Article Text

Statistics from Altmetric.com
Introduction
IBD, which primarily includes UC and Crohn's disease (CD), is a progressive, chronic and relapsing condition. This debilitating disease is steadily becoming a worldwide medical concern, with increasing prevalence and incidence in both industrialised and developing countries.1 While the exact aetiology of the disease remains unknown, genetic predisposition and various environmental and immunological causes have been identified as contributing factors.2 Generally, IBD is characterised by a dysregulated excessive immune response and tissue damage in the GI tract.3 ,4 This aberrant and sustained immune response is thought to be mainly facilitated by defects in the function of the intestinal epithelial barrier and in the regulation of mucosal immunity.5 Yet, tissue damage associated with IBD is commonly considered solely a downstream effect and not a contributing factor. This view has led to a concentrated focus on the development of IBD treatments that target inflammatory pathways, but all, thus far, have exhibited limited efficacy. In contrast, none of the IBD drugs released to date were designed to specifically target the tissue-destructive processes associated with the disease.
When it comes to basic and clinical research of IBD-associated tissue destruction, most groups have directed their efforts to investigating cellular responses during inflammation, neglecting altogether the main component undergoing physical rupture, namely, the extracellular matrix (ECM). In a similar manner, while the regulatory role of the ECM in the progression of invasive diseases, such as cancer, is becoming acknowledged, its function in IBD is often overlooked despite the common notion that tissue destruction is imperative to disease progression.
In this article, we highlight ECM remodelling as an integral part of directional pathological signalling in IBD rather than, as is often considered, a passive bystander. We argue that the ECM, in the context of IBD, is not only a static scaffold holding cells in place and maintaining tissue architecture but a dynamic participant in intestinal immune responses capable of determining cell fate. Through our review of some common facts and new studies in the field of IBD, we will promote the new concept of the ECM immune signalling response.
Why study ECM biology in IBD?
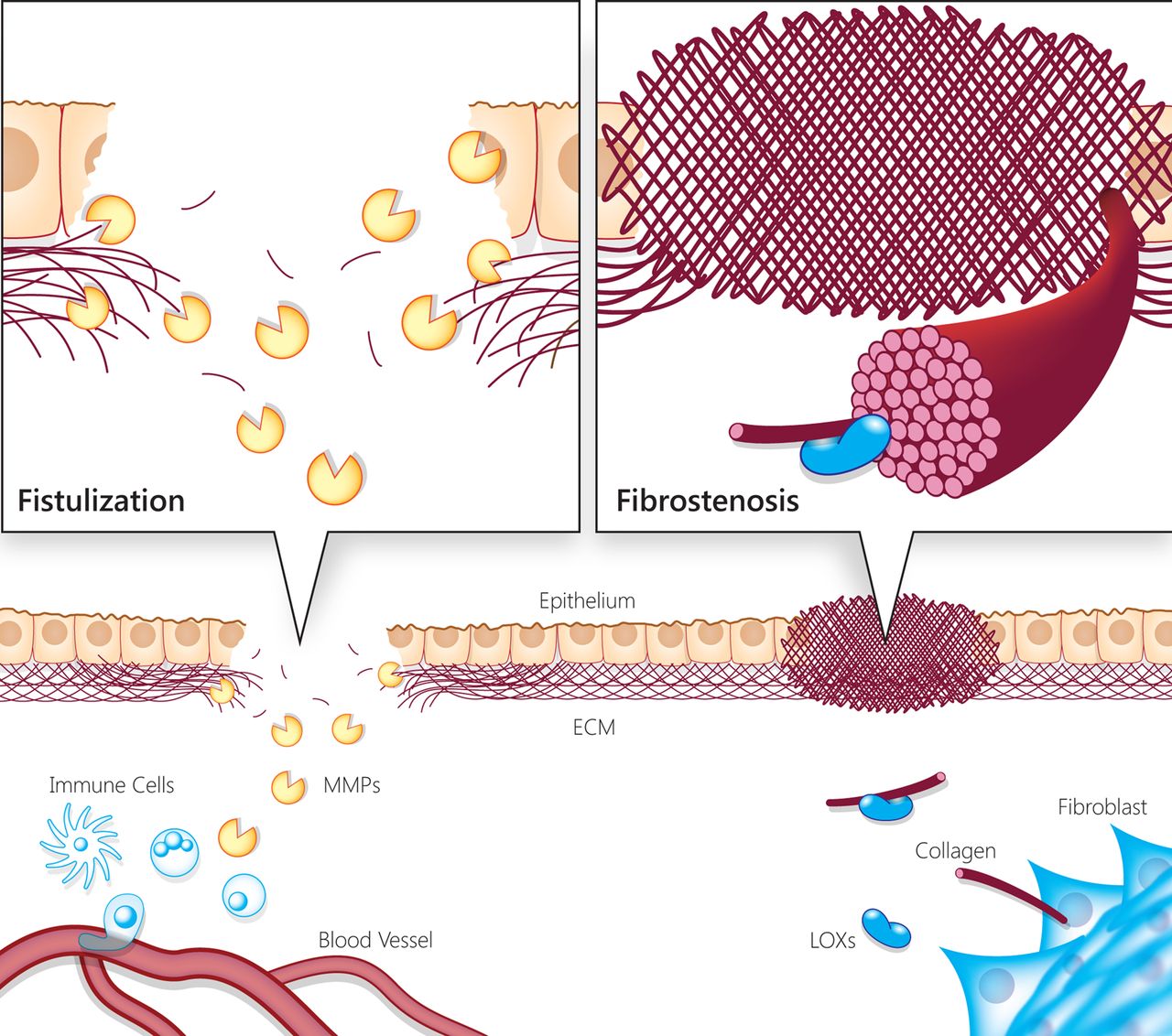
Tissue damage is a hallmark of IBD progression and severity. It is known, for instance, that CD progression can advance (in some cases simultaneously) towards two seemingly opposing directions—stricturing or penetrating disease (figure 1). The stricturing type of disease results from fibrosis in the intestinal tissue,6–8 while the penetrating type is characterised by the formation of fistulae.9 Whereas the first type is a phenomenon of excess in deposition of ECM components by myofibroblasts, the second type is an outcome of ECM destruction that eliminates the boundaries between tissues. Microscopic colitis, a less common form of IBD, has an ECM component as well, the thickening of the collagenous layer in the subepithelium, which is thought to be the cause of diarrhoea in this condition.10 These examples demonstrate that ECM build-up and/or destruction play a central role in IBD, and its clinical classification and complications.

Illustration of IBD-associated progressive tissue damage and complications due to extracellular matrix (ECM) remodelling imbalance and dysregulation. The illustration represents the two faces of progressive tissue damage and complications associated with IBD, resulting from imbalanced and dysregulated ECM remodelling (ie, fistulising vs fibrostenotic disease). During chronic intestinal inflammation, the ECM is remodelled due to secretion of enzymes and structural components by immune, epithelial and stromal cells. Matrix metalloproteinases (MMPs) contribute to epithelial and endothelial barrier disruption and enable immune cells to infiltrate into the tissue. Extracellular proteolysis is a propagator of inflammation via cytokine processing, and release of bioactive molecules from the ECM. On the other hand, fibrotic processes also take place simultaneously by fibroblast activation and secretion of ECM components that assemble via lysyl oxidase (LOX) activity. In turn, the increased stiffness of this fibrotic tissue leads to further fibrogenesis. Therefore, both the destructive and fibrogenic processes in the ECM are self-amplifying and contribute to the tissue damage and excess inflammatory response characteristic of IBD.
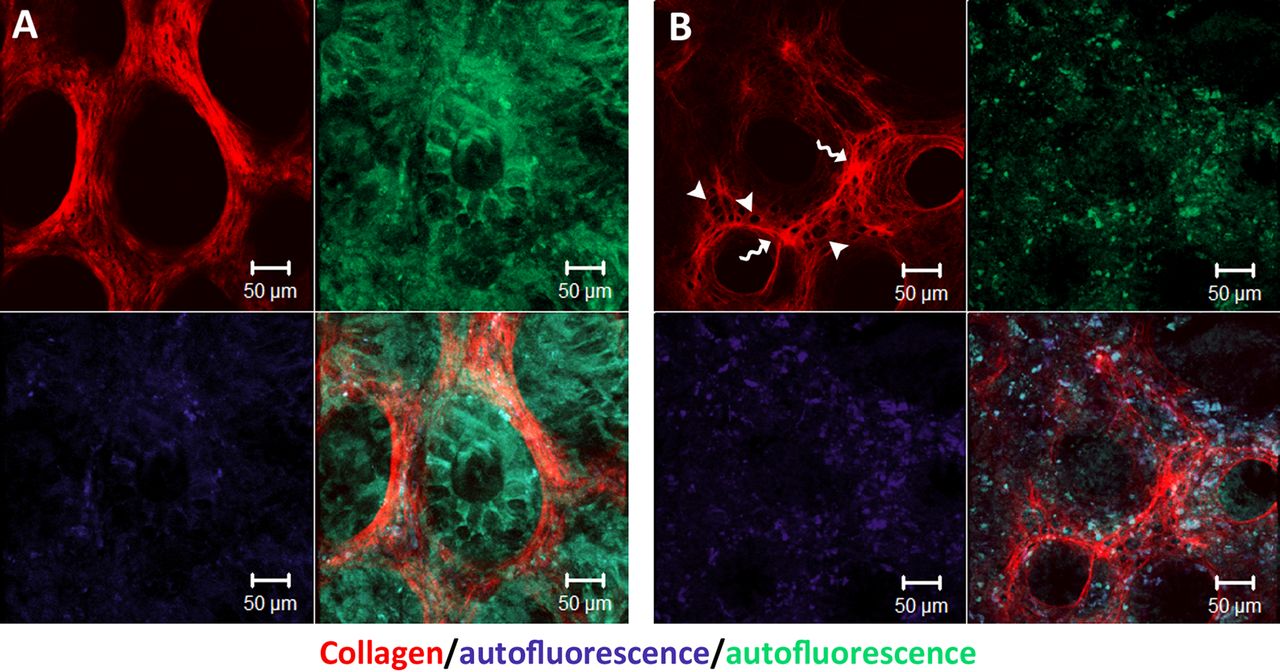
Tissue damage associated with IBD is most likely preceded by molecular events within the ECM. In figure 2, we provide evidence, for such ECM remodelling processes taking place in IBD-afflicted human colonic mucosa, revealed by second-harmonic imaging of the collagen scaffold. Second-harmonic imaging uses a two-photon microscope in order to induce an optical process in which two excitation photons are effectively combined in non-centrosymmetric materials, such as fibrillar collagen, to generate a photon with twice the excitation energy. This technique enables us to image the structure and morphology of non-labelled collagen in native tissues in high spatial resolution.

{kind=link}
{kind=link}
Second-harmonic imaging of native human colon biopsies revealing extracellular matrix (ECM) remodelling in IBD. All samples were thawed in phosphate buffered saline and immediately imaged under two-photon microscope (×20 objective). (A) Healthy colon biopsy from a patient without IBD. (B) Inflamed colon biopsy from a patient with IBD. Note the thickening of ECM barrier between crypts (indicated by curvy arrows), the formation of holes within this barrier (indicated by arrowheads) and changes in collagen microstructure.
The excessive inflammatory response taking place in the intestinal tissue involves not only cellular processes but cellular processes and molecular interactions that take place in the context of a dynamically modifying ECM. Therefore, it is reasonable to assume that ECM remodelling contributes to IBD pathogenesis and inflammation sustenance and is not merely a secondary by-product.
ECM as a pivotal biological entity in homeostasis and disease
The ECM is a substantial component of tissues and thus essential for tissue function, architecture and homeostasis, yet the investigation of ECM function presents many technical challenges.
Composed of an intricate mesh of fibrous proteins and glycosaminoglycans (GAGs), the ECM serves as a scaffold for cells within tissues. It is mainly composed of three types of proteins with distinct roles: structural proteins (eg, collagen and elastin), specialised glycoproteins (eg, fibronectin) and proteoglycans (eg, lumican and decorin).11 Accumulating evidence suggests that, in contrast to previous conception, the ECM is not merely a supportive platform for cells but a dynamic tissue component involved in many cellular processes, such as proliferation, migration and adhesion.12 ECM morphology and structure are constantly undergoing remodelling; components are deposited, degraded or otherwise modified by the cues that cells convey to the matrix. In turn, this dynamic and physically variegated molecular landscape, through its interactions with cell surfaces, is able to induce directional signalling and changes in gene expression. Increased ECM stiffness has already been shown to precede fibrosis in the liver.13 Specifically regarding IBD with fibrostenotic phenotype, a recent study indicates that intestinal fibrosis is autopropagated independent of inflammation, as human colonic fibroblasts cultured on stiff matrices, corresponding to CD strictures, develop a fibrogenic phenotype via mechanotransduction signalling.14
The ECM also serves as a reservoir for signalling molecules, which are exposed upon proteolysis.15 ,16 ECM remodelling processes are executed by matrix proteinases (eg, matrix metalloproteinases (MMPs) and cathepsins), lysyl oxidases (LOX/LOXL) and heparanases. These ECM remodelling enzymes are differentially expressed and contribute to various biological processes.17–19 In many cases, in a somewhat counterintuitive manner, enzymes with contradicting activities (eg, collagen cross-linking vs proteolysis) exhibit a synergistic effect on the same physiological or cellular process, as demonstrated by the attenuation of breast carcinoma cell invasion following combined inhibition of LOX and MMPs.20 Hence, this net enzymatic activity appears to assist the cellular crosstalk in IBD. Of note, in contrast to other post-translational modifications, the processes catalysed by ECM remodelling enzymes (eg, covalent cross-linking and proteolysis) are irreversible. This introduces a higher level of control, generating master switches in the regulation of the immune system and other physiological processes.
Accordingly, the activity of ECM remodelling enzymes is regulated on several levels, including transcription, translation, secretion, activation by cleavage of the pro-domain and inhibition by the endogenous tissue inhibitor of metalloproteinase (TIMP) family. Thus, in the study of the ECM and its remodelling enzymes, gene expression data are far from sufficient in order to assess the actual situation in the extracellular space. This issue, along with the biochemical and physical complexity of the ECM, poses a challenge when approaching ECM involvement in biological processes including IBD pathogenesis.
ECM remodelling and integrity are integral factors in IBD
Various studies over the past two decades have found ECM remodelling enzymes to be upregulated in human IBD. In a complementary manner, the key role of ECM composition and remodelling in the onset, progression and severity of IBD has been implicated in various rodent models. Specifically, animal models genetically modified so as to produce an altered composition of ECM enzymes and/or components exhibit differential phenotypes in response to intestinal inflammation induction. However, the difficulty in performing mechanistic follow-up studies has left the role of ECM remodelling in IBD uncharted territory. A survey of the limited data from rodent models and human studies, presented below, reveals a clear link between ECM remodelling and IBD.
This link has been exhibited in several rodent knockout models. Heparanase, for instance, was shown to be abundant in the colonic epithelium of patients with IBD in contrast to normal colonic tissue or colonic tissue afflicted by infectious colitis,21 and its overexpression in mice led to an increase in colitis via enhanced macrophage activation.22 Elevated levels of collagenase-1 (MMP-1) and stromelysin-1 (MMP-3) have likewise been associated with IBD,23 ,24 as has collagenase-3 (MMP-13), which contributes to intestinal barrier dysfunction, with collagenase-3-deficient mice less susceptible to experimental colitis.25 Matrilysin (MMP-7) deficiency in mice contributes to defence against luminal bacteria and re-epithelialisation,26 while TIMP-3 deficiency results in higher susceptibility to experimental colitis.27 However, some MMPs produce a counter-effect on disease progression. For example, stromelysin-2 (MMP-10)-deficient mice are more susceptible to colitis and inflammation-associated colonic dysplasia.28
Another revealing example of the differential function of MMPs is the highly homologous gelatinases A (MMP-2) and B (MMP-9). The former is more closely associated with homeostasis, while the latter is expressed predominantly in pathological scenarios. Interestingly, gelatinase B has been found to be the predominant upregulated MMP in inflamed intestinal tissue of patients with IBD.29
Gelatinase B is secreted mainly by infiltrating neutrophils and the intestinal epithelium. Remarkably, there is a correlation between this protease's activity in the intestine and the tissue's condition, with the greatest amount exhibited in the inflamed tissue of patients with UC and CD, a lesser amount in the non-inflamed areas in the intestine of these patients, as well as intestinal tissue of patients in clinical remission, and the least amount in healthy controls. This increase in dysregulated proteolytic activity correlates with endoscopic and histological disease scores and with the extent of morphological tissue damage.29–33 Specifically, in fistulae of patients with CD—one of the most debilitating tissue-destructive complications of the disease—gelatinase B presents a marked increase in transcript, protein and activity levels.34 ,35
Gelatinase B has also been recently implicated as a serological, urinary and faecal biomarker for IBD in several studies, which can be used as a tool for IBD diagnosis and monitoring. Its levels in patient bodily fluids were shown to positively correlate with other known IBD indicators and to be influenced by immunosuppressive treatments.36–39 Moreover, recent studies propose gelatinase B to be the superior serum biomarker for CD and UC in a group of acceptable IBD markers exhibiting a strong association with endoscopy and imaging-defined inflammation, as well as with clinical disease activity.24 ,40
Taken together, these data suggest that gelatinase B is a reliable biomarker for human IBD, not a trivial statement considering that gelatinase B is not a canonical inflammatory molecule, such as proinflammatory cytokines. They also imply that ECM remodelling enzymes are prominent factors that characterise and strongly correlate with IBD.
The most striking findings concerning gelatinase B on disease onset demonstrate that gelatinase B-deficient mice are less susceptible to experimental colitis41–43 and preserve a high degree of microfloral diversity associated with protection against colitis,44 whereas gelatinase B overexpression in the gut leads to increased susceptibility.45 These results strengthen the view that stimulated ECM remodelling by gelatinase B is a key mechanism in IBD pathogenesis rather than just a by-product of inflammation.
It is important to note that, in addition to their ECM maintenance duties, the structural components of the ECM also actively take part in intestinal inflammation. For example, the ECM proteoglycan Lumican was shown to contribute to innate immunity via its interaction with toll-like receptor 4 (TLR-4). Consequently, Lumican-deficient mice display a different reaction to colitis induction compared with wild-type mice, exhibiting an attenuated immune response, but also increased morbidity and tissue damage. This phenotype resembles that of myeloid differentiation primary response gene 88 (MyD88)−/− mice and TLR4−/− mice, highlighting the importance of Lumican in innate immunity.46 ,47 Thus, the enzymatic remodelling of the ECM and its composition are important in IBD development and, presumably, also its morphology and biomechanical properties.
In view of this body of evidence, it is plausible that the ECM is responsible for maintaining tissue regulation under normal conditions. Therefore, perturbation of ECM homeostatic flexibility inevitably influences the sensitivity of the intestinal tissue to inflammation. This leads us to postulate that ECM remodelling processes taking place within the intestinal tissue are indeed pivotal in IBD pathogenesis and, furthermore, are capable of steering the tissue towards health or disease.
Postulated IBD pathogenic mechanisms involve ECM remodelling events
Several pathogenic mechanisms are thought to contribute to the chronic intestinal inflammation characteristic of IBD. Among these are (1) increased permeability of the intestinal epithelium, (2) elevated endothelial permeability and (3) self-feeding proinflammatory loops. All three mechanisms require some sort of destruction of ECM tissue barricades, which opens the way to encounters between microbial antigens and the immune system.
Changes in endothelial and epithelial barrier integrity have been associated with MMP activation, which prompts the cleavage and redistribution of cell–cell junction and adhesion proteins.48–54 The migration and invasion of immune cells into the gut mucosa is enabled by proteolysis of the ECM and the basement membrane, which is predominantly performed by matrix proteases expressed by these cells.55 ,56
In addition to their role in barrier function, certain ECM proteins have been identified as factors in inflammation. Heparanase, for example, was shown to increase macrophage sensitivity to stimulation by lipopolysaccharide, and the secretion of proinflammatory cytokines by these cells.22 Another example is the gene for LOX, which was found to be upregulated in rat colitis, and the enzyme was shown to mediate vascular inflammation induced by increased matrix stiffness.57 ,58 Gelatinase B was found to be secreted upon inflammatory stimulation, while its levels decrease in response to anti-inflammatory stimulation by interleukin (IL)-10 in a number of cell types, including immune, epithelial and stromal cells.59–61 Furthermore, this enzyme was shown to potentiate chemokines and cytokines such as IL-8 and IL-1β by proteolytic processing.62 ,63
Only one recent study suggests a specific disease-promoting mechanism for gelatinase B participation in human IBD, revealing how ECM proteolysis-generated fragments directly propagate immune signalling. This mechanism involves a proinflammatory cycle in the gut in which gelatinase B activity induces the formation of a collagen-derived fragment, proline-glycine-proline, which is a chemoattractant for neutrophils, and an inducer of gelatinase B expression.64 This represents the first report of ECM fragment signalling in IBD pathogenesis resulting from specific proteolysis by MMPs.65 This finding highlights the notion that elevated gelatinase B secretion to the extracellular space is not simply a result of the inflammatory process, but also an amplifier of it.
Another key mechanism indicating the regulatory role of ECM integrity in IBD pathogenesis is hyaluronan (HA)-mediated signalling. HA is an abundant GAG ECM component, and its level of polymerisation indicates matrix integrity, in contrast to fragmented HA molecules. Elevated HA fragment deposition in the intestine is associated with inflamed tissue in IBD and has been shown to induce leucocyte infiltration into the intestine and innate immune activation.66 ,67 Also, fragments of HA promote wound healing, but also fibrotic, processes by contributing to fibroblast proliferation and myofibroblast differentiation.68 ,69 Along the same lines, intact HA, formed by high-molecular-weight HA chains, promotes differentiation of anti-inflammatory regulatory T-cells.70
In conclusion, these findings join together like pieces of a puzzle to indicate the active mechanistic role that ECM remodelling, as well as other types of extracellular proteolytic events, plays in the commonly accepted pathogenic pathways responsible for IBD development and progression.
Therapeutic opportunities in the study of ECM function in IBD
Immunosuppressive treatments (eg, corticosteroids and antitumour necrosis factor α (TNF-α)) have had a limited effect on mucosal healing.71 Furthermore, these medications have not shown much promise in preventing or treating long-term complications of IBD (ie, fibrosis and fistulisation). There is, therefore, a pressing need to devise pharmaceutical means to prevent and treat the tissue damage in IBD.
In this respect, we now return to gelatinase B since it has been vastly explored in relation to IBD tissue damage and, as mentioned in previous sections, appears to be a prominent factor in the pathology. This notion is highlighted by several studies showing that decreased gelatinase B transcript, protein and activity levels in the intestinal tissue of patients with IBD correlate with clinical improvement and mucosal healing, facilitated by immunomodulatory treatments, such as TNF-α blockers.72–74 In a similar manner, antigelatinase neutralising antibodies developed in our laboratory exhibit great effectiveness in attenuating IBD in murine models75 and, most recently, a humanised antigelatinase B antibody has entered phase I clinical trials (ClinicalTrials.gov identifier: NCT01831427). This development outlines a remarkable conceptual transformation to one that envisions the role of remodelling enzymes as therapeutic targets in IBD.
Manipulation of HA polymerisation may also be a worthy therapeutic option as this disaccharide polymer has many immunoregulatory roles. In fact, it was recently reported that the injection of high-molecular-weight HA has a beneficial effect on experimental colitis.76
Another medical condition that may benefit from ECM remodelling factors-based therapeutics is IBD-associated fibrosis, considered an irreversible self-propagating process, currently treated mainly by mechanical means (eg, surgical resection or balloon dilation). Unravelling the molecular processes and markers preceding bowel strictures in the early inflammatory state will assist in mitigating this destructive process. As increase in matrix stiffness seems to be an early event in tissue fibrosis, targeting collagen cross-linking enzymes, such as the LOX family, may be of therapeutic significance.
In our view, early treatment of patients with IBD consisting of a combination of immunomodulation and manipulation of ECM remodelling protocols aimed at reaching homeostatic balance in the intestinal tissue holds great promise to preventing inflammation-associated tissue damage. Furthermore, in patients already suffering from tissue destructive complications, ECM homeostatic restoration therapy may very well be the means to reverse these processes.
Summary
The current outlook on IBD biology portrays ECM remodelling and processing in the pathology as a bystander effect of inflammation. We wish to challenge this notion and suggest investigating the extracellular point of view. IBD involves both tissue destruction and fibrosis. These two devastating phenomena are a direct consequence of ECM remodelling events—the first involves ECM degradation and the second, accumulating extracellular fibrogenesis. Therefore, ECM remodelling is a key event and an active participant in IBD pathophysiology.
The ECM tightly interacts with the stromal and parenchymal cells, and ensures the separation between the two groups under homeostatic conditions. Hence, any modification of the ECM influences cellular processes and, since the ECM also acts as a reservoir for signalling molecules, also induces signalling pathways. Therefore, considering that ECM biology is such a large, integral and dynamic part of the tissue, it should not be neglected when approaching any disease, especially IBD. For these reasons and in light of recent evidence from our laboratory, including the results presented herein, targeting ECM remodelling in a specific and fine-tuned manner may contribute to the treatment of IBD by preventing both propagated inflammation and tissue damage.
Acknowledgments
The authors thank Vyacheslav Kalchenko for his assistance in acquiring the second-harmonic imaging data. The research leading to these results has received funding from the Israeli Scientific Foundation, EU FP7, Helmsley Trust and SaveMe project ( to I.S.) and The Leona M. and Harry B. Helmsley Charitable Trust (to ID). IS is the incumbent of the Maurizio professorial chair.
References
Footnotes
-
Contributors ES, DY, ID and IS wrote the manuscript and contributed to discussion. ES and DY collected the literature and analysed it. ES and LB analysed ECM samples.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/