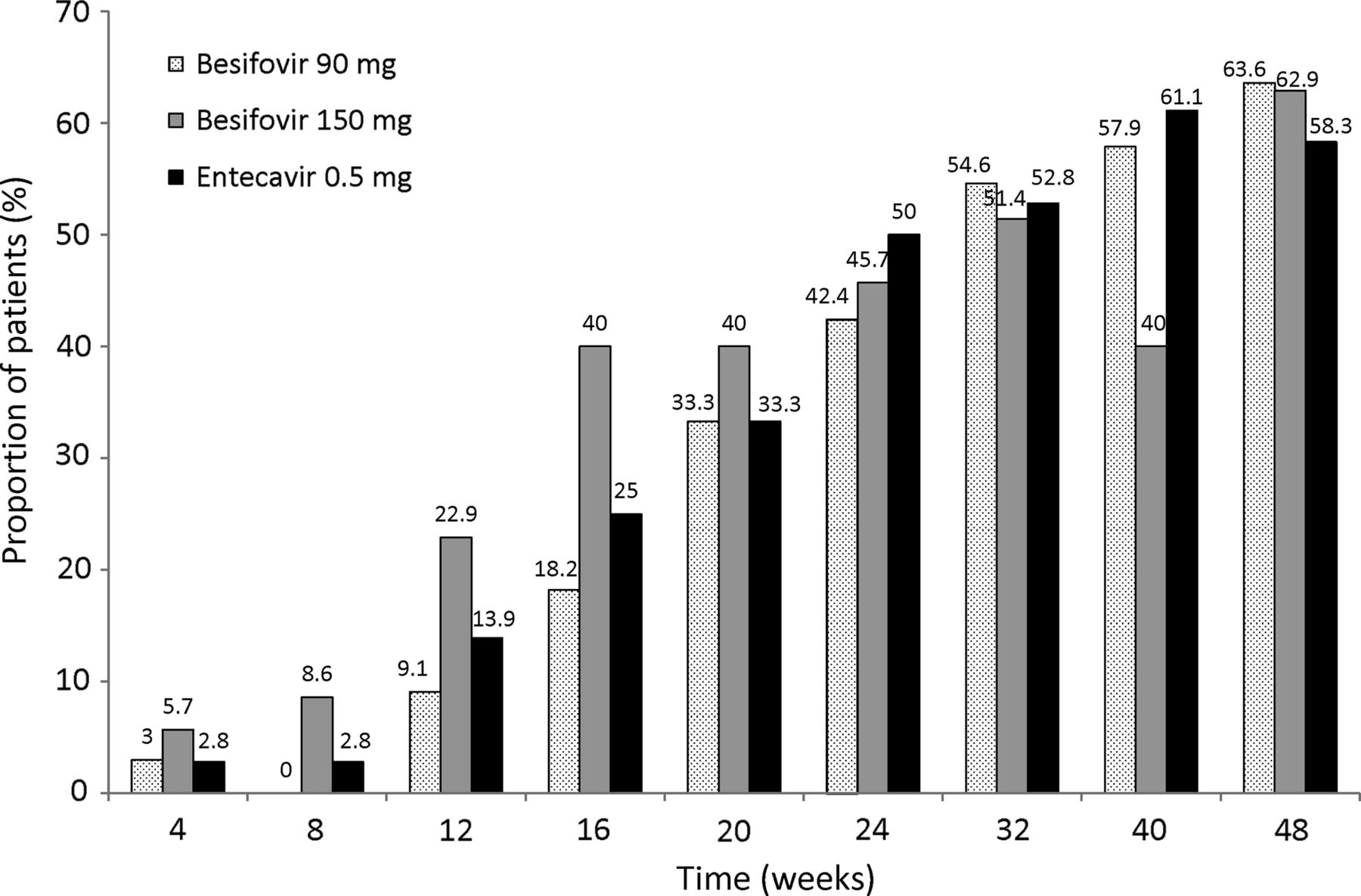
Proportion of patients with undetectable hepatitis B virus DNA (<20 IU/mL) at each time point in the three groups of patients.
None of the patients had primary treatment failure, defined as <2 log10 copies/mL reduction in HBV DNA at week 24 in the protocol.
ALT normalisation and breakthrough
The proportion of patients with normalisation of ALT is shown in figure 4. Five patients in each of the besifovir groups and seven in the entecavir group had ALT breakthrough, defined as rebound of ALT levels to above ULN after initial normalisation. These ALT breakthroughs occurred between weeks 8 and 48 with the highest ALT levels ranging from 41 to 373 IU/mL. All the breakthroughs were transient, and treatment was continued in all 17 patients.

Proportion of patients with normalisation of serum alanine aminotransferase levels at week 48.
The number of patients with HBV DNA levels <60 IU/mL (approximately 300 copies/mL) and normal ALT at week 48 were 22 (66.7%) for the besifovir 90 mg group, 24 (68.6%) for the besifovir 150 mg group, and 25 (69.4%) for the entecavir 0.5 mg group.
Response in the HBeAg-positive groups
Table 3 shows the proportion of patients with HBeAg loss and seroconversion to antibody to HBeAg (anti-HBe) as well as the proportion with HBeAg loss and HBV DNA <20 IU/mL (approximately 116 copies/mL) and <60 IU/mL (approximately 300 copies/mL) by both ITT and PP analyses.
Proportion of patient with HBeAg loss and HBeAg seroconversion at week 48 in the HBeAg-positive patients
Virologic breakthrough and HBV reverse transcriptase mutations
Virological breakthrough was defined as an increase in serum HBV DNA level 10 times above nadir after achieving virologic response (less than 20 IU/mL (116 copies/mL)) during continued treatment in the protocol. Up to week 48, only one patient in besifovir 90 mg group showed the virological breakthrough. This patient had a baseline HBV DNA level of 1.30×107 IU/mL (7.57×107 copies/mL). After decreasing to undetectable level (<20 IU/mL) at week 24, the HBV DNA level rose to 2.90×103 IU/mL (1.69×104 copies/mL) and 1.25×102 IU/mL (7.28×102 copies/mL) at weeks 40 and 48, respectively. There were no specific mutations in the whole RT region identified during the HBV DNA breakthrough.
Sequencing of the full RT region of HBV DNA to detect any besifovir-related was done at baseline and week 48. At baseline, it was observed that 9, 5 and 10 patients in 90 mg, 150 mg and entecavir groups, respectively, had diverse kinds of mutations even though the enrolled patients had not been treated with any antiviral agent for more than 12 weeks. The details of the mutations are depicted in table 4. At week 48, no mutation was detected in the patients whose HBV DNA titre higher than 2000 IU/mL (approximately 104 copies/mL). Therefore, the rate of resistance to both doses of besifovir and to entecavir at week 48 was 0.
Details of the mutations in the RT region identified at the baseline
Safety and adverse events
The AE and serious adverse events (SAE) are shown in table 5. L-carnitine levels were only measured in the two besifovir groups, as mentioned in the Materials and Methods section.
Number of adverse drug reactions and number of patients with adverse drug reactions (including serum L-carnitine abnormality) till week 48
Five cases of transaminase increase (hepatic flare) at weeks 2 or 4 of three patients in the besifovir 150 mg group and two patients in the entecavir group were reported as SAEs, and were considered not to relate with the treatment. The patient with hepatocellular carcinoma was on 150 mg besifovir and was diagnosed by computerised tomography 10 months after recruitment. He was treated with transarterial chemoembolisation. The one patient, on 90 mg besifovir, with ventricular tachycardia, had radiofrequency ablation performed. This was considered unrelated to the study treatment.
As markers of nephrotoxicity, increases of serum creatinine and decreases of serum phosphorus were assessed using statistical method of Kaplan–Meier estimation. No patient had serum creatinine increase of 0.5 mg/dL or greater from baseline. The median-estimated glomerular filtration rates using Cockcroft–Gault formula at each time point for the study patients are depicted in figure 5. Two patients (each 1 in 90 and 150 mg groups, respectively) had hypophosphataemia (defined as decrease below than 2.0 mg/dL) during the study. They had the phosphorus level of 1.9 mg/dL at week 16 and 1.6 mg/dL at week 32, respectively. The phosphorus levels return to normal in the next follow-up (4 weeks later) in both patients without additional treatment. No clinical signs were observed due to the hypophosphataemia. The safety set data (the population who took at least one investigational drug during the study) of the phosphorus level at each time point in patients receiving besifovir 90 mg and 150 mg are listed in table 6.
Proportion of patients receiving besifovir 90 or 150 mg daily with carnitine (normal 34–78 µmol/L) and phosphorus drug toxicity

Estimated glomerular filtration rate at each time point in the three groups of patients.
The serum total and free L-carnitine values in the patients enrolled in besifovir groups were measured at every visit from baseline. The serum total L-carnitine is the sum of free and acyl L-carnitine, and the normal ranges of total and free L-carnitine are 34–78 μmol/L and 25–54 μmol/L. The changes in the serum total L-carnitine are shown in figure 6. The safety set data of the carnitine level at each time point in patients receiving besifovir 90 mg and 150 mg are listed in table 6.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes of serum total (upper diagram) and free L-carnitine (lower diagram) value throughout the study.
L-carnitine supplement was given to patients with levels below the normal ranges from week 4 onwards. A total of 64 (94.1%) patients (26 in 90 mg and 38 in 150 mg) had depletion of serum L-carnitine. The proportion of patients with carnitine depletion was higher in the group of patients taking besifovir 150 mg daily compared with the group taking 90 mg daily (p=0.002 by χ2 test throughout the study period). Among them, 24 (90 mg) and 37 (150 mg) patients took the supplement. During the period when no supplements were given (baseline to week 4), the degree of depletion in 150 mg was larger than in 90 mg group. The mean levels of serum L-carnitine returned to the normal range after supplementation, though statistically significant decline from baseline levels of total and free L-carnitine levels were observed till week 32. After treating with carnitine supplement, the serum carnitine levels were sometimes below normal in 12 patients (1 in the 90 mg group and 11 in the 150 mg group): during the initial few weeks, or when the patients were non-compliant.
Discussion
Following a phase Ib/IIa study of besifovir in treatment-naive HBeAg-positive subjects9 and a phase IIa study in lamivudine-resistant subjects,10 this study was designed to show the non-inferiority of besifovir when compared with entecavir in nucleoside/nucleotide-naive patients. This was found to be true with either dose of besifovir. The 90 mg daily dose and the 150 mg daily dose showed almost identical reduction in HBV DNA levels and ALT normalisation as 0.5 mg entecavir. The rates of HBV DNA decline as well as the proportions of patients with HBV DNA <20 IU/mL (approximately 116 copies/mL) in both groups of patients on besifovir were also comparable with entecavir, especially with 150 mg of besifovir. The proportions of patients with HBeAg loss and HBeAg seroconversion were all similar in all three groups of patients.
It has been shown that guanosine analogues, such as entecavir, unlike other nucleoside analogues, can very effectively suppress the priming reaction of the HBV polymerase, the first step in the conversion of the greater-than-genome-length pregenomic RNA into the minus strand of the HBV DNA.11 This step involves the linking of a deoxynucleotide residue, usually deoxyguanosine triphosphate (dGTP) via a phosphodiester bond to a specific tyrosine residue of the polymerase.12 This is one of the reasons why entecavir is so potent. One of the other reasons is the rapid intracellular phosphorylation of entecavir to its active triphosphate metabolite. Besifovir is also a guanosine analogue with rapid intracellular phosphorylation. This may partly explain why its efficacy in viral suppression is comparable with entecavir.
The one major side effect of besifovir is serum L-carnitine depletion. In living cells, L-carnitine is required for the transport of fatty acids from the cytosol into the mitochondria during the breakdown of lipids for the generation of metabolic energy. Besifovir, similar to adefovir, has a pivalic moiety. It has been reported that compounds containing the pivalic moiety can give rise to reduction in serum L-carnitine levels even when given for a relatively short period of time.13 Pivalate generated from such compounds enters the cells to form pivaloyl-CoA, and the pivaloyl moiety may be transferred from coenzyme A to carnitine to form pivaloylcarnitine which is directly excreted in the urine. In the case of adefovir, supplementation of L-carnitine was administered to patients receiving 30 mg and 60 mg of adefovir in several clinical studies.14 ,15 ,16 No adverse effects related with carnitine deficiency had been noted in the adefovir studies.
In the current study, lowering of serum L-carnitine levels occurred in the majority of patients on besifovir (72% in Group 1 and 95% in Group 2). However L-carnitine levels returned to normal levels in all the patients after L-carnitine supplement. There were also no symptoms or signs related to L-carnitine depletion.
There were no significant changes in renal function in any of the patients and only two patients on besifovir developed transient hypophosphataemia.
With its high potency (comparable with entecavir), its effectiveness in lamivudine-resistant patients,10 and its absence of renal toxicity up to 48 weeks of treatment, besifovir (taken together with carnitine supplement) is a potential alternative agent for the treatment of CHB. It is also an effective agent for patients with lamivudine or telbivudine resistance.
In conclusion, 48 weeks of besifovir, for the 90 mg daily and 150 mg daily doses, was non-inferior to entecavir 0.5 mg daily in treatment-naive patients with CHB. Further studies are required to determine which of these doses is the optimal dose.
References
Footnotes
-
C-LL and SHA contributing equally.
-
Correction notice This article has been corrected since it was published Online First. The authors' first names and surnames have been updated.
-
Contributors C-LL and SHA involved in study concept and design, acquisition of data, analysis and interpretation of data and drafting of manuscript. KSL, SHU, MC, SKY, J-WL, NHP, Y-OK, JHS, JL and J-AK involved in analysis and interpretation of data and revision of manuscript. K-HH and M-FY involved in study concept and design, critical revision of manuscript and overall study supervision.
-
Funding The present study was sponsored by LG Life Sciences, Ltd, South Korea.
-
Competing interests C-LL and M-FY are the consultants of LG Life Sciences, Ltd. JL and J-AK are the employees of LG Life Sciences, Ltd. All the other authors, SHA, KSL, SHU, MC, SKY, J-WL, NHP, Y-OK, JHS, K-HH, had none to declare.
-
Ethics approval The study was approved by the institutional review boards in all the study centres in Hong Kong and Korea.
-
Provenance and peer review Not commissioned; externally peer reviewed.