Article Text

Abstract
Objective Inflammation and oxidative stress drive disease progression in chronic hepatitis C (CHC) towards hepatocellular carcinoma. HCV is known to increase intracellular levels of reactive oxygen species (ROS), but how it eliminates ROS is less well known. The role of the ROS scavenger glutathione peroxidase 4 (GPx4), induced by HCV, in the viral life cycle was analysed.
Design The study was performed using a replicative in vitro HCV infection model and liver biopsies derived from two different CHC patient cohorts.
Results A screen for HCV-induced peroxide scavengers identified GPx4 as a host factor required for HCV infection. The physiological role of GPx4 is the elimination of lipid peroxides from membranes or lipoproteins. GPx4-silencing reduced the specific infectivity of HCV by up to 10-fold. Loss of infectivity correlated with 70% reduced fusogenic activity of virions in liposome fusion assays. NS5A was identified as the protein that mediates GPx4 induction in a phosphatidylinositol-3-kinase-dependent manner. Levels of GPx4 mRNA were found increased in vitro and in CHC compared with control liver biopsies. Upon successful viral eradication, GPx4 transcript levels returned to baseline in vitro and also in the liver of patients.
Conclusions HCV induces oxidative stress but controls it tightly by inducing ROS scavengers. Among these, GPx4 plays an essential role in the HCV life cycle. Modulating oxidative stress in CHC by specifically targeting GPx4 may lower specific infectivity of virions and prevent hepatocarcinogenesis, especially in patients who remain difficult to be treated in the new era of interferon-free regimens.
- HEPATITIS C
- OXIDATIVE STRESS
- LIPID PEROXIDATION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject
HCV induces oxidative stress.
Oxidative stress and inflammation drive disease progression towards hepatocarcinogenesis.
Data on the type of reactive oxygen species (ROS) as well as ROS scavenging enzymes induced by HCV remain contradictory and are mostly based on non-replicative HCV infection models.
The role of ROS scavenging members of the family of glutathione peroxidases in HCV replication remains unknown.
What are the new findings?
Infectious HCV virions (genotype 2a) elevate superoxide anion but reduce peroxide levels.
NS5A of HCV induces expression and activity of glutathione peroxidase (GPx) 1 and 4 in a phosphatidylinositol-3-kinase/protein kinase B-dependent manner, but only GPx4 is required for HCV infection.
The physiological activity of GPx4 is required for maintenance of high specific infectivity of progeny virions, by preventing accumulation of lipid peroxides in virion membranes and ensuring efficient viral fusion.
The HCV-induced increase of GPx4 expression is reversible and returns to baseline upon viral eradication in vitro and in vivo, following interferon-based or interferon-free regimens.
How might it impact on clinical practice in the foreseeable future?
Our results, generated with original and relevant models, show that oxidative stress is detrimental for HCV infection and that HCV depends on the induction of ROS scavengers, and particular scavenging of lipid peroxides.
Antioxidant therapies may alleviate the detrimental effects of ROS on liver physiology, but they will favour viral replication as well. Our findings give a plausible explanation why complementation of antiviral therapies with antioxidants has not shown much benefit to patients.
Targeted induction of lipid peroxidation using pharmacological approaches may be of benefit to patients with chronic hepatitis C.
Introduction
HCV infection is thought to induce oxidative stress in vitro and in vivo; for example, increased levels of lipid peroxidation products have been detected in livers1 ,2 and sera3 of chronic HCV-infected patients. HCV-induced oxidative stress is thought to be caused by a combination of virus–host cell interactions, iron overload and host immune reactions1 and to correlate with markers of inflammation, fibrosis and hepatic iron storage.4 However, accumulation of oxidative stress markers has also been reported in patients with mild, moderate or no liver disease,2 pointing to a direct role for HCV in reactive oxygen species (ROS) production. In vitro, several HCV proteins, expressed alone or in the context of the polyprotein, induce oxidative stress and antioxidant enzymes (ref. 5 and references therein). However, studies quantifying ROS using replicative HCV remain few and the results contradictory, particularly with respect to the induction of peroxides.6–9 Peroxides are second messengers for inflammatory signalling pathways, oxidise lipids, proteins and DNA and trigger the formation of hydroxyl radicals, the most damaging form of ROS.5 Peroxides are eliminated by different enzymatic families, including catalase (CAT), glutathione peroxidases (GPx) or peroxiredoxins. While HCV is known to induce ROS scavengers such as superoxide dismutase (SOD) and CAT and to induce accumulation of oxidised glutathione,5 the role of GPx enzymes in stress defence in HCV infection remains unexplored. So far, only a decline of GPx2 and 3 transcripts and an increase of GPx8 transcripts have been observed in HCV-infected cells.10–12
The GPx family consists of eight isoforms.13 GPx1, 2 and 3 are cytoplasmic or secreted, and share a common tetrameric structure with strong affinity for soluble peroxides. In contrast, GPx4, a monomer with its active site embedded in a hydrophobic surface,14 ,15 is thought to be the predominant scavenger of phospholipid or cholesterol hydroperoxides, even if these are incorporated in lipoproteins or biomembranes.16–18 GPx5 to GPx8 remain ill characterised and GPx7 and 8 mediate protein folding at the endoplasmic reticulum (ER).13
We have used HCVcc-infected Huh7.5 cells and liver biopsies to re-assess and further explore ROS production and elimination in HCV infection. We show that HCV induces GPx4 in vitro and in vivo and that the catalytic role of GPx4 in the scavenging of lipid peroxides is required for the production of HCV virions with high specific infectivity. The fact that, upon successful viral elimination, GPx4 levels reverse back to baseline in patients and in tissue culture furthermore underlines the importance of GPx4 in HCV infection.
Experimental procedures
HCVcc production
An adapted JFH1 strain (F172C, P173S, N534K) and a selectable version of the same strain were in vitro transcribed and electroporated into Huh7.5 cells.19 Supernatants were harvested upon three passages, 0.45 µM filtered, quantified using the 50% tissue culture infective dose (TCID50) method and used for infections as previously described.19 To quantify infection, intracellular and extracellular RNA was harvested as described in online supplementary methods. For determination of intracellular and extracellular TCID50 values, virions were harvested by repeated freeze/thaw cycles or from the supernatant, respectively, 0.45 µM filtered, calculated as previously described20 and normalised to cell numbers.
Dichlorofluorescein diacetate, dihydroethidium, glutathione and BODIPY 581/591 C11 staining
H2O2, superoxide and lipid peroxide production were measured using 2′,7′–dichlorofluorescein diacetate (FluoProbes), dihydroethidium (DHE) (FluoProbes) and BODIPY 581/591 C11 (Life Technologies), respectively, according to the manufacturer's instructions. Total and oxidised glutathione were quantified using a commercial kit (Enzo). Glutathione content was normalised to protein content.
RNA silencing
Huh7.5 cells were transfected with siRNA mixes targeting GPx1 (Sigma EHU218531), GPx4 (Sigma EHU119391, Dharmacon LQ-011676-00-0002), SOD1 (Sigma EHU050511), SOD2 (Sigma EHU075891), GSR (Sigma EHU160161) or mock (Sigma SIC001, Dharmacon D-001810-01) using DharmaFECT1 (Dharmacon) according to manufacturer's recommendations, or were transduced with a lentivirus pLKO.1 expressing an shRNA targeting GPx4 (Sigma). Medium was changed 15 h later. Viability tests were performed with the CellTiter96 AQueous One Solution Cell Proliferation Assay (Promega). Unless otherwise mentioned in the study, the Sigma siRNA mixture targeting GPx4 was used.
GPx activity assay
Total GPx-specific and GPx4-specific activities were measured with tert-butyl hydroperoxide (tBOOH) and cumene hydroperoxide (CHP), respectively, as substrates.16 ,21 Huh7.5 cells lysates (100 mM Tris–HCl pH 7.6, 5 mM EDTA, 1 mM NaN3, 0.1% Triton-X100, 30 s sonication 3×, centrifugation (5 min, 14000 g)) were complemented with 0.6 U/mL glutathione reductase (GR) (Sigma), 0.2 mM nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), 3 mM reduced glutathione (GSH), 100 µM tBOOH or CHP (Sigma) and NADPH consumption quantified on an Infinite 200PRO Reader (Tecan) at 340 nm for 5 min at 37°C. GPx activities were calculated by subtracting absorbance decay obtained with buffer without cell lysates, by using NADPH extinction coefficient of 6220/M/cm and by normalising to total protein content.
Density gradient separation
Huh7.5 cells were infected at a multiplicity of infection (MOI) of 0.1 and transfected with siRNA the same day. The medium was changed 15 h after transfection. Supernatants were harvested 3 days later, spun at 11 000 g for 5 min to remove cell debris, filtered (0.45 μm) and loaded on Amicon Ultracel 100 K centrifugal filter units (Millipore) to concentrate them 10-fold, according to the manufacturer's recommendations. Concentrated supernatants (1 mL) were loaded on precast iodixanol–sucrose density gradients and ultracentrifuged for 16 h at 35 000 rpm at 4°C using SW 41 Ti rotor in an Optima L-90K centrifuge (Beckman). Fractions were harvested from the top of the tube (first four fractions=500 µL, the rest of the fractions=1 mL) and their density was determined with a refractometer. Discontinuous iodixanol–sucrose density gradients were prepared by successive freezing of fractions ranging from 35% or 54% iodixanol to 6% iodixanol. Each fraction contained 10 mM Tris–HCl pH 8, 2 mM EDTA, iodixanol (OptiPrep density medium) and sucrose (each fraction was filled up to 1 mL with 0.25 M sucrose to keep osmolarity). Precast density gradients were thawed at 4°C for 5 h before use in order to obtain continuous density gradients.
GC-MS analysis
Gas chromatography-mass spectrometry (GC-MS) analysis was carried out on a Hewlett–Packard quadruple mass spectrometer interfaced with a Hewlett–Packard gas chromatograph as previously described.22 The gas chromatograph was equipped with an HP-5MS fused-silica capillary column (60 m×0.25 mm inner diameter, 0.25 µm film thickness; Agilent Technologies). The oven temperature gradient program used was 2 min at 57°C, then increased to 180°C at 20°C/min, followed by an increase to 280°C at 4°C/min. Samples were injected with a pulse splitless injector with a head pressure of 7.9 p.s.i. The interface, injector and ion source temperatures were set at 280, 260 and 150°C, respectively. Electron energy was set at 70 eV. Helium and methane were used as carrier and reagent gases, respectively. Negative ion chemical ionisation mode was used, and the mass spectra were acquired from 100 to 800 Da. Phosphatidylethanolamine (PE)-Michael adducts were quantified in the selected ion monitoring mode using the main specific characteristic fragments of derivatised ethanolamine-alkenal adducts at /m/z/ 561 and 541 for PE-4-hydroxy-2E-hexenal (4-HHE), 603 and 583 for PE-4-hydroxy-2E-nonenal (4-HNE), 643 and 623 for PE-4-hydroxydodeca-(2E,6Z)-dienal (4-HDDE), and 564 and 544, 606 and 586, and 646 and 626 for their deuterated counterparts used as internal standards, respectively. For internal standardisation, deuterated 4-HNE, 4-HDDE and 4-HHE were synthesised as previously described22 and added to the samples.
Fusion assay
To obtain R18-labelled liposomes R18 and lipids were mixed as ethanol and chloroform solutions, respectively (5 mol% R18 final).23 Viruses suspended in 0.85% (w/v) NaCl, 10 mM Tricine-NaOH, pH 7.4 and R18-labelled liposomes (final lipid concentration 15 μM) were mixed in a cuvette. After temperature equilibration, fusion was initiated by acidification with an appropriate volume of HCl. Kinetics were recorded on a PicoFluor hand-held fluorimeter (Turner Biosystems, excitation and emission wavelengths 540±20 and >570 nm, respectively). Maximal R18 dequenching was measured after addition of 0.1% TritonX-100 (final concentration).
Liver biopsies and clinical data
Liver biopsies from control (Hodgkin's lymphoma, n=12, autoimmune hepatitis n=12 and fatty liver n=3; total n=27) and patients with chronic hepatitis C (CHC) (n=116) were acquired during routine diagnostic work at the University of Cologne and stored in liquid nitrogen whenever sufficient material was available. The patient's written informed consent had been obtained in accordance with the local ethical policies. Among the 116 HCV biopsies, 53 were derived from male patients, 36 from female patients. Mean age of patients was 44±14 years and mean body mass index (BMI) 24.8±3.7 kg/m2. Detailed patient data including DESMET scoring for fibrosis and inflammation were available for 89 HCV biopsies (median inflammation grade: 2, range 1–3; median fibrosis stage: 2, range 0–4). No information on genotype was available for nine biopsies, 50 were infected with genotype 1 and 30 with genotypes 2 or 3.
Paired biopsies from patients with CHC before and after antiviral treatment were used under the French IRB ‘Comité de Protection des Personnes (CPP) Sud-Est 287 IV’ agreement #11/040 obtained in 2011. Written informed consent had been obtained. Among the paired biopsies obtained from 16 different patients with CHC before and after antiviral treatment, seven were derived from male patients and nine from female patients. Mean age of patients was 61.5±21.5 years. Fourteen patients were infected with genotype 1 and two with genotype 2. METAVIR scores revealed a median inflammation grade of 1, range 0–3 and median fibrosis stage of 2, range 0–3.5. Mean transaminase scores were 1.6 N±0.4, range 1–4 N.
Statistics
Data were analysed with the Wilcoxon Mann–Whitney test and by calculating Pearson correlation coefficients using GraphPad Prism software.
Additional procedures are described in the online supplement.
Results
HCV induces intracellular oxidative stress
Huh7.5 cells were infected with HCVcc, produced from an adapted JFH1 strain,19 at various MOIs. Three or four days postinfection (dpi) cells were harvested and stained either with DHE, a stain that fluoresces upon interaction with superoxide anions (figure 1A, B), or with an anti-HCV serum in order to validate HCV expression in the cultures (see online supplementary figure S1A). HCV increased superoxide levels between 1.3-fold and 1.7-fold depending on the MOI, and close to twofold in cells constitutively replicating a selectable version of the same JFH1 strain (figure 1B). To quantify peroxide levels, infected Huh7.5 cells were stained with 2′,7′-dichlorodihydrofluorescein diacetate, which is oxidised in a peroxide-dependent manner to fluorescent 2′,7′-dichlorofluorescein (DCF). Three dpi, an MOI-dependent decrease in DCF fluorescence, was observed, suggesting that HCV actively mobilises peroxide scavenging (figure 1C, online supplementary figure S1B). In constitutively infected cultures, DCF fluorescence was reproducibly increased by 30%, suggesting that ROS accumulate in the long term in HCV-infected cells, although activated ROS scavenging. Finally, in HCVcc cells infected with an MOI of 0.1, no changes in the ratio of oxidised to GSH were observed at 3 dpi, when over 90% of cells actively replicated the virus (figure 1D, online supplementary figure S1C) nor at 4 or 5 dpi, when HCV had saturated the culture (data not shown). However, an increase in oxidised glutathione was observed in constitutively infected cells (figure 1D).

HCV infection induces intracellular oxidative stress. Reactive oxygen species (ROS) quantification in Huh7.5 cells, infected with HCVcc at the indicated multiplicity of infections (MOIs) for 3 or 4 days, or infected constitutively with a selectable HCV genome (const.inf.). (A) An overview of ROS species and scavengers. Superoxide (O2•−) is produced by electron leakage at the electron transport chain in mitochondria or by cytoplasmic sources and converted by superoxide dismutase into peroxides. Peroxides are scavenged by the family of glutathione peroxidases (GPx) or catalase (CAT). These enzymes require reduced glutathione (GSH) as cofactor which is oxidised to glutathione disulfide (GSSG) in the process. GSSG is recycled and reduced into GSH using nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) by glutathione reductase (GR). Glutathione is synthesised by glutathione synthase (GS). (B) Superoxide anion was quantified by dihydroethidium (DHE) staining and FACS. Results are expressed as fold change in comparison to naïve Huh7.5 cells (n=4). (C) Hydrogen peroxide was quantified by dichlorofluorescein diacetate (DCFDA) staining and calculated as fold change compared to uninfected Huh7.5 cells. As negative and positive controls, 500 mU/mL of CAT and 50 µM H2O2 were added for 15 min to the cultures (n=4). (D) The ratio of oxidised versus total glutathione was quantified (n=3). (B–D) Graphs are depicted as means±STD. Duplicate wells were analysed in parallel by immunofluorescence with HCV-specific antibodies at the time of ROS analysis (see online supplementary figure S1A–C).
HCV induces glutathione peroxidases
To investigate how HCV decreases peroxide levels, the expression levels of peroxide scavenging enzymes were quantified by RTqPCR in Huh7.5 cells at 3 dpi (MOI 0.1) (figure 2A). Consistent with previous reports obtained with HCVcc, transcript levels of SOD2, glutathione synthase (GS) and GR were increased by 1.7-fold, 1.4-fold and 2.8-fold, respectively. However, a previously reported induction of CAT could not be confirmed.11 ,24 Among the family of GPx, a 2.8-fold and threefold induction of GPx1 and GPx4, respectively, was observed. Furthermore, a decrease of GPx3 transcripts (0.7-fold), but not of GPx2 mRNA was confirmed.11 Instead, GPx2 transcript levels were slightly increased.
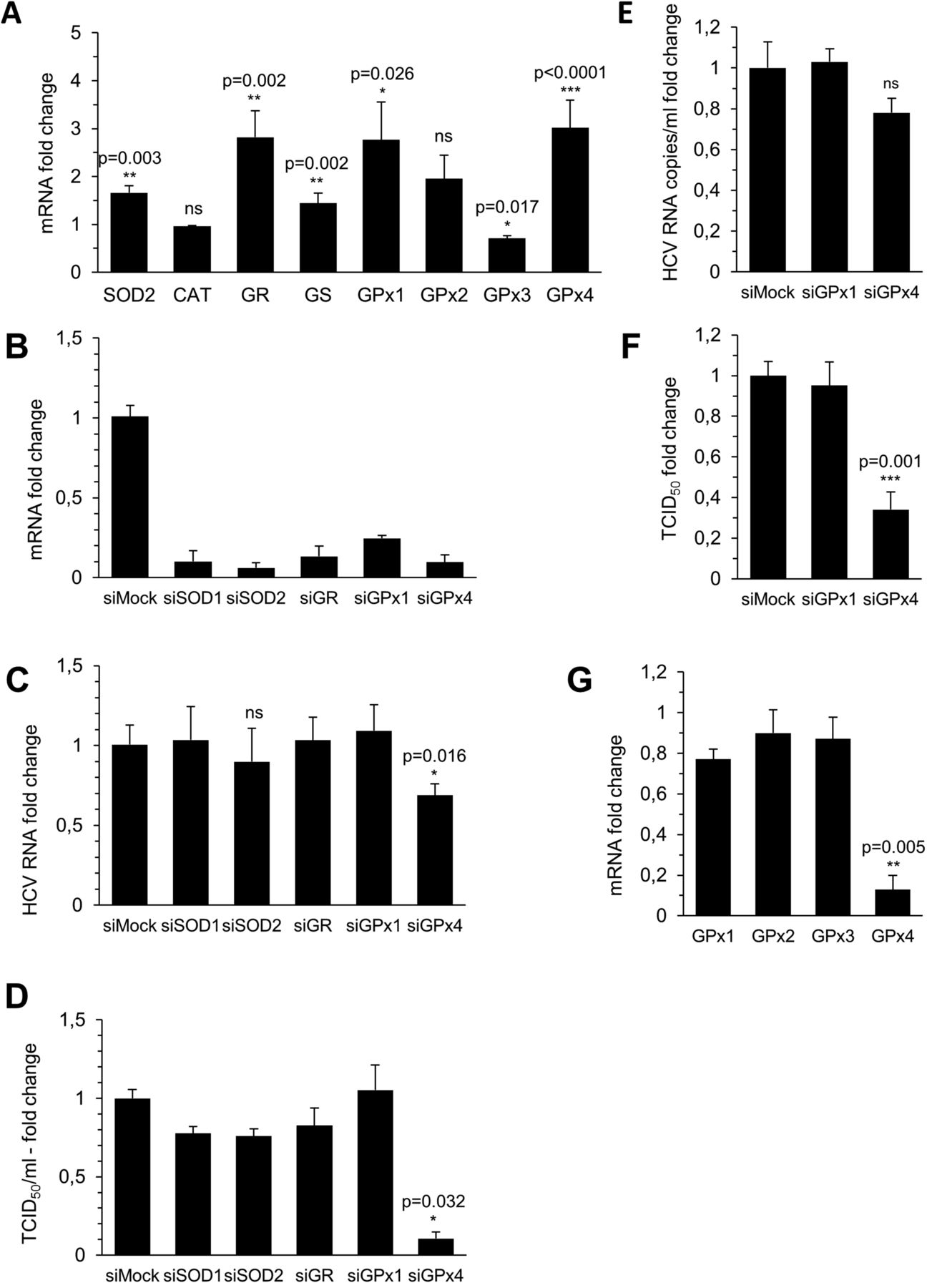
HCV infection induces glutathione peroxidase 4 (GPx4). (A) Transcript levels of key reactive oxygen species (ROS) scavengers were quantified by RTqPCR (normalisation to β-glucuronidase (GUS)) in Huh7.5 cells infected at a multiplicity of infection (MOI) of 0.1 3 dpi (SOD2, manganese superoxide dismutase; CAT, catalase; GR, glutathione reductase; GS, glutathione synthase; GPx, glutathione peroxidase) (n=6 for CAT, GR and GS; n=13 for GPx1 and 4 and SOD2). (B) Huh7.5 cells were transfected with 50 nM of the indicated siRNAs (Sigma) and transcript expression of ROS scavengers quantified by RTqPCR 3 days later (normalised to GUS) (n=3). (C–F) Huh7.5 cells were infected at an MOI of 0.1 and transfected with 50 nM of the indicated siRNAs the same day. Three dpi intracellular and extracellular HCV RNA and infectivity were determined. (C) Intracellular HCV RNA copies (normalised to GUS) are expressed as fold change in comparison with control siRNA transfected cells (SOD1 and 2, GR n=3; GPx1 n=5, GPx4 n=8). (D) Supernatant infectivity was determined using 50% tissue culture infective dose (TCID50) assays, and is depicted as fold change in comparison with control siRNA transfected cells (n=3). (E) Extracellular HCV RNA was quantified using OneStep RTqPCR. Results are expressed as fold change in respect to untransfected cells (n=5). (F) Infectivity of intracellular pools of HCV was determined using TCID50 assays and is depicted as fold change in comparison with control siRNA transfected cells (n=3). (G) Huh7.5 cells were transfected with 50 nM of siGPx4 or control siRNA and transcript expression of GPx isoforms was quantified by RTqPCR 3 days later (normalised to GUS) (n=3). (A–G) Data are shown as means±STD.
Impact of GPx4 silencing on HCV replication
To assess the respective importance of these ROS scavenging genes in the HCV life cycle, Huh7.5 cells were transfected with gene-specific or control siRNAs and infected with HCVcc at an MOI of 0.1. Three days later, the silencing efficiency was validated (figure 2B, online supplementary figure S2) and the effect on intracellular HCV RNA levels assessed. Only silencing of GPx4 slightly (by 32%), but reproducibly inhibited accumulation of intracellular HCV RNA (figure 2C). However, silencing of GPx4 strongly reduced the infectivity of virions in the supernatant by up to 10-fold (figure 2D). This loss of infectivity was neither due to cell toxicity (data not shown) nor due to reduced virion secretion, as extracellular HCV RNA levels were only slightly reduced by 22% in siGPx4 transfected cells, and this reduction did not reach statistical significance (figure 2E). Assessment of the infectivity of intracellular virion pools, liberated by repeated freeze/thaw cycles, showed that silencing of GPx4 reduced also the infectivity of not-yet-secreted virions (figure 2F).
To exclude off-target effects of siRNAs targeting GPx4 for the drop of HCV infectivity, the isoform specificity of these siRNAs targeting GPx4 was validated (figure 2G), and the effect of additional siRNAs (Dharmacon) and shRNAs (Sigma) targeting GPx4 on HCV infectivity was tested with comparable results (data not shown).
The induction of GPx1 and 4 by HCV was validated by immunoblotting with a protein induction of 2.1-fold and 2.4-fold, respectively (figure 3A). In constitutively infected cultures, GPx4 mRNA and protein levels remained induced at 1.6-fold and 1.4-fold, respectively, compared with uninfected cultures (figure 3B). To assess the catalytic activities of GPx1 and GPx4 in HCV infection, peroxidation assays were performed using the isoform unspecific hydrophilic peroxide substrate tBOOH as well as the lipophilic CHP, which is predominantly catalysed by GPx4 and allows distinction between GPx1 and GPx4 activities.16 ,21 Using CHP, a 1.5-fold increase in GPx4 activity was observed in lysates derived from infected versus uninfected cells (figure 3C).

HCV infection induces glutathione peroxidase 4 (GPx4) activity. (A) Cell lysates of Huh7.5 cells infected with HCVcc at a multiplicity of infection (MOI) of 0.1 for 3 days were probed with anti-NS3, anti-b-actin, anti-GPx1 or anti-GPx4 antibodies. Signals were quantified using ImageJ (n=4). (B) GPx4 levels were quantified by RTqPCR (normalised to GUS) and western blotting in constitutively infected and uninfected Huh7.5 cells. The western blot was quantified using ImageJ (n=3). (C) Total GPx-specific and GPx4-specific enzymatic activities were quantified using tert-butyl hydroperoxide (tBOOH) and cumene hydroperoxide (CHP) in naïve or infected cells (MOI 0.1) 3 dpi. (n=4). (A–C) Data are depicted as means±STD.
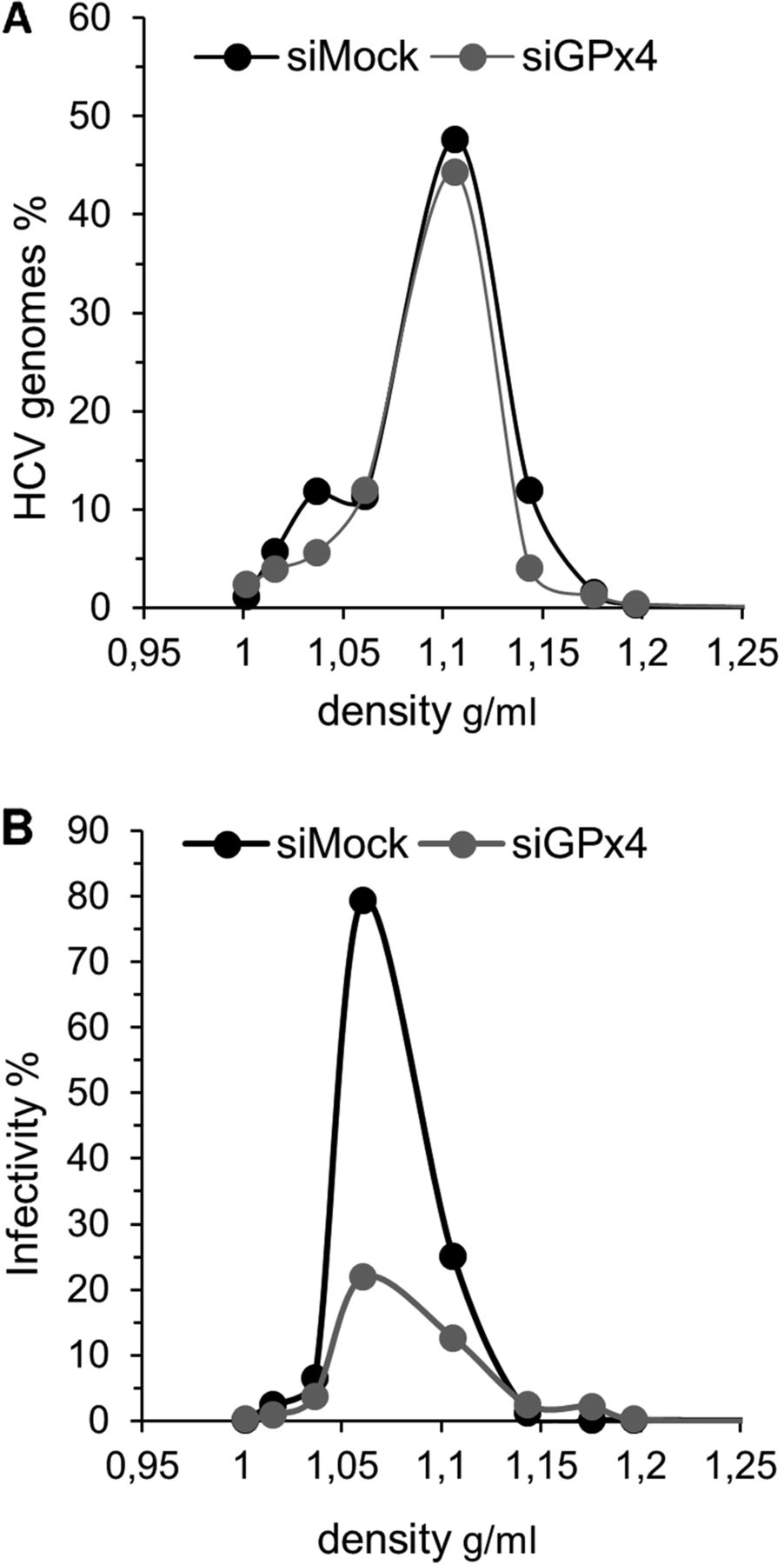
To further confirm the importance of GPx4 in HCV infection, supernatants harvested from GPx4 or control siRNA silenced cells were run on density gradients, and RNA and infectivity (TCID50) were quantified in all fractions. Knock down of GPx4 affected neither secretion nor the density profile of virions (figure 4A). However, the specific infectivity of the major RNA-containing fractions was significantly reduced with a 75% decrease in the 1.06 g/mL fraction compared with supernatant from control siRNA transfected cells (figure 4B). These data show that GPx4, but not GPx1, is a host cell factor that regulates the specific infectivity of HCV.

Glutathione peroxidase 4 (GPx4) silencing decreases specific infectivity of HCVcc. Huh7.5 cells were infected at a multiplicity of infection of 0.1 and transfected with 25 nM of control or GPx4-specific siRNA the same day. Supernatants were collected 3 dpi, filtered, concentrated and run on iodixanol–sucrose gradients. (A) HCV RNA was quantified in each fraction by RTqPCR and standardised to the total amount of HCV loaded onto the gradient. (B) Infectivity of each fraction was quantified using 50% tissue culture infective dose (TCID50) assays and standardised to the infectivity present in the inoculum loaded onto the column. A representative experiment is shown (n=2).
Effect of lipid peroxidation on HCV
To analyse the effect of HCV on the lipid peroxidation status, the lipid peroxidation products 4-HHE, 4-HNE and 4-HDDE were quantified in neo-infected and constitutively infected cells by GC/MS. In comparison with uninfected cells, in cells infected at an MOI of 0.1 and harvested at 3 dpi, the levels of 4-HHE, 4-HNE and 4-HDDE were increased by 41%, 18% and 33%, respectively, suggesting that despite of the induction of GPx4, lipid peroxides accumulate as a result of elevated ROS at early stages of infection (figure 5A). This result was confirmed with the lipid peroxide-specific stain BODIPY 581/591 C11 (figure 5B). In constitutively infected cells, levels of all lipid peroxidation products had returned to baseline (figure 5A), consistent with the observation that GPx4 protein levels remain elevated in these cells (figure 3B).

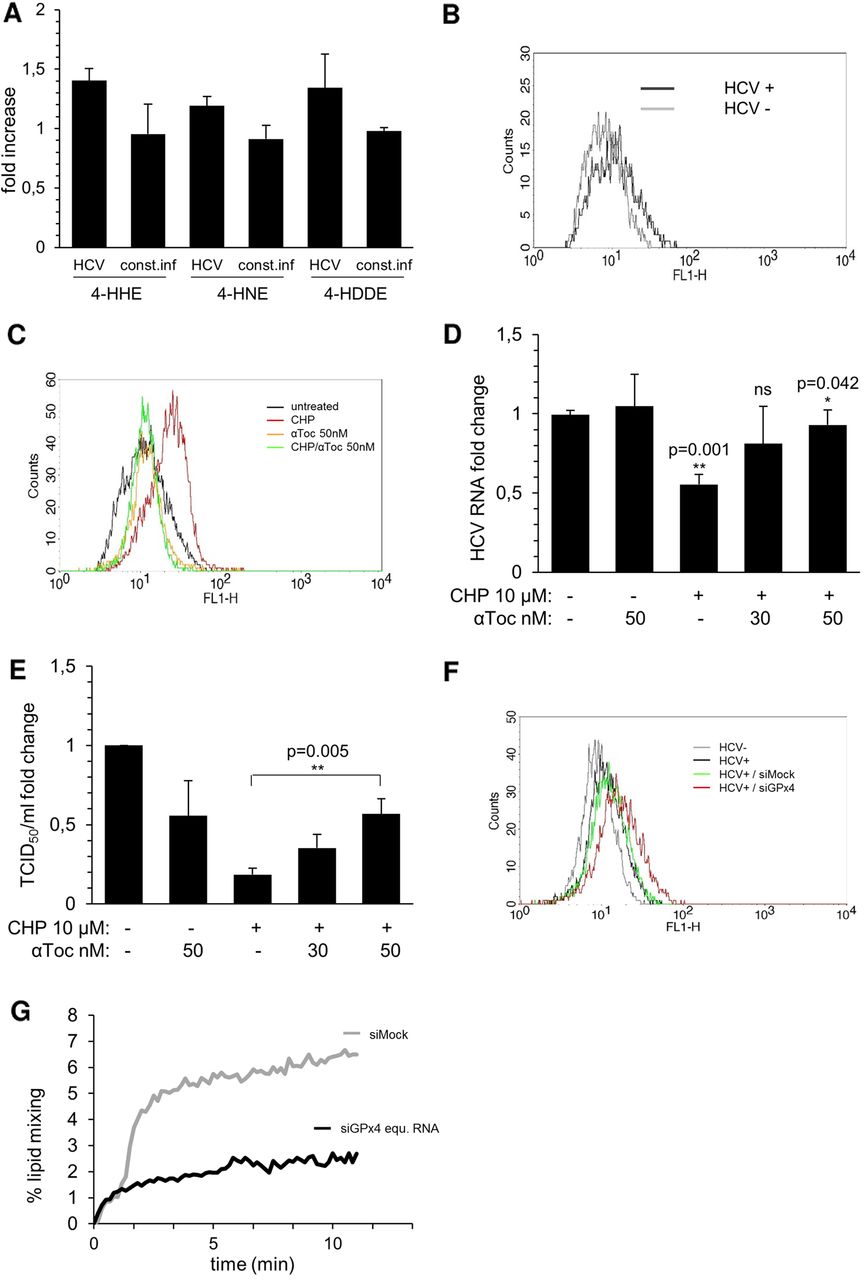
HCV infection and lipid mixing is sensitive to lipid peroxidation. (A) The indicated lipid peroxidation products were quantified in naïve and Huh7.5 cells infected at a multiplicity of infection (MOI) of 0.1 for 3 days by gas chromatography and mass spectrometry. Results were standardised to values obtained in naïve Huh7.5 cells (n=3). (B) HCV infected and uninfected Huh7.5 cells (MOI 0.1) were analysed by BODIPY C11 staining 3 dpi. A representative graph is shown (n=3). (C–E) Huh7.5 cells were infected (MOI of 0.1), 10 µM of cumene hydroperoxide (CHP) added immediately after infection and the indicated concentrations of α-tocopherol (α-toc.) 24 h later. Analysis was performed 3 dpi. (C) Cells were analysed by BODIPY C11 staining. A representative graph is shown (n=3). (D) Intracellular HCV RNA was quantified by RTqPCR (normalised to GUS) (n=4) (E) Supernatant infectivity was analysed in 50% tissue culture infective dose (TCID50) assays (n=3) (A, D and E) Data are depicted as means±STD. (F and G) Huh7.5 cells were transfected with 25 nM glutathione peroxidase 4 (GPx4) or control siRNA, infected and analysed 72 h later. (F) Cells were analysed by BODIPY C11 staining (n=3). (G) Supernatants of infected and control/GPx4 siRNA treated Huh-7.5 cells were harvested, filtered, concentrated and tested in lipid mixing assays with R18 labelled liposomes. Viral input was standardised to equal input of RNA copies. Lipid mixing was induced by low pH (n=3). Data of one representative experiment are depicted.
To find out whether the requirement for GPx4 activity is linked to its physiological activity, the elimination of membrane- associated or lipoprotein-associated lipid peroxides,16 ,17 the effect of the exogenous lipid peroxide CHP on HCV infection was tested. Addition of a final concentration of 10 µM CHP to Huh7.5 cells for 15 h did not affect cellular viability (data not shown), but increased lipid peroxides 4-HHE, 4-HNE and 4-HDDE by 17%, 36% and 69%, respectively (data not shown) and caused a shift using BODIPY C11 (figure 5C). CHP treatment of Huh7.5 cells infected for 3 days with HCVcc at an MOI of 0.1, decreased intracellular HCV RNA levels by 45% (figure 5D), and this decrease could be reversed by the addition of α-tocopherol (figure 5D), a highly lipophilic anti-oxidant, known to block lipid peroxidation of membranes25 (figure 5C). CHP treatment also reduced the infectivity of supernatant by up to 80% (figure 5E), and the decrease could again be rescued in a dose-dependent fashion by the addition of α-tocopherol (figure 5E).
Lipid peroxidation is known to alter membrane fluidity,26 and increased incorporation of lipid peroxides may impact the fusiogenic activity of virions. To test this, lipid mixing assays were performed using virions and liposomes loaded with a fluorescent marker. Upon low pH-induced fusion, lipid mixing between virions and liposomes leads to dilution and thence de-quenching of the fluorescent marker and can be quantified as increase in fluorescence over time.23 HCV virions were harvested from cells transfected with GPx4 or control siRNAs. Accumulation of lipid peroxides in GPx4 siRNA transfected producer cells was verified by BODIPY C11 staining (figure 5F). Virions were then mixed with fluorescently labelled liposomes using equal HCV RNA input and lipid mixing was monitored (figure 5G). A 70% decrease in lipid mixing activity was observed for HCVcc derived from GPx4-silenced cells compared with virions harvested from control cells. Thus, the elimination of lipid peroxides, and hence the catalytic activity of GPx4, plays an important role in the HCV life cycle, probably by preventing the accumulation of lipid peroxides in the virion membrane, thereby ensuring proper membrane fluidity and efficient lipid mixing capacity of progeny virions.
NS5A induces GPx4
Among the HCV proteins predominantly Core, NS3/4 and NS5A have been associated with the induction of oxidative stress.27 Doxycycline-inducible Huh7.5 cell lines expressing these viral proteins as well as NS5B were used to identify the viral protein that induces GPx4. Seventy-two hours after induction, expression of the various HCV proteins was verified by western blotting or RTqPCR (figure 6A, B). A 2.2-fold increase of GPx4 transcripts was detected by RTqPCR in cells expressing NS5A, but not in any other cell line (figure 6C). These results were confirmed by western blotting (up to 1.9-fold induction, figure 6D,E). GPx enzymes have been shown to be induced by several different pathways, including phosphatidylinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR).28 PI3K/AKT/mTOR is also known to be modified by direct interaction with NS5A,29 and thus may play a functional role in GPx induction in HCV-infected cells. Pharmacological inhibition of PI3K with the small molecule inhibitor LY294002 suppressed Akt phosphorylation and GPx4 induction by NS5A (figure 6D, E). Finally, the NS5A-induced increase in GPx4 protein levels correlated with a twofold increase of GPx4 activity using CHP as substrate (figure 6F).

NS5A induces glutathione peroxidase 4 (GPx4) expression. Huh7.5 cells were induced with 5 µg/mL doxycycline (dox) for 3 days to express the indicated V5-tagged HCV proteins. (A) Core, NS34A and NS5A protein induction was verified by immunoblotting with anti-core, anti-V5 or anti b-actin antibodies (n=3) (B) NS5B expression was validated by RTqPCR (normalised to β-glucuronidase (GUS)) (n=3) (C) GPx4 mRNA was quantified by RTqPCR (normalised to GUS) in the induced and uninduced cell lines. The fold change in GPx4 mRNA is shown (n=3). (D) GPx4, NS5A, p-Akt and b-actin levels were quantified by western blotting in the induced and uninduced NS5A V5 and green fluorescent protein (GFP) inducible cell lines, treated or not with LY294002 (50 µM) (n=3). (E) Signals obtained in (D) were quantified using ImageJ. (F) GPx4 activity levels were quantified in the indicated cell lines using cumene hydroperoxide as substrate (n=3). (B,C,E and F) Data are depicted as means±STD.
GPx4 levels are elevated in biopsies of patients with chronic HCV
Transcript levels of the ROS scavengers that were most strongly induced by HCV in vitro, including GPx4, GPx1 and GR, as well as GS, which is only weakly induced by the virus in vitro, were quantified by RTqPCR in liver biopsies derived from patients with CHC (n=116) and patients diagnosed with non-virally induced liver diseases (n=27). GPx1, GPx4 and GS, but not GR, transcript levels were elevated in the HCV compared with the control cohort (figure 7). For GPx4, the increase in transcript levels was the most elevated (2.2-fold) and correlated with intrahepatic HCV RNA levels (R=0.2684; p=0.0036; data not shown). In contrast, no correlations could be found between intrahepatic HCV RNA and GPx1, GS or GR transcript levels (data not shown). Further statistical analysis was performed on 89 of the 116 HCV biopsies, where patient data were available. No correlation between GPx1, GPx4 or GS mRNA levels and fibrosis stage, inflammation scores, age or BMI could be detected. Finally, induction of GPx4, GPx1 or GS transcripts could not be linked to the presence of a particular genotype, with genotypes 1, 2 and 3 being present.

Glutathione peroxidase 4 (GPx4) transcript levels are elevated in chronic hepatitis C. GPx1, GPx4, glutathione synthase (GS) and glutathione reductase (GR) transcripts were quantified by RTqPCR in 116 HCV and 27 control biopsies (normalised to PPM1). 2−ΔCp data are presented as box plots. Whiskers represent the 5th and 95th percentiles, lower and upper boundaries represent the 25th and 75th percentiles and the horizontal lines represent the median values.
HCV-induced GPx4 induction is reversible
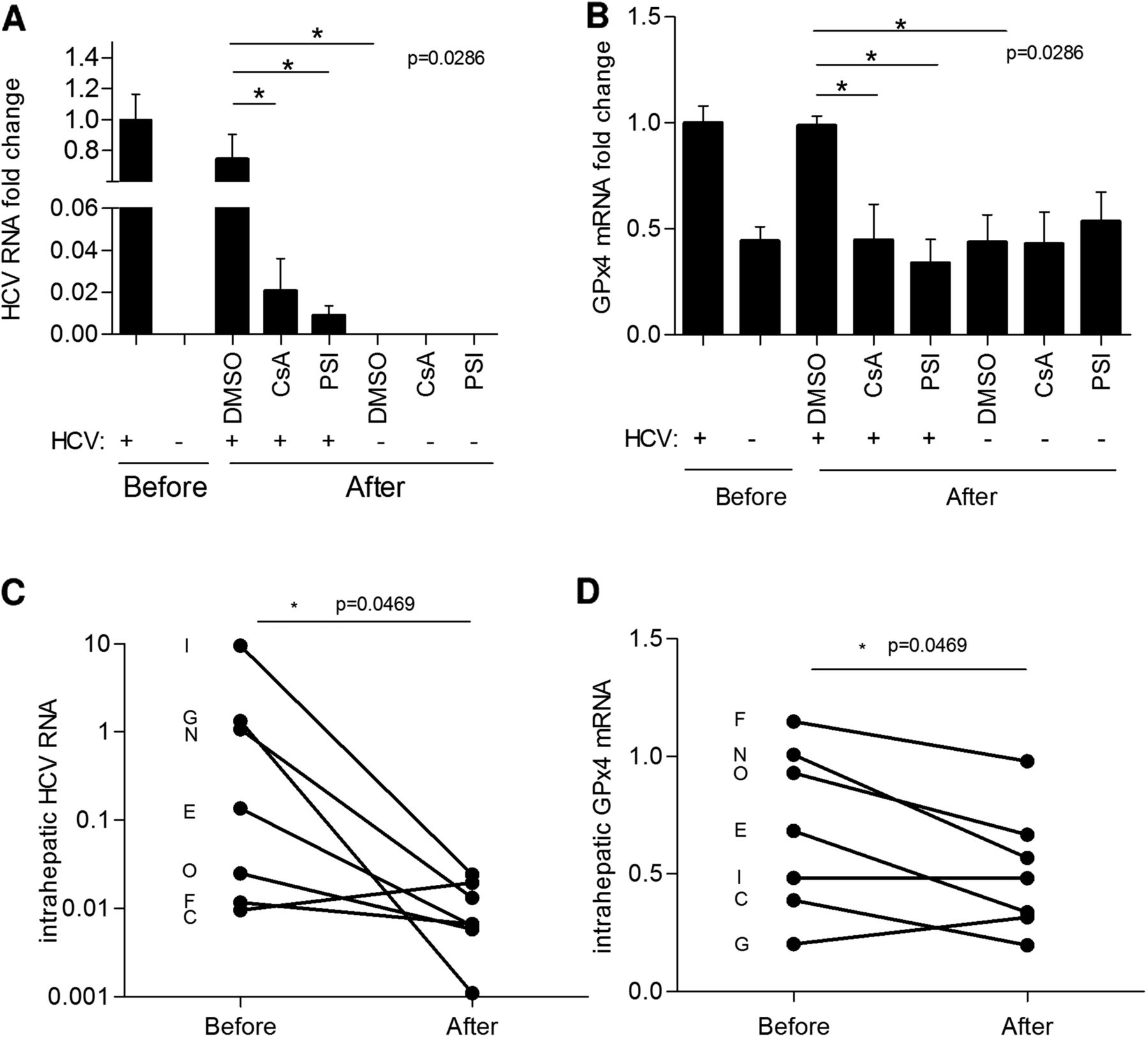
To investigate whether induction of GPx4 by HCV is reversible, Huh7.5 cells were infected at an MOI of 0.1 for 5 days, split and then treated with sofosbuvir (PSI) or ciclosporin A (CsA) for 3 days. HCV and GPx4 levels were quantified in infected and uninfected cells before start of treatment to verify the replication levels and induction of GPx4 by HCV (figure 8A, B). Treatment with either CsA or PSI but not solvent control eliminated HCV replication considerably and caused GPx4 levels to drop to the baseline levels observed prior to start of treatment. Comparable results were detected after drug treatment for 6 days, which reduced HCV replication to undetectable levels (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HCV-induced glutathione peroxidase 4 (GPx4) induction is reversible in vitro and in vivo. (A) Huh7.5 cells were infected at multiplicity of infection of 0.1 for 5 days and then treated with sofosbuvir (PSI, 5 μM), ciclosporin A (CsA, 10 μM) or dimethyl sulfoxide (DMSO) solvent control for 3 days. HCV and GPx4 levels were quantified by RTqPCR (GUS normalised) before and after treatment. Data were standardised to HCV and GPx values obtained before treatment (n=4; means±STD). (B) GPx4 transcripts were quantified by RTqPCR in seven paired biopsies obtained from chronic HCV carriers (identified by letters) before and after successful antiviral treatment (PPM1 normalised).
In order to demonstrate the reversibility of GPx4 induction in vivo, paired biopsies were obtained from 16 patients with CHC that had responded to interferon/ribavirin combination therapy. GPx4, HCV and β-glucuronidase mRNA could be successfully amplified by RTqPCR in biopsies sampled before and after treatment in seven patients. Reversibility of GPx4 induction was observed in most HCV-infected patients that responded to antiviral treatment with interferon/ribavirin, with the exception of the two patients who displayed the lowest levels of GPx4 transcripts (patients G and C). Overall, statistical analysis confirmed a significant drop of both HCV and GPx4 RNA levels in these patients, suggesting that with the elimination of HCV, GPx4 levels return to normal (figure 8C,D).
Discussion
Increased ROS levels are thought to play an important role in the pathogenesis of CHC, but findings regarding the induction of superoxide anions and peroxides by HCV remain contradictory.6–9 ,27 Our data confirm an increase of superoxide levels in infected Huh7.5 cells. However, peroxide levels have predominantly been described to be increased by HCV,8 ,9 but a contradictory report exists7 and the latter is consistent with our findings, which show a dose-dependent decrease of peroxides in infected cells (figure 1C). This decrease is, at least in part, due to the fact that HCV induces several members of the peroxide scavenging family of GPx, which leads to a net drop in peroxides at the initial stages of infection. However, in cells constitutively replicating HCV, peroxide levels are increased, suggesting that the virus produces more ROS than it eliminates on the long term. The fact that GSH levels remain constant throughout the first 5 days of infection points to a sufficient reductive capacity of the infected cell in the early stage of infection (figure 1D). However, the reductive capacity may diminish over time, which could explain the observed drop of GSH in constitutively infected cells and subsequent accumulation of peroxides notwithstanding the continued presence of GPx enzymes (figure 3B).
Among ROS scavenging enzymes, we found transcript levels, protein and catalytic activity of GPx1 and GPx4 to be induced by the virus. GPx1 is known to prevent oxidative DNA mutations and induction of proinflammatory mediators.13 GPx4 has been characterised as a lipid peroxidation scavenger due to its unique ability to reduce soluble hydrogen peroxides and particularly peroxides of complex lipids such as phospholipid, cholesterol and cholesteryl ester hydroperoxides, even when present in biomembranes and lipoproteins.13 ,18 Interestingly, silencing of GPx4, but not GPx1, decreased the specific infectivity of progeny virions by average 10-fold. This finding, together with the fact that addition of the exogenous lipid peroxide CHP inhibits HCV infection, suggests that HCV is highly sensitive to lipid peroxidation. This is consistent with the finding that several steps of the viral life cycle are known to depend on formation of cellular membrane compartments at the ER and the hepatic lipoprotein metabolism.30 In the replicon system, polyunsaturated fatty acids, which are precursors of oxygenated lipids, inhibit HCV replication.31 ,32 HCV replication is also inhibited by hydrogen peroxide and arsenic trioxide, which can in turn activate lipid peroxidases and trigger non-enzymatic lipid peroxidation.33 ,34 Interestingly, hydrogen peroxide, which is highly membrane permeable, is known to inhibit replication of HCV replicons by disturbing the intracellular localisation of replication complexes.33 However, in our hands, silencing of GPx4 had a major impact only on virion infectivity and did not, or only slightly decrease intracellular HCV RNA levels, suggesting that replication of the HCV strain used in this study is not very sensitive to lipid peroxide accumulation, but further tests using subgenomic replicons or negative RNA strand quantification would be needed to confirm this.
In biopsies of patients with CHC, we found GPx4 and GPx1 and GS levels to be elevated. The fact that GPx4 induction seems to occur in vivo in the context of genotypes 1, 2 and 3, and declines to baseline upon successful antiviral treatment, are additional findings that suggest that ROS-scavenging and, particularly, lipid peroxide-scavenging is an important feature of HCV replication. Reversibility of GPx4 induction was observed in all HCV patients who responded to antiviral treatment with interferon/ribavirin, with the exception of two patients with low levels of GPx4. Reversibility of GPx4 induction was also observed upon use of direct acting antivirals such as ciclosporin A or the polymerase inhibitor sofosbuvir in tissue culture. Thus, HCV may actively induce GPx4 in order to prevent accumulation of peroxides and particularly lipid peroxides because they are counterproductive for viral infectivity and because minimising oxidative stress and its pathophysiological effects on the liver is likely to favour viral persistence.
Direct quantification and identification of lipid peroxides contained in HCV virions derived from parental or GPx4-silenced producer cells is technically very difficult due to the vast amounts of material needed. However, we have been able to show that accumulation of lipid peroxides in producer cells leads to formation of progeny virions with reduced fusogenic activity. Increased levels of lipid peroxides or of oxidised lipoproteins in virions may simply alter membrane fluidity26 and may also have a negative impact on cell attachment. Several of the HCV entry factors belong to the family of the lipoprotein receptors, notably low-density lipoprotein receptor (LDLR), scavenger receptor B1 and syndecan-1. Indeed, oxidised LDL is known to block HCV cell entry, while the LDLR is considered to mediate non-productive uptake of HCV.35 The impact of the lipid peroxidation status of HCV on the interaction with these receptors remains to be established.
We identified NS5A as the viral factor responsible for induction of GPx4 mRNA, protein and activity levels. GPx enzymes have been shown to be induced by several different pathways, including PI3K/AKT/mTOR and p53.28 ,36 The latter two pathways are known to be also modified by direct interaction with NS5A,29 ,37 and thus may play a functional role in GPx induction in HCV-infected cells. Our finding that the PI3K inhibitor LY294002 blocks GPx4 induction confirms a role for this pathway in GPx4 regulation, but further details remain to be explored.
Interestingly, NS5A has been shown to regulate apoptosis (ref. 5 and references therein), a process in which GPx4 could be involved.13 One of the roles of GPx4 is to prevent oxidation of cardiolipin, a complex lipid that resides on the inner mitochondrial membrane where it forms complexes with and retains cytochrome C to prevent caspase activation and apoptosis. Thus, elevation of GPx4 activity may attenuate HCV-induced apoptosis and thus play an important role not only in controlling inflammatory processes and fibrosis progression but also by predisposing to neoplastic transformation in chronic HCV infection. However, the impact of GPx4 induction in chronic HCV infection on apoptosis remains to be elucidated.
In conclusion, we have identified the lipid peroxide scavenger GPx4 as a host factor with a critical role in HCV infection. Targeted interference with GPx4 and possibly other ROS scavenging enzymes, or induction of particular lipid peroxidation products to which HCV is highly sensitive, may be developed into future therapeutic strategies that will decrease or eliminate HCV replication and prevent hepatocarcinogenesis, particularly in patients at advanced stages of disease who remain difficult to be treated in the new era of interferon-free regimens.
Acknowledgments
The authors thank H. Eischeid, J. Molle, A. Kennel, N. Guichard, C. Villiers, M. Guichardant and the lipidomics platform (UCBL) for expert technical assistance and advice, and T. Wakita, C. M. Rice and C. Wychowski for reagents.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors CB, PL, SD, MM, AS, E-IP, M-LP, AVI and BB generated and collected data. M-LP, RP, MD, H-MS, MO and FZ provided clinical samples and data. EV and AIV contributed reagents. CB, FZ and BB designed the study. BB wrote the paper.
Funding French National Agency for AIDS and Viral Hepatitis Research (grant 13444), Comité de Savoie de la Ligue contre le cancer, Agence Nationale de Recherche, the Region Rhone-Alpes, DevWeCan French Laboratories of Excellence Network (Labex, Grant ANR-10-LABX-61), OpeRa IHU programme (GRANT ANR-10-IBHU-004).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethical committee University of Cologne, French IRB ‘Comité de Protection des Personnes (CPP) Sud-Est 287 IV’ agreement #11/040 obtained in 2011.
Provenance and peer review Not commissioned; externally peer reviewed.