Article Text

Abstract
Background and aim Hepatocellular carcinoma (HCC) is the most common liver cancer. We characterised HCC associated with infection compared with non-HBV-related HCC to understand interactions between viral and hepatocyte genomic alterations and their relationships with clinical features.
Methods Frozen HBV (n=86) or non-HBV-related (n=90) HCC were collected in two French surgical departments. Viral characterisation was performed by sequencing HBS and HBX genes and quantifying HBV DNA and cccDNA. Nine genes were screened for somatic mutations and expression profiling of 37 genes involved in hepatocarcinogenesis was studied.
Results HBX revealed frequent non-sense, frameshift and deletions in tumours, suggesting an HBX inactivation selected in HCC. The number of viral copies was frequently lower in tumour than in non-tumour tissues (p=0.0005) and patients with low HBV copies in the non-tumour liver tissues presented additional risk factor (HCV, alcohol or non-alcoholic steato-hepatitis, p=0.006). P53 was the most frequently altered pathway in HBV-related HCC (47%, p=0.001). Furthermore, TP53 mutations were associated with shorter survival only in HBV-related HCC (p=0.02) whereas R249S mutations were identified exclusively in migrants. Compared with other aetiologies, HBV-HCC were more frequently classified in tumours subgroups with upregulation of genes involved in cell-cycle regulation and a progenitor phenotype. Finally, in HBV-related HCC, transcriptomic profiles were associated with specific gene mutations (HBX, TP53, IRF2, AXIN1 and CTNNB1).
Conclusions Integrated genomic characterisation of HBV and non-HBV-related HCC emphasised the immense molecular diversity of HCC closely related to aetiologies that could impact clinical care of HCC patients.
- HEPATITIS B
- HEPATOCELLULAR CARCINOMA
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Hepatocellular carcinoma (HCC) is the fifth most common cancer and hepatitis B is the most common underlying cause of HCC worldwide.
-
HCC is a group of heterogeneous tumours with several molecular subclasses.
-
HBV infection synergises with exposure to aflatoxin B1 to promote HCC with R249S TP53 mutation in subtropical countries.
What are the new findings?
By analysing resected HCC in France, we identified specific molecular features related to HBV infection:
-
HBX inactivating mutations are selected in HCC tissues suggesting specific pressure of selection during hepatocarcinogenesis.
-
In patients with a low number of HBV DNA copies per cell in the liver, we identified additional risk factor (HCV infection, alcohol intake or non-alcoholic steato-hepatitis) suggesting a cooperative effect for HBV-induced carcinogenesis.
-
TP53 mutations demonstrate different prognostic consequences according to the viral status; relation with poor prognosis was restricted to HBV-infected resected patients.
-
HBV-related tumours demonstrate more frequent progenitor phenotype than non-HBV HCC with an upregulation of genes involved in cell-cycle regulation and encoding oncofetal/progenitor proteins.
How might it impact on clinical practice in the foreseeable future?
-
HBV and non-HBV-related HCC are molecularly different as a result of a different selection of genomic and transcriptomic alterations. Consequently, identification of prognostic markers and therapeutic targets in HCC should be validated in HBV and non-HBV cohorts of patients.
-
A molecular classification of HCC is required to design a more personalised care of the patients taking into account the molecular diversity of the tumours.
Introduction
Hepatitis B is a potentially life-threatening liver disease affecting approximately 5% of world population (350–400 million people).1 Persistent HBV infection causes chronic liver disease and is thought to be responsible for approximately 50% of all hepatocellular carcinoma (HCC) cases and virtually all HCC in childhood. Overall, HCC occur predominantly in male patients and is the fifth most common cancer worldwide.2 ,3
HBV is a small enveloped DNA virus and contains four overlapping reading frames: S (encoding the viral surface proteins, HBS), P (encoding viral polymerase), X (encoding the regulatory X protein, HBX, which is capable of transactivating the expression of numerous cellular and viral genes) and, finally, pre C (encoding the antigens ‘e’ and ‘c’).
HBV is thought to play a key role in HCC development by increasing chromosomal instability and promoting cell proliferation.4 In infected liver tissues, several variants of HBV proteins and genes have been described, including truncated HBS,5 ,6 mutated or deleted HBX,7 ,8 and a novel spliced transcript of HBV, referred to as the hepatitis B spliced protein.9 ,10 HBV integration into the host cell genome has also been reported to be responsible for gene mutations, insertions, deletions or rearrangements of the host genome.11 ,12 Recently, using next generation sequencing technics, a large number of insertion sites were identified.13–15 Two insertion sites of HBV were most frequent: in proximity to TERT, activating telomerase, and in the MLL4 gene. These alterations are suspected to play a major role in HCC carcinogenesis.13–15
However, the underlying nature of viral–host interaction remains elusive. A synergistic effect of HBV infection and exposition to aflatoxin B1 (AFB1) has been reported specifically in subtropical countries in Asia and Africa. AFB1 is a mycotoxin produced by the fungus Aspergillus flavus growing on improperly stored grains; it is metabolised in the liver by cytochrome P450 and induces the formation of highly promutagenic DNA adducts. Patients with both HBV and AFB1 risk factors display a 5–10-fold higher risk of HCC development as compared with HBV or AFB1 alone.16–18 A common site for adduction of AFB1 metabolites is the third base of codon 249 in the TP53 tumour suppressor gene, leading to G to T transversions (AGG to AGT, R to S). This base substitution is identified in 50% of HCC cases from areas with AFB1 exposure, but not in HCC without AFB1 exposure.19 ,20 Therefore, the R249S mutation is considered to be a signature of AFB1 exposure.
HCC is also a heterogeneous group of tumours with variable clinical outcome and molecular features. We formerly reported a robust classification of HCC that comprised six main subgroups (G1–G6) according to their transcriptomic profile.21 These subgroups were closely associated with clinical and molecular features: G1–G2 subgroup demonstrated overexpression of fetal stage-associated genes and were controlled by parental imprinting; G3 tumours were characterised by TP53 mutations and demonstrated adverse clinical outcome;21 ,22 G4 was a heterogeneous subgroup of tumours; and, finally, G5–G6 subgroups were strongly related to β-catenin mutations, leading to Wnt pathway activation.21 We observed that HBV infection was associated with G1–G2 tumours.21
However, the relationship between molecular classification and HBV infection remains to be elucidated. We therefore analysed two groups of tumours related (n=86) or unrelated (n=90) to HBV infection, and investigated the molecular diversity between the two cohorts by screening mutations in genes frequently mutated in HCC14 ,23 ,24 and by profiling of gene expression. Finally, our data were correlated with clinical features and HBV characteristics.
Materials and methods
Patients and tissue samples
The study population consisted of 86 HBV-HCC cases (83 AgHBs-positive and three AgHBs-negative/AbHBc-positive and viral DNA-positive liver samples) and 90 cases of HBV-negative HCCs. Tissue samples were obtained after surgical resection from two French medical care centres in Bordeaux and Creteil. All tumour specimens and their matched adjacent non-tumour liver tissue samples were immediately cryopreserved at −80°C after surgical treatment.25 Demographic features and clinical-pathological records of these patients were prospectively collected in dedicated databases. These data were blindly extracted and are summarised in table 1 and online supplementary table S1. Patients who presented a fatal outcome within 2 months after surgery were censored from the survival analysis. Follow-up was stopped at 60 months (mean 33±20). All patients had given their informed consent according to French law and the Saint Louis Hospital's ethic committee approved the study.
Clinical, histological and pathological data of 86 HBV- and 90 non-HBV-related HCC
HBS/HBX sequencing and genotyping
Viral DNA was amplified by PCR techniques using the Expand High Fidelity PCR System (Roche Diagnostics, Mannheim, Germany), according to manufacturer's instructions, and oligonucleotide primers specific for HBV DNA sequences flanking the S and X genomic regions. In several cases, HBV full-length genomes were amplified by a nested PCR approach using two different primer pairs (see online supplementary table S2) to obtain the amplification of HBS and HBX fragment.21 ,26 Each purified PCR product was sequenced using Applied Biosystems BigDye terminator sequencing chemistry and run on an ABI Prism 3500Genetic Analyzer (Applied Biosystems, California, USA) according to manufacturer’s instructions. The sequences were analysed with Sequencher V.4.7 software (Gene Codes Corporation, Ann Harbor, USA). HBV genotyping (A to H) was then performed according to the sequence of HBS by multiple sequence alignments using the Clustal Omega (http://www.clustal.org/).27 ,28
Virus quantification
Absolute quantification of HBV genome was performed by qPCR in 86 HBV-related tumour samples and in 84 counterpart non-tumour tissues using a set of primers which target HBS gene (HBS4F: GTCCTGGTTATCGCTGGATG; HBS2R: CAAACGGGCAACATACCTTG)29 via the SYBR Green (Applied Biosystems)-based expression analysis method. Results were reported to a gene (ANKRD49) that is located in a chromosomal region (11q21) usually not altered by copy number variation in HCC.25 HBV cccDNA (copy number/ cell) was quantified by a specific real-time PCR, according to previous protocol.30
Mutation screening
DNA was extracted from frozen tissue, in accordance with standard procedures.21 For each tumour, nine genes were screened for somatic mutations: TP53 (exons 2–11), CTNNB1 (exons 2–4, 6–8), PIK3CA (exons 2, 9 and 20), RPS6KA3 (exons 1–22), ARID1A (exons 1–20), IRF2 (exons 2–9), ARID2 (exons 1–20), NFE2L2 (exons 1–5) and AXIN1 (exons 1–10). DNA sequencing was performed by Sanger method. Primers and protocols are available on request. In all cases, the somatic origin of the mutation found in a tumour sample was verified by sequencing the corresponding adjacent, non-tumour liver sample.
Quantitative RT-PCR
RNAs were extracted with RNeasy kit and RT-PCR was performed, as previously described, with the 2-delta delta CT method using the ribosomal 18S as calibrator gene and the mean level expression in normal liver tissues as calibrator sample.21 Expression levels of a set of 37 genes, known in the literature to be involved in tumourigenesis pathways,21 ,31–35 were analysed using TaqMan predesigned gene expression assay (Applied Biosystems). This includes a combination of 16 genes used in the G1–G6 HCC transcriptome classification.21 TaqMan gene expression assays are listed in online supplementary table S3.
Statistical analysis
We used a hypercube algorithm technique to explore model parameter space. HyperCube is an alternative and original approach for the discovery of subgroups derived from least general generalisation algorithms (Torre, Globo). The key characteristic of HyperCube is that it uses non-parametric modelling without any metric and without making any assumption on the distribution of variables or the relationship between the outcome and the input variables.36 ,37 Here we apply this methodology and consider 89 explanatory variables for the series of HBV-related HCC and 80 variables for the series of non-HBV-related HCC (the variables between these two series are similar except for those nine variables about the virus) to identify key factors underlying the risk of HCC of different aetiologies. Quantitative data were presented as mean±SD, and non-parametric Mann–Whitney tests were used to compare quantitative values. All differences were considered significant when the p value was less than 0.05. The interdependence between numerical variables was performed by the use of the Spearman rank correlation test. χ2 And Fisher exact tests were used for given genetic or clinical variable. We used log-rank test and Kaplan–Meier method to assess survival.
Results
Studied cohort characteristics
In this study, we included 86 patients with HBV-HCC and 90 patients with non-HBV-HCC between 1999 and 2009 after tumour resection. In both cohorts, male patients were predominant (80%), and aggressiveness of the tumours were similar as shown by non-significant differences in the frequency of vascular invasion, satellite nodules and early relapse. In contrast, HBV-related patients were younger (p<0.0001), frequently migrants from Africa or Asia (63%, p<0.0001), presented higher α-fetoprotein (AFP) serum levels (p=0.008), had less differentiated and smaller tumours (p=0.05 and p=0.03) than non-HBV patients. Other risk factors of HCC were associated with HBV infection in almost 26% of the cases, whereas aetiologies in non-HBV-related HCC were mainly alcohol intake (40%), HCV infection (23%), haemochromatosis (7%) and non-alcoholic steato-hepatitis (NASH, 4%), with a non-negligible percentage of patients who had a cryptogenic aetiology (26%) (table 1 and see online supplementary table S1).
Viral characterisation in tumour and non-tumour tissues
Sequencing the HBS gene, we identified HBV genotypes according to the Viral Genotype Tool of the NCBI databases in 82 HCC tumours and in 80 of their corresponding non-tumour tissues (four HCCs could not be determined for genotypes because of HBS gene deletion). Genotypes A, B, C, D and E were found in 40%, 12%, 10%, 23% and 15% of non-tumour tissues and in 43%, 10%, 16%, 23% and 8% of tumour samples, respectively (see online supplementary figure S1). In 13 cases (15%), two different genotypes were identified in the same sample (nine only in HCC, three in non-tumour tissue and one in both HCC and non-tumour counterpart). Genotypes were similar in the tumour when compared with the corresponding non-tumour liver tissues, except in eight patients (9% of the cases) with non-tumour to tumour genotypes E:A (n=3), A:C (n=3), B:C (n=1) and D:C (n=1). Genotypes identified in the non-tumour tissues were selected to search for correlations with clinical features. As described in the literature, genotypes A and D were common in patients from Africa and Europe, genotypes C and B in patients from Asia and genotype E in patients from Africa (p<0.0001) (see online supplementary table S4).38 ,39 Genotype D was also more frequent in old patients (p=0.01) and genotype E was associated with hepatitis D virus co-infection in our limited series (p=0.01, n=3/4) (see online supplementary table S4).
Then, we sequenced HBS and HBX genes in 86 HCC and 84 non-tumour counterparts’ samples and compared the spectrum of nucleotide variants in tumour and non-tumour samples (figure 1A, B). Missense mutations were defined as sequence variants modifying amino acid translation and found in less than 1% of the 7822 HBS sequences available in public databases (Hepatitis Virus Database). Inactivating mutations were defined as nucleotide variants leading to stop or frameshift; they also included truncated deletions. Among the tumour and non-tumour tissues, frequency of HBS mutations (88% and 88%, respectively) were similar and the ratio of missense/stop and frameshift mutants were 1.8 and 2.5 (48/27 and 53/21, respectively; figure 1A, B). In contrast, in HBX gene, inactivating alterations were significantly more frequent in tumours (71%) than in non-tumour tissues (33%, p<0.0001, figure 1A, B). The I127N/T/L, K130M/K/Q, V131I/L/T amino acid substitutions in HBX gene, which have been previously identified in HCC tumours,40 ,41 were frequently detected in the present study both in non-tumour and tumour tissues at a similar frequency. These observations suggest that whereas mutations inactivating HBX are selected in tumours, substitution at residues I127, K130 and V131 are not.

HBV virus profile in tumour and non-tumour adjacent samples. (A and B) Spectra of mutation in HBS and HBX gene in tumour and paired non-tumour samples. (C) Distribution of tumour and non-tumour samples according to quantification of HBV copy/cell and determination of two groups characterised by low (<0.5 HBV copy/cell) and high (>0.5 HBV copy/cell) copy number. (D) Correlation of HBV quantification (log10 copy/cell) in tumour and paired non-tumour samples. p Values obtained from χ2 (*) and Willcoxon signed rank (**) tests are shown. MHL, major hydrophilic loop; RD, regulatory domain; PSR, proline serine rich hypervariable region; KI, Kunitz domain-like; TD, transactivation domain.
Using quantitative PCR, we quantified the total number of viral copies by determining an absolute quantification of HBS DNA corresponding to the different types of HBV DNA in the tissues, that is, episomal, integrated or the covalently closed circular DNA (cccDNA). According to the median number of HBS copies/cell in the non-tumour liver tissues, we defined two groups of patients with high total HBS copies (>0.5 copy/cell) or with low total HBS copies (<0.5 copy/cell) in the non-tumour tissues (figure 1C). Overall, in 53 out of 84 (63%) of the paired samples, we found a lower amount of total HBS DNA copy in the tumour tissues compared with their corresponding non-tumour tissues (p=0.0005; figure 1D). In contrast, cccDNA number of copies is similarly distributed in both compartments (see online supplementary figure S2A,B). Consequently, the lower amount of HBV DNA observed in tumours is mainly related to a decrease of non-replicative viral genome. Thus, in tumours, the mutations observed in HBV sequences should be mainly related to non-replicative form of the virus.
Interestingly, we showed a correlation between viral copy numbers in non-tumour tissue, viral genotype in non-tumour tissue and the presence of other risk factors. Patients with low HBS DNA copy number presented more frequently an additional risk factor (HCV, alcohol or NASH) than patients with high viral quantification (38% vs 12%, p=0.006). Moreover, patients with a low viral quantification were more frequently infected by HBV genotypes D and E (p=0.01).
Spectrum of somatic gene mutations correlates with HBV infection and survival
In the overall series of 176 HBV and non-HBV-related HCC, we searched for somatic mutations by screening the coding sequence of nine genes previously described as frequently altered in HCC23 (table 2, figure 2 and see online supplementary tables S5 and S6). Comparing the spectrum of somatic mutations in HBV and non-HBV HCC, we identified significant differences. NFE2L2, coding NRF2 a key transcription factor involved in oxidative stress response, was only mutated in non-HBV HCC (7/90, 8%) and not in HBV-infected tumours (0/86, p=0.008; table 2). CTNNB1 activating mutations were also less frequent in HBV-HCC (13/86, 15%) compared with non-HBV HCC (40/90, 44%, p<0.0001; table 2). These results suggested that the WNT/ß-catenin and the oxidative stress pathways are significantly less frequently activated by gene mutation in HBV-related carcinogenesis than in HCC associated with other risk factors.
Gene mutations of 86 HBV- and 90 non-HBV-related HCC

Somatic mutation spectra of TP53 and CTNNB1 in 86 HBV-related and 90 non-HBV-related HCC. Functional domains are coloured boxes. ARM, armadillo repeat; DBD, DNA binding domain; Dim, dimerisation domain; NES, nuclear export signal; NLS, nuclear localisation signal; NTD, negative transactivation domain; TD, transactivation domain; ser, serine; HCC, hepatocellular carcinoma.
In contrast, P53 pathway was more frequently altered in HBV-HCC and the TP53 gene was the most frequently mutated gene in HBV-related HCC (41%), whereas it was rarely mutated in non-HBV tumours (16%, p=0.0002). R249S mutation in TP53, specific of the AFB1 exposure, was exclusively found in HBV-HCC (16%, p<0.0001; table 2) particularly with genotype B (p=0.005; table 3). According to the lack of AFB1 exposure in France, all the patients with R249S mutations were migrants coming from Asia or sub-Saharan Africa (p=0.02; table 3). Interestingly, we have observed a correlation between R249S mutations and poor tumour differentiation (p=0.05; table 3); tumours with R249S mutations were also more frequently developed in non-cirrhotic liver (p=0.02) in contrast to other HBV-infected HCC.42 Overall, most of the TP53 mutations were described to inactivate P53 according to the IARC database (see online supplementary table S5). Alterations inactivating IRF2, a tumour suppressor gene controlling p53 protein activation, were also exclusively identified in HBV-HCC (7%, p=0.01; table 2). According to their function in the same pathway, IRF2 and TP53 mutations were mutually exclusive in tumours (p=0.002).
Clinical, histological and viral data of 86 HBV-related HCC, classified by the TP53 R249S mutation or not
Strikingly, TP53 mutations were significantly associated with shorter disease-specific survival (tumour-related death) in HBV-HCC (p=0.03; figure 3A) but not in non-HBV-HCC (figure 3B). In HBV-HCC, Edmondson grade III–IV (p=0.01), the presence of vascular invasion (portal invasion p<0.0001 and microvascular invasion p=0.009) and satellites nodules (p=0.02) were also significantly associated with a shorter disease-specific survival using univariate analysis (table 4). In multivariate analysis, TP53 mutations (p=0.004), the presence of portal invasion (p=0.001) and of microvascular invasion (p=0.09) were independently associated with an increased risk of death (table 4).
Univariate and multivariate analysis for the selected clinical features of HBV- and non-HBV-related HCC

Significant prognostic values of TP53 mutations. (A) Disease-specific survival in patients infected by HBV with HCC mutated or wild-type to TP53 and (B) in patients with HCC, mutated or wild-type to TP53, related to other aetiologies. HCC, hepatocellular carcinoma.
Interestingly, among HBV-HCC patients, European origin was associated with good prognosis in univariate and multivariate analyses (p=0.02 and p=0.02, respectively) and R249S hot spot mutations were not associated with survival (see online supplementary figure S3). Finally, TP53 mutations were also significantly associated with poor prognosis in the subgroups of HBV patients with small (<55 mm; p=0.05) or well-differentiated HCC (Edmondson grade I–II; p=0.04; see online supplementary figure S4).
Altogether these data underlined that TP53 mutations have a prognostic value in early HCC independent of classical clinical features in HBV-related HCC but not in other aetiologies.
Integration of transcriptomic profiling in HBV-related HCC classification
We selected 37 genes differentially expressed among the different HCC subgroups previously defined by transcriptomic profiling and we classified the 176 HCC samples according to the six defined transcriptomic subgroups G1–G6.21 HBV-HCC retains a genomic diversity and they were distributed in all G1–G6 subgroups. However, the genomic distribution according to the G1–G6 classification was significantly different between HBV and non-HBV-related HCC (p=0.001) with an enrichment of G2 and G3 HCC in HBV patients compared with non-HBV HCC (figure 4A). We further were able to demonstrate that the genes associated with progenitor features (EpCAM, AFP, KRT19 and CCNB1) were globally significantly overexpressed in HBV-HCC compared with HCC related to other aetiologies (figure 4B). This result showed that HBV-HCC more frequently demonstrates a progenitor profile than the other tumours.

Integration of transcriptomic profiling. (A) Transcriptomic groups (G1–G6) in HBV (white) and non-HBV-related (grey) HCC (mean with 95% CI). p Values obtained from χ2 test are shown. (B) Gene's expressions in HBV- and non-HBV-related HCC (mean with 95% CI). p Values obtained from Mann–Whitney test are shown. EPCAM, epithelial cell adhesion molecule; KRT19, keratin 19; AFP, α-fetoprotein; CCNB1, cyclin B1; HCC, hepatocellular carcinoma.
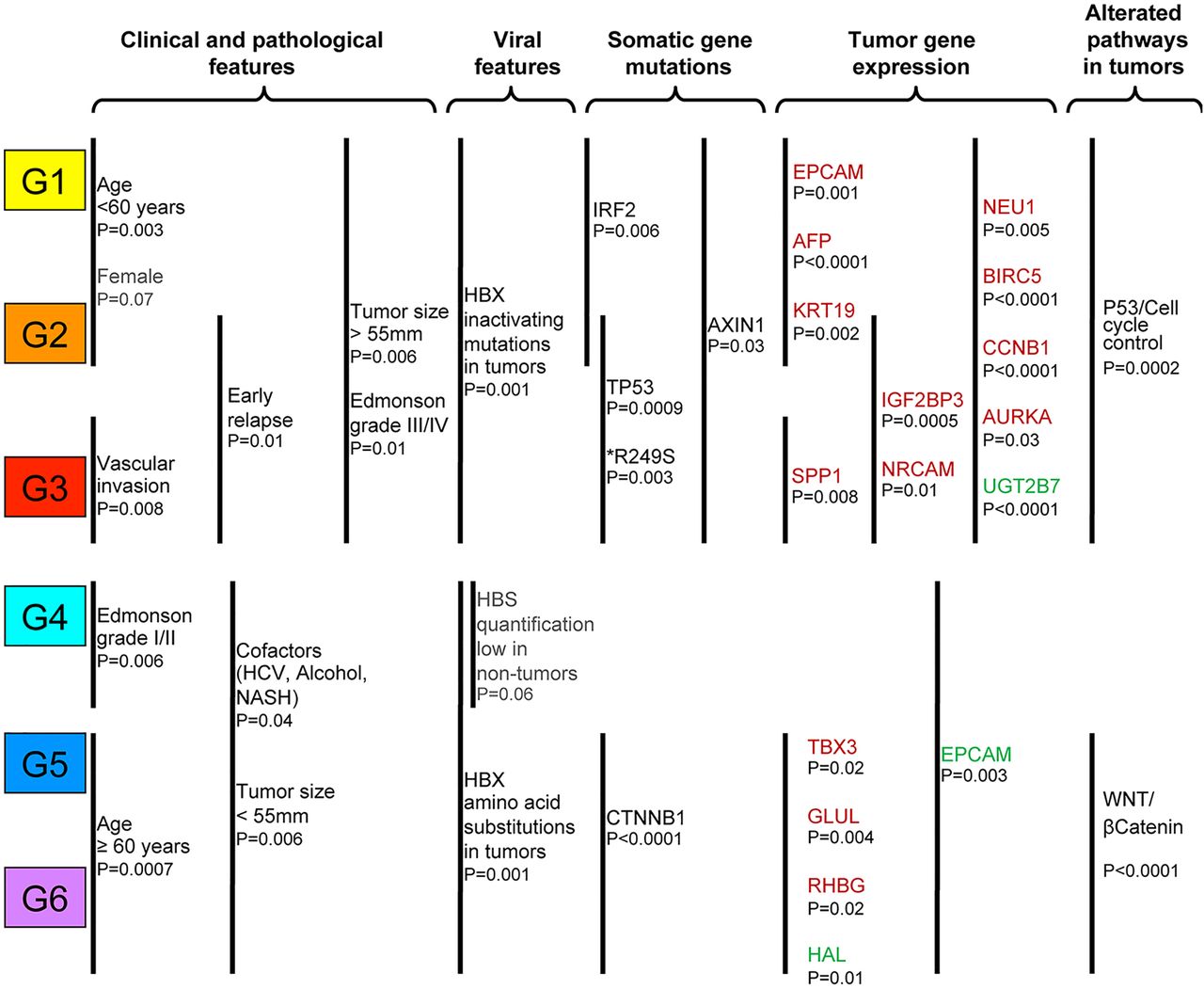
Within HBV-HCC, using transcriptome classification, we defined subgroups of patients in terms of clinical, pathological, viral and genetic features (figure 5). Partition in the two major groups (G1–G3/G4–G6) showed that G1–G3 subgroups included 57% of HBV-HCC and were enriched in large tumours (>55 mm; p=0.006; figure 5), characterised by frequent HBX gene inactivating mutations (p=0.001; figure 5) and frequent somatic AXIN1 mutations (p=0.03; figure 5). Similar to HCC based on various aetiologies, HBV-HCC classified in G1–G3 was also characterised by the proliferative/stem cell features with an overexpression of a large number of genes implicated in cell-cycle control (AURKA p=0.03, BIRC5 p<0.0001, NEU1 p=0.005, CCNB1 p<0.0001; figure 5) and a down-expression of genes involved in differentiation like UGT2B7 (p<0.0001; figure 5). These associations were confirmed at the histological level, demonstrating that tumours categorised in G1–G3 are less differentiated (p=0.01; figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematisation of the different HBV-HCC subgroups (G1–G6) defined by transcriptome analysis with their related clinical and genetic alteration. Red and green indicate overexpression and under-expression of gene, respectively, in the corresponding transcriptomic subgroup(s). p Values obtained from χ2 tests are shown. IRF2, interferon regulatory factor 2; EPCAM, epithelial cell adhesion molecule; KRT19, keratin 19; AFP, α-fetoprotein; SPP1, osteopontin; TBX3, T-box protein 3; GLUL, glutamate-ammonia ligase; RHBG, Rh family, B glycoprotein; HAL, histidine ammonia-lyase; IGF2BP3, insulin-like growth factor 2 MRNA binding protein 3; NRCAM, neuronal cell adhesion molecule; NEU1, sialidase 1; BIRC5, baculoviral IAP repeat containing 5; CCNB1, cyclin B1; AURKA, aurora kinase A; UGT2B7: UDP glucuronosyltransferase 2 family, polypeptide B7; HCC, hepatocellular carcinoma.
Among the G1–G3 group of HBV-HCC, in G1–G2 classes, we identified a specific subgroup of tumours enriched with IRF2 inactivating alterations (p=0.006; figure 5) and characterised by a high overexpression of genes encoding oncofetal/progenitor proteins like epithelial cell adhesion molecule (EPCAM) (p=0.001), AFP (p<0.0001) and KRT19 (p=0.002; figure 5). Moreover, tumours classified in G3 showed frequent vascular invasion (p=0.008) and SPP1 (gene target of DNA replication, coding osteopontin) overexpression (p=0.008) underlining their aggressiveness (figure 5).21 ,43 Next, in G2–G3, we confirmed the association with frequent TP53 mutations (p=0.0009), all including R249S substitution (p=0.003) and a high rate of early recurrence (p=0.01; figure 5).
Interestingly, HBV-HCC classified in G4–G6, the second major transcriptomic class, was frequently associated with an additional risk factor (p=0.04) like HCV, alcohol intake or NASH, suggesting that HBV genome interaction would be less important for carcinogenesis in G4–G6 HCC than for those classified in G1–G3 subgroups (figure 5). Accordingly, HCC group G4 was characterised by a lower number of HBS DNA copy/cell in their corresponding non-tumour liver tissues (p=0.06) and by a good histological HCC differentiation (p=0.006; figure 5). G5–G6 groups consisted of a homogeneous group of HCC developed in older patients (≥60 years; p=0.0007; figure 5) with CTNNB1 activating mutation related to WNT/β-catenin pathway activation (p<0.0001), demonstrating an overexpression of GLUL (p=0.004), TBX3 (p=0.02) and RHBG (p=0.02) and a down-expression of HAL (p=0.01), which are four well-known WNT/β-catenin target genes (figure 5). In this line, the low number of HCC classified in the G5–G6 subgroup in HBV patients is closely related to the low number of CTNNB1 mutations observed in these tumours.
Discussion
In this study, we have undertaken a comprehensive and integrative approach to elucidate the molecular characteristics of HBV-HCC. Using transcriptome classification, gene sequencing, viral HBV characterisation and gene expression analysis, we showed that HBV-related HCC differs from non-HBV-related tumours. Moreover, among HBV-related HCC, we identified subgroups of tumours revealing specific viral, genetic and transcriptomic features.
We showed that genomic features were significantly different in the group of HBV-HCC compared with non-infected patients. These results suggest that HBV-related HCC use alternative mechanisms for tumourigenesis to some extent. A high frequency of p53 inactivation and stem cell genes overexpression provide a potential pathogenic link between impaired cell reprogramming and HBV infection. Notably, these observations have clinical implication since TP53 mutations were associated with poor prognosis only in HBV-related tumours. These results could at least partly explain the discrepancies in the literature regarding the prognostic predictive value in HCC patients. Indeed, most frequently, TP53 mutations were shown to be associated with prognosis in Asian populations infected by HBV,44–46 whereas in a series of patients with mixed aetiologies, TP53 mutations were usually not an independent prognostic marker.43 ,47
According to our previous study,21 CTNNB1 mutations defined a homogenous subtype of HCC classified in G5–G6 transcriptomic groups and overall G4–G6 groups of HCC were less frequently HBV infected. However, the tumours classified in G4–G6 subgroups also represent a small subset of tumours among the HBV-related HCC, characterised by frequently older patients, with other cofactors such as HCV, alcohol consumption or NASH. In the same line, we observed an association between low HBS DNA copy and presence of other risk factors. Altogether, these results suggest that among the group of HBV-infected patients, the association with an additional cofactor could interfere with the mechanism of carcinogenesis and the molecular features of this tumour type. We could hypothesise a different time frame of genetic alterations with a minor role of direct viral carcinogenesis, absence of AFB1 mutagenesis, and a predominant role of cofactor and stochastic accumulation of mutations during ageing. This carcinogenic pathway could promote the occurrence of CTNNB1-mutated HCC and HCC classified in the G4 subgroup that is otherwise unusual in HBV-related HCC.
Among the tumours related to HBV infection, genomic alterations can be associated with specific subgroups of tumours with particular clinical and epidemiological features. The R249S variant, the hot spot of mutation induced by AFB1 in TP53, is the result of the synergistic effects of AFB1 and HBV on tumour development.16 In this regard, migrants coming from AFB1 exposed regions were treated and their tumours analysed similar to the non-migrant and non-infected patients. In our study, we were able to demonstrate that R249S mutation is associated with HBX inactivation and that this association is detected essentially, if not exclusively, in patients who developed HCC without clinical evidence of pre-existing or simultaneous liver cirrhosis. This observation supports the hypothesis that R249S and HBX could cooperate in promoting a pathway of hepatocarcinogenesis that does not require liver cirrhosis as an underlying, preneoplastic state.
Despite a large number of published studies, the function of HBX in liver carcinogenesis is still debated.8 ,48 ,49 Here, by viral profiling of more than 80 cases, we identified frequent inactivating mutations in HBX gene in more than 70% of the tumours. We identified deletion of the COOH terminal part similar to that described by Iavarone and collaborators in their first report of six analysed tumours.40 Interestingly, in the present work, by analysing a larger number of cases we also identified premature stop codon and large deletions leading to a complete inactivation of the HBX gene. It seems that wild type HBX or overexpression of truncated HBX proteins could demonstrate oncogenic function and promote tumourigenesis by abrogating cell-cycle arrest and apoptosis inhibition.40 ,50–52 Our results suggest that mutations inactivating HBX are selected in the tumours in contrast to the non-tumour liver tissues. Consequently, we can hypothesise that HBX inactivation could participate in liver carcinogenesis. Surprisingly, we also identified a correlation between inactivating mutations of HBX gene and G1–G3 transcriptomic groups suggesting a close association between viral genome alterations and transcriptome expression during carcinogenesis that remains to be further investigated. Interestingly, transcriptomic classification revealed that these tumours belong to the group of patients with specific features: younger female patients frequently coming from Africa or Asia with tumours characterised by abnormal expression of oncofetal genes (EPCAM, AFP and KRT19).
In conclusion, our data collectively suggest that HBV infection leads to a specific type of HCC characterised by immature features with stem cell features when compared with HCC related to other risk factors. These results suggest that HBV-related HCC is associated with both HBX and p53 inactivation in a subset of tumours, proposing that these two factors could cooperate. Moreover, TP53 mutation is associated with outcome for HBV-infected patients and it could have a notable clinical implication in risk stratification and treatment decision in this population of patients. Moreover, we identified in HBV-related HCC a high variability that probably reflects molecular heterogeneity and this aspect may also explain the high level of resistance again in a large number of therapeutic agents.
Acknowledgments
We thank Kornelius Schulze for critical reading of this manuscript. We warmly thank all physicians who have participated in this work and particularly Jean Saric, Brigitte Le Bail, Anne Rullier, Antonio Sa Cunha, Daniel Cherqui and Jeanne Tran Van Nhieu. We thank Cecile Guichard and Ichrafe Ben Maad for the help in sequencing genes. We thank the tumour bank of CHU Henri Mondor (Creteil, Paris) and CHU Bordeaux for providing tumour samples. This work was supported by the ANRS, INCa with the ICGC project, the Réseau national CRB Foie, HEPTROMIC (FP7), BioIntelligence and INSERM. GA is supported by a fellowship from the ANRS and QC is supported by a fellowship from HEPTROMIC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
-
GA and QC contributed equally.
-
Contributors GA, QC and YL performed molecular characterisations. J-CN contributed to survival analysis. SI, DJ and YGM performed statistical analyses. CL and AL provided samples and clinical information. JC and PB-S provided samples and pathological reviewing. JZ-R designed and coordinated the overall study. All authors contributed to writing the manuscript.
-
Funding ANRS; INCa; FP7 (HEPTROMIC); OSEO (BIOINTELLIGENCE); INSERM.
-
Competing interests None.
-
Patient consent Obtained.
-
Data sharing statement The authors commit to assisting any investigators seeking to replicate or follow up this study with advice and sharing data.
-
Ethics approval Saint Louis Hospital's ethic committee (Paris, France).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/