Article Text

Abstract
Objective The endocannabinoid system (ECS) exerts key roles in the development of liver fibrosis and fatty liver, two diseases that promote the development of hepatocellular carcinoma (HCC). Although cannabinoids exert potent antitumour effects in vitro, the contribution of the ECS to carcinogenesis in vivo remains elusive.
Design Expression of key components of the ECS, including endocannanabinoids, endocannabinoid-degrading enzymes and endocannabinoid receptors, was determined in healthy liver and tumours. Diethylnitrosamine-induced hepatocarcinogenesis was determined in mice deficient in fatty acid amide hydrolase (FAAH), the main anandamide (AEA)-degrading enzyme, in cannabinoid receptor (CB)1, CB2, or transient receptor potential cation channel subfamily V member 1 (TRPV1)-deficient mice.
Results Murine and human HCCs displayed activation of the ECS with strongly elevated expression of CB1 and CB2 but only moderately altered endocannabinoid levels. Contrary to the antitumour effects of cannabinoids in vitro, we observed increased hepatocarcinogenesis in FAAH-deficient mice, a mouse model with increased AEA levels. Accordingly, inactivation of CB1, the main receptor for AEA, in wild-type or FAAH-deficient mice suppressed hepatocarcinogenesis. In contrast, inactivation of CB2 increased hepatocarcinogenesis. CB1 was strongly expressed within HCC lesions and its inactivation suppressed proliferation and liver fibrosis. CB2 was predominantly expressed in macrophages. CB2 inactivation decreased the expression of T-cell-recruiting chemokines and inhibited hepatic T-cell recruitment including particular CD4+ T cells, a population with known antitumour effects in HCC. TRPV1 deletion did not alter HCC development.
Conclusions Similar to their role in fibrogenesis, CB1 and CB2 exert opposite effects on hepatocarcinogenesis and may provide novel therapeutic targets.
- HEPATOCELLULAR CARCINOMA
- LIVER
- INFLAMMATORY MECHANISMS
- FIBROSIS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
A wide body of literature suggests anticancer effects of endocannabinoids in cancer of other organs.
In the liver, endocannabinoid receptors CB1 and CB2 regulate fibrosis in opposite fashion.
In the liver, endocannabinoid receptor CB1 regulates cell proliferation and steatosis.
What are the new findings?
We demonstrate that the endocannabinoid system is activated during hepatocarcinogenesis, and that its hyperactivation increases hepatocarcinogenesis.
CB1 drives hepatocarcinogenesis through increased hepatocyte proliferation and liver fibrosis.
CB2 inactivation enhances hepatocarcinogenesis by altering hepatic immune cell infiltration including infiltration of CD4+ T cells, a population with a key role in antitumour immunity in hepatocarcinogenesis.
How might it impact on clinical practice in the foreseeable future?
Our data suggest that the endocannabinoid system can be employed for hepatocellular carcinoma prevention or treatment by receptor-specific approaches, for example, CB1 antagonists or CB2 agonists.
Our data suggest the need for further investigation of the effects of recreational cannabis consumption on hepatocarcinogenesis.
Introduction
Liver cancer has recently been declared the second leading cause of worldwide cancer mortality, causing 800 000 deaths annually.1 In virtually all patients, liver cancer develops as a consequence of chronic liver disease with 80% of hepatocellular carcinomas (HCCs) arising in patients with liver fibrosis and cirrhosis. Hence, HCC is viewed as the final consequence of the chronic wound healing response, with inflammation, fibrosis and chronic regeneration being driving forces.2
The endocannabinoid system (ECS) consists of endocannabinoids such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG), endocannabinoid-degrading enzymes such as fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase, cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2), as well as several additional endocannabinoid-binding receptors such as G protein-couple receptor 55 (GPR55) and transient receptor potential cation channel subfamily V member 1 (TRPV1).3 Although best known for its role in the central nervous system, the ECS has been shown to exert a major role in the development of liver disease.4 ,5 As such, CB1, the main receptor for AEA, promotes the development of liver fibrosis and fatty liver.6 ,7 In contrast, CB2 suppresses the development of liver fibrosis.8 Accordingly, inhibition of CB1 signalling by non-selective or peripherally acting CB1 receptor antagonists has been shown to reduce liver fibrosis, fatty liver development and hepatocyte proliferation following partial hepatectomy.6 ,9–11 Based on these findings, it is conceivable that components of the ECS might contribute to the progression of chronic liver disease to HCC and provide a therapeutic target to halt this progression. On the other hand, a large number of studies have found that cannabinoids and endocannabinoids exert potent antitumour effects in vitro as well as in xenograft models.12–16 Hence, an alternative hypothesis could be that endocannabinoids actively suppress HCC development despite their role in the promotion of liver disease development. Here, we tested these hypotheses by various genetic approaches that allowed us to assess the effects of increased endocannabinoid levels or decreased cannabinoid receptor signalling in vivo. By these approaches, we found that the endocannabinoid AEA and its receptor CB1 acted as potent tumour promoters in vivo, whereas CB2 exerted tumour-suppressive effects.
Materials and methods
Mice
All animal procedures were in accordance with guidelines by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee at Columbia University. Wild-type, CB2-deficient (B6.129P2-Cnr2tm1Dgen/J, Jax stock number 005786) and TRPV1-deficient mice (B6.129X1-Trpv1tm1Jul/J, Jax # 003770) were purchased from Jackson Laboratories. FAAH-deficient mice were a gift from Benjamin F. Cravatt, Scripps Research Institute, La Jolla, USA.17 CB1-deficient mice were generated by breeding CB1fl/fl mice (a gift from Beat Lutz, University of Mainz, Germany) with germline-deleter Ella-Cre (B6.FVB-Tg (EIIa-cre) C5379Lmgd/J, Jax # 003724). All knockout mice were intercrossed with C57Bl/6 mice, followed by establishment of knockout mice and wild-type controls from heterozygote mice.
HCC induction and evaluation
HCC was induced in male offspring by intraperitoneal injection of diethylnitrosamine (DEN (25 mg/kg), Sigma) given at day 15 post partum. Mice were sacrificed between 10 months (FAAHko mice) and 11 months (all other mice) after initial DEN injection. Animals were sacrificed by inhalation of isoflurane and subsequent exsanguination. Tumour number and largest tumour size were determined by counting the number of visible tumours (at least 1 mm in diameter) and measuring the size of the largest tumour with a calliper as described.18 Liver tissue (non-tumour or tumour) was snap-frozen in liquid nitrogen and stored at −80°C for RNA and protein analysis as well as fixed in 10% formalin for 24 h and subsequently paraffin-embedded for further analysis. Whole blood samples were centrifuged for 5 min to collect serum and stored at −80°C.
Human samples
Human liver samples (10 controls and 10 sets of non-tumour/tumour samples) were obtained from patients who underwent liver resection because of colorectal liver metastasis and from patients with HCC caused by chronic hepatitis B virus infection. Samples were provided by the Ajou and Keimyung Human Bio-Resource Bank, a member of the National Biobank of Korea, supported by the Ministry of Health and Welfare. Human sera were obtained from five healthy controls and five HCC patients at Chuncheon Sacred Heart Hospital, Chuncheon, South Korea.
Statistical evaluation
Statistical analysis was performed using Prism V.5.0 (GraphPad, San Diego, California, USA) and SPSS, V.19.0 (SPSS, Chicago, Illinois, USA). Differences between two groups were calculated by Student's t test or Mann–Whitney U test. Differences between multiple groups were determined by one-way analysis of variance, followed by Dunnett's post hoc test. All data are expressed as means±SEM. Additional methods are described in the online supplementary materials.
Supplemental material
Results
Activation of the ECS in murine and human HCC
Based on previous studies showing a key role of the ECS in hepatic responses to chronic injury,6 ,9–11 we sought to investigate the hypothesis that the ECS may be involved in the regulation of hepatocarcinogenesis. To test this hypothesis, we first compared the expression of key components of the ECS between normal mouse liver and DEN-induced HCCs. Microarray analysis revealed changes of several components of the ECS including enzymes involved in endocannabinoid synthesis, endocannabinoid receptors, as well as endocannabinoid-degrading enzymes (figure 1A). These data were further confirmed by qPCR, which demonstrated significant upregulation of cannabinoid receptors CB1 (99.5-fold vs normal liver, p<0.01, 24.8-fold vs non-tumour, p<0.001), CB2 (6.98-fold vs normal liver, p<0.01; 3.63-fold vs non-tumour, p<0.01) and GPR55 (11.7-fold vs normal liver, p<0.01; 4.99-fold vs non-tumour, p<0.01), a putative cannabinoid receptor,19 but unaltered expression of TRPV1, a receptor that is activated by AEA in addition to various other ligands.20 Moreover, the expression of endocannabinoid-degrading enzymes FAAH (0.63-fold vs normal liver, p<0.01; 0.6-fold vs non-tumour, p<0.01) and monoacylglycerol lipase (MGLL) (0.31-fold vs normal liver, p<0.01; 0.35-fold vs non-tumour, p<0.001) was reduced in HCCs (figure 1B). Of note, glypican 3 mRNA expression was increased several hundred folds in tumour samples, thus confirming tumours as HCCs (figure 1B). Similar findings were made in human HCCs, with significant upregulations of CB1 (307-fold vs normal liver, p<0.01; 143-fold vs non-tumour, p<0.01), CB2 (5.44-fold vs normal liver, p<0.05) and GPR55 (9.73-fold vs normal liver, p<0.05), unaltered TRPV1 expression and downregulation of FAAH (0.45-fold vs non-tumour, p<0.05), MGLL (0.49-fold vs non-tumour, p<0.05), as well as increased levels of glypican 3 mRNA in tumours (figure 1C). Decreased expression of FAAH in HCC was confirmed by western blot analysis (figure 1D). High protein expression of CB1 in HCC versus normal liver was confirmed by immunohistochemical staining in mice and patients (figure 1E). The endocannabinoid AEA was moderately decreased in murine HCCs while 2-AG was moderately increased (figure 1F). Endocannabinoids, including AEA and 2-AG, were increased in human HCCs (figure 1G). In sera of patients with HCC, there was no statistically significant change in endocannabinoid levels (see online supplementary figure S1). Taken together, these data demonstrate local activation of the ECS that predominantly occurs at the level of endocannabinoid receptor expression.

Activation of the endocannabinoid system in murine and human hepatocellular carcinoma (HCC). (A) Microarray comparison of components of the endocannabinoid system between normal liver and HCC induced by diethylnitrosamine (DEN) (given at day 15 post partum) in mice. (B) qPCR confirmation of the HCC microarray data in mice normal liver (N, n=5), non-tumour (NT, n=17) and tumour (T, n=17). (C) qPCR confirmation of the HCC microarray data in human N (n=5), NT (n=8) and T (n=8). (D) Fatty acid amide hydrolase (FAAH) was determined by western blot analysis in non-tumour and tumour livers of DEN-injected mice. (E) Immunohistochemical confirmation of CB1 expression in murine and human HCCs. (F) Endocannabinoids levels in normal (N, n=5), non-tumour (NT, n=5) and tumour tissue (T, n=5) from DEN-induced murine HCC. (G) Endocannabinoids levels in normal (N, n=5), non-tumour (NT, n=5) and tumour tissue (T, n=5) from patients. *p<0.05, **p<0.01. AEA, anandamide; 2-AG, 2-arachidonoylglycerol; LEA, linoleoylethanolamide; OEA, oleoylethanolamide; PEA, palmitoylethanolamide; SEA, stearoylethanolamide.
Hyperactivation of the ECS enhances DEN-induced hepatocarcinogenesis
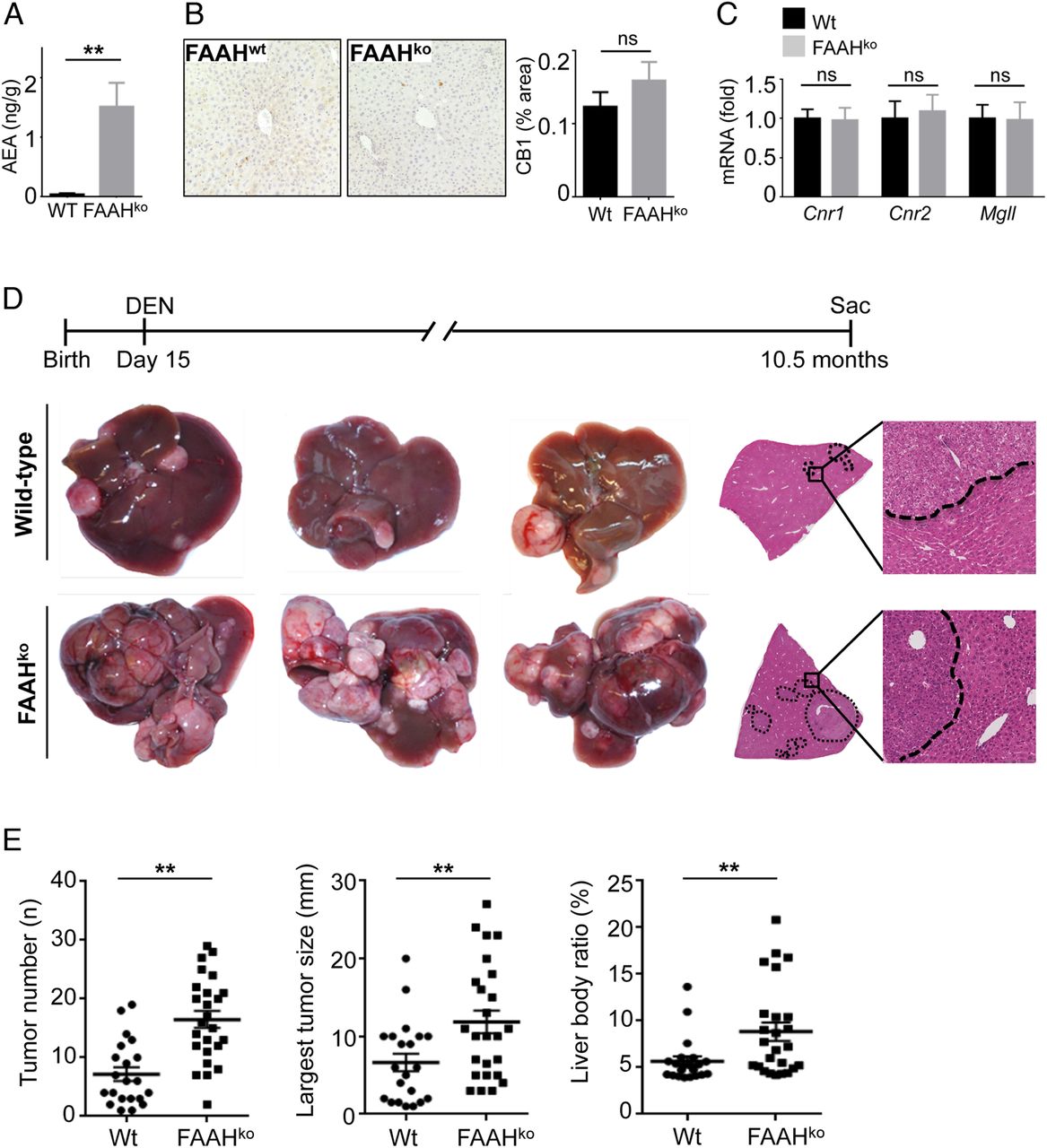
Next, we sought to determine whether activation of the ECS would affect hepatocarcinogenesis. Based on our finding that the ECS in murine hepatocarcinogenesis was predominantly activated via increased endocannabinoid receptor expression, we reasoned that increasing endocannabinoid levels rather than further enhancing the already upregulated endocannabinoid receptors would represent the best approach to hyperactive endocannabinoid signalling. As such, FAAH-deficient mice have strongly elevated hepatic levels of the endocannabinoid AEA due to decreased AEA degradation (figure 2A)17 without alterations of CB1 protein or CB1, CB2 or Mgll mRNA expression (figure 2B, C). To test the effect of increased endocannabinoid levels on HCC formation, we therefore subjected wild-type and FAAH-deficient mice to DEN-induced hepatocarcinogenesis. In comparison to wild-type mice, we observed a striking increase of three different parameters of tumour load, that is, tumour number, tumour size and liver body weight ratio in FAAH-deficient mice in comparison to wild-type mice (figure 2D, E). Of note, we did not find any differences in acute responses to DEN, including upregulation of inflammatory and p53-dependent genes (see online supplementary figure S2), indicating that the initial response to DEN was not altered by FAAH status.
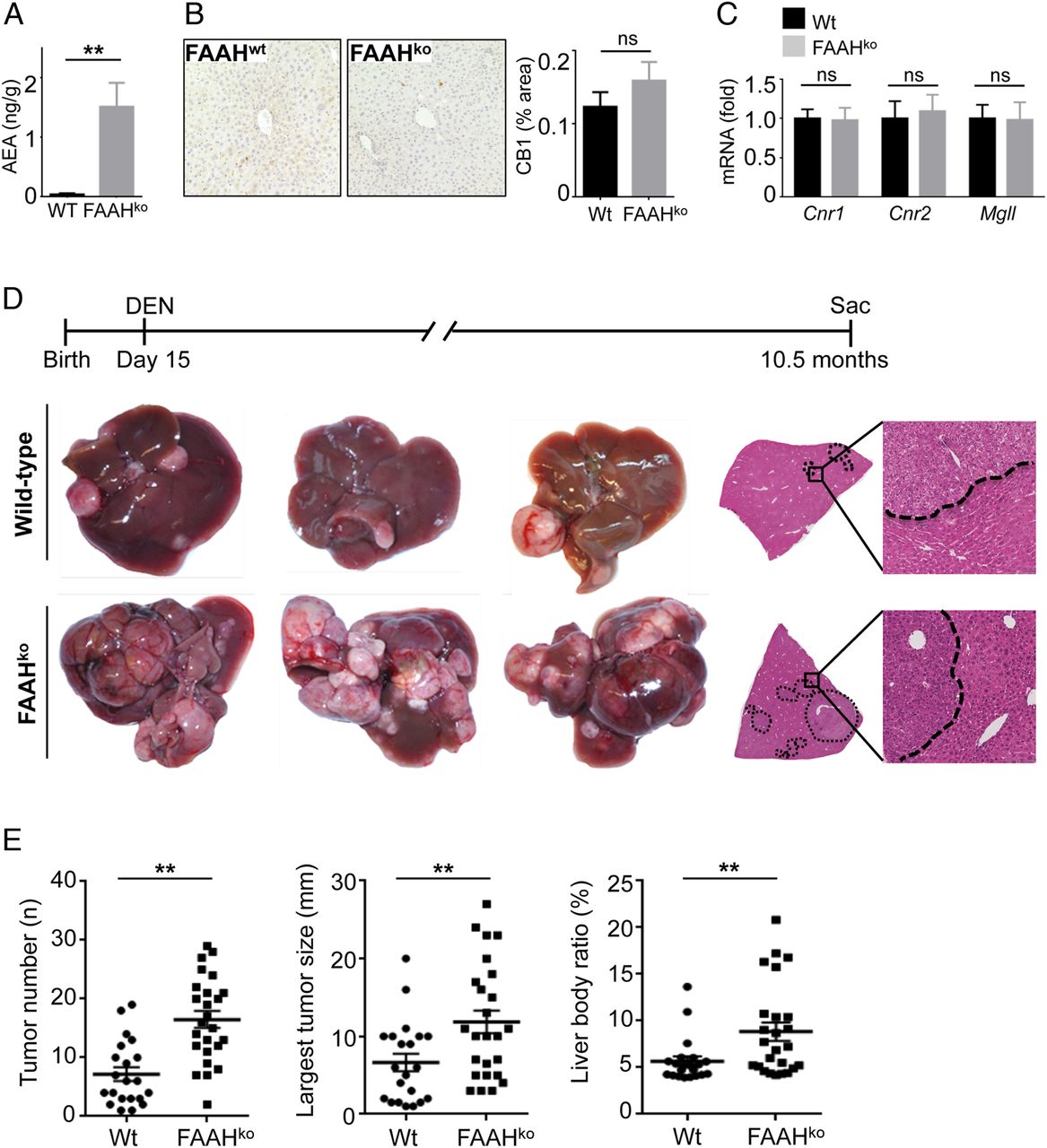
Hyperactivation of the endocannabinoid system promotes hepatocellular carcinoma (HCC) development. (A) Anandamide (AEA) was determined in liver extracts by liquid chromatography-tandem mass spectrometry (LC-MS/MS). (B and C) CB1 protein expression (B) and expression of Cnr1, Cnr2 and Mgll mRNA (C) were compared between livers of wild-type (Wt) and fatty acid amide hydrolase (FAAH)-deficient mice by immunohistochemistry and qPCR, respectively. (D and E) Wild-type mice (n=21) and FAAHko mice (n=25) were injected with diethylnitrosamine (DEN) (25 mg/kg intraperitoneally) at the age of 15 days and sacrificed 10.5 months after DEN. Shown are tumour number, largest tumour size, liver/body weight ratio, H&E sections and representative images. **p<0.01.
Opposite functions of endocannabinoid receptors CB1 and CB2 in hepatocarcinogenesis
Next, we sought to determine which cannabinoid receptors were involved in endocannabinoid-mediated modulation of hepatocarcinogenesis. To determine the role of CB1, the endocannabinoid receptor with the strongest upregulation in murine and human HCC (figure 1), we subjected wild-type and CB1-deficient mice to DEN-induced hepatocarcinogenesis. Of note, CB1 is the main receptor for AEA, the endocannabinoid that is elevated in FAAH-deficient mice. We observed a significant reduction of tumour number, and liver body weight ratio, and a trend towards decreased largest tumour size in CB1-deficient mice in comparison to wild-type mice (figure 3A, B). Next, we compared DEN-induced hepatocarcinogenesis between CB2-deficient and wild-type mice. In contrast to our results in CB1-deficient mice, we observed significant increases in tumour number, largest tumour size and liver body weight ratio in CB2-deficient mice (figure 4A, B). These results are remarkably similar to the opposite functions of CB1 and CB2 in hepatic fibrogenesis, where CB1 promotes and CB2 inhibits liver fibrosis development.6 ,8

CB1 promotes hepatocellular carcinoma (HCC) development. Wild-type (Wt) mice (n=20) and CB1ko mice (n=11) were injected with diethylnitrosamine (DEN) (25 mg/kg intraperitoneally) at the age of 15 days and sacrificed 10.5 months after DEN. Shown are tumour number, largest tumour size, liver/body weight ratio, H&E sections and representative images. *p<0.05, **p<0.01.

CB2 suppresses hepatocellular carcinoma (HCC) development. Wild-type (Wt) mice (n=15) and CB2ko mice (n=15) were injected with diethylnitrosamine (DEN) (25 mg/kg intraperitoneally) at the age of 15 days and sacrificed 10.5 months after DEN. Shown are tumour number, largest tumour size, liver/body weight ratio, H&E sections and representative images. *p<0.05, **p<0.01.
TRPV1 does not regulate hepatocarcinogenesis
TRPV1 represent another receptor that has been reported to be activated by AEA.20 Although we had not found alterations in TRPV1 expression in our microarray and qPCR data, we next compared hepatocarcinogenesis between wild-type and TRPV1-deficient mice. We did not find any difference in DEN-induced tumour number, tumour size or liver body weight ratio between wild-type and TRPV1-deficient mice, suggesting that TRPV1 does not regulate DEN-induced hepatocarcinogenesis (see online supplementary figure S3).
FAAH deficiency-induced increase in hepatocarcinogenesis is mediated by CB1
To further determine mechanisms by which increased endocannabinoid signalling in FAAH-deficient mice promotes HCC development, we generated double knockout mice in which CB1, CB2 or TRPV1 were deleted in addition to FAAH, and subjected these mice to DEN-induced hepatocarcinogenesis. We found that CB1/FAAH double-deficient mice had significantly reduced development of HCC in comparison to FAAH single knockout mice (figure 5A), whereas there was no reduction in DEN-induced hepatocarcinogenesis in CB2/FAAH double-deficient (figure 5B) and TRPV1/FAAH double-deficient mice (figure 5C). These data confirm our data on the key role of CB1 in tumour promotion and are consistent with the fact that AEA, which is significantly increased in FAAH-deficient mice, is known to predominantly act via CB1.

CB1 mediates anandamide (AEA)-induced hepatocarcinogenesis. (A) Fatty acid amide hydrolase (FAAH )ko (n=11) and FAAH/CB1dko mice (n=6), (B) FAAHko (n=15) and FAAH/CB2dko mice (n=15) and (C) FAAHko (n=12) and FAAH/TRPV1dko mice (n=12) were injected with diethylnitrosamine (DEN) (25 mg/kg intraperitoneally) at the age of 15 days and sacrificed 10.5 and 11 months after DEN. Shown are tumour number, largest tumour size, liver/body weight ratio, H&E sections and representative images. *p<0.05, **p<0.01.
The AEA-CB1 axis promotes proliferation and fibrogenesis during hepatocarcinogenesis
To determine mechanisms by which the AEA-CB1 axis promotes HCC development, we analysed several parameters that are known to critically influence hepatocarcinogenesis, including inflammation, cell death, proliferation and fibrosis. While we observed no differences in inflammation and cell death, as determined by qPCR for Il6 and Tnf (see online supplementary figure S4A) and cleaved caspase-3 staining (see online supplementary figure S4B), there was a profound increase in proliferation, as determined by Ki-67 staining (figure 6A) and qPCR for Mki67 and Ccnb2 (figure 6C) in FAAH-deficient mice. The increased proliferation was observed in non-tumour regions, suggesting that FAAH deficiency promoted proliferation in premalignant lesions rather than in large established tumours, or that the endocannabinoid signalling in these tumours was already at maximum levels that could not be further enhanced by additional FAAH deficiency-mediated increase in AEA. These data are further supported by our finding that FAAH deficiency increased hepatocyte proliferation following two-thirds partial hepatectomy (see online supplementary figure S5A, B). Conversely, CB1 deficiency decreased Ki-67 protein and mRNA expression in non-tumour areas (figure 6B, D). Moreover, deletion of CB1 in FAAH-deficient mice reduced proliferation, assessed by Ki-67 immunohistochemistry and mKi67 qPCR, in tumour and non-tumour areas (figure 6E, F). Since cannabinoids including AEA have been shown to induce cancer cell proliferation via transactivation of epidermal growth factor receptor (EGFR),21 we additionally investigated the levels of phosphorylated extracellular signal-regulated kinases (pERK), a key downstream target of EGFR. Consistent with above findings, we observed increased levels of pERK in tumours of FAAH-deficient mice and decreased levels in tumours from CB1-deficient and FAAH-/CB1-double-deficient mice (figure 6G, H and online supplementary figure S5C–F). Since CB1 has been also shown to promote the development of liver fibrosis,6 a risk factor for HCC development, we next investigated how ablation of FAAH and CB1 affected liver fibrosis. While FAAH deficiency increased the deposition of fibrillar collagen as well as the mRNA expression of Acta2 and Col1a1 in non-tumour and Col1a1 in tumour tissue (figure 7A, C), there were no significant differences in fibrillar collagen deposition and only a trend towards reduced Acta2 and Col1a1 mRNA expression in CB1-deficient mice (figure 7B, D). Most likely, this finding reflects the low grade of fibrosis observed in the DEN-induced HCC model, which can be exacerbated by increased endocannabinoid signalling but not much further reduced by inhibition of CB1-mediated signals. This hypothesis is further supported by our findings that CB1 deficiency reduced fibrillar collagen deposition and the expression of Col1a1 in FAAH-deficient mice (figure 7E, F). Of note, FAAH deficiency also resulted in increased fibrogenesis in response to hepatotoxin CCl4 (see online supplementary figure S6). The more profound effect of CB1 deficiency in FAAH-deficient than in wild-type mice may not only be due to the overall higher levels of fibrosis but also the strong increase of CB1 agonist AEA in this model. Together, our data suggest that proliferation and fibrogenesis are key mechanisms by which the AEA-CB1 axis promotes HCC development.
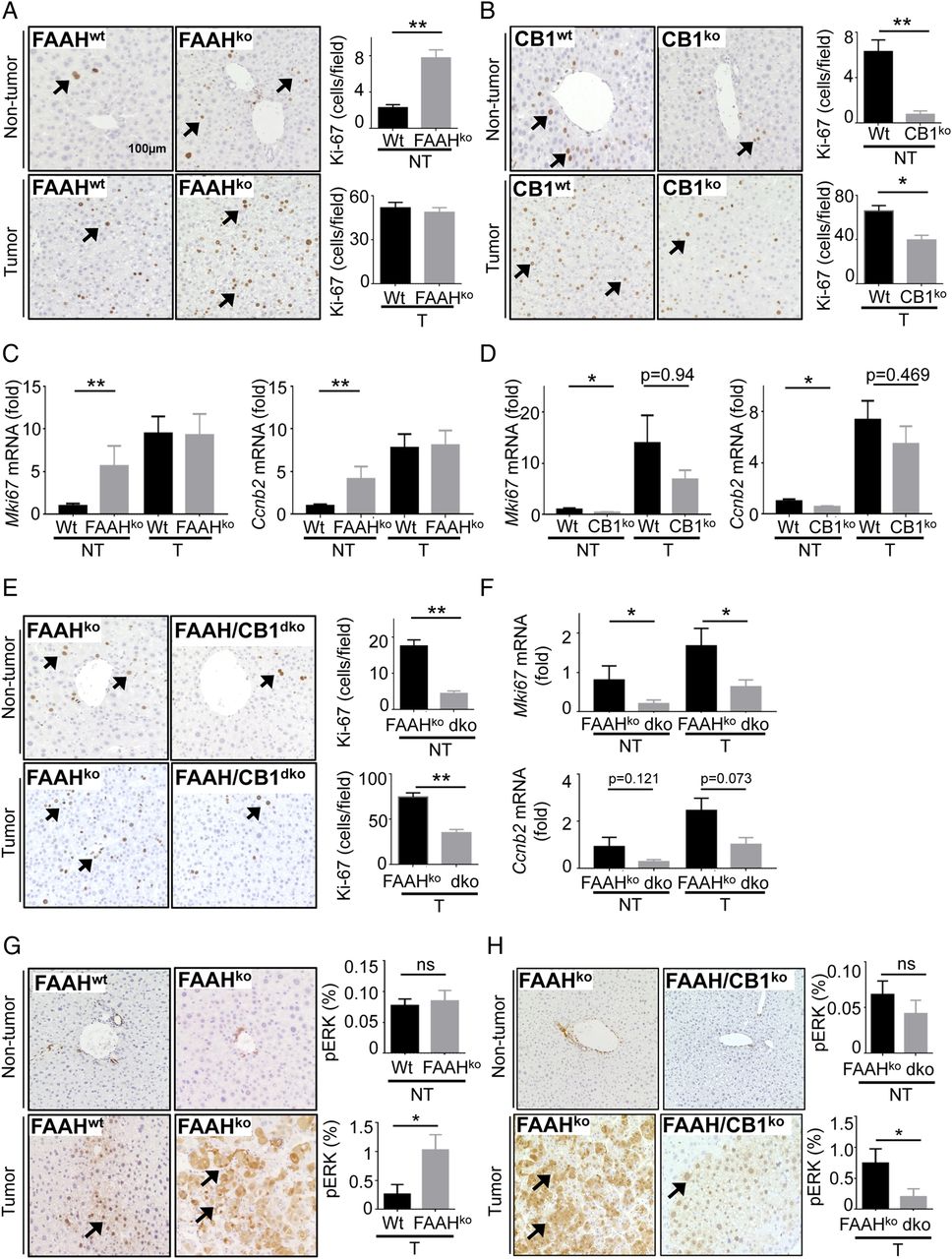
Fatty acid amide hydrolase (FAAH) and CB1 promote hepatocyte proliferation in hepatocarcinogenesis. (A and B). Proliferation was determined by Ki-67 staining and morphometric quantification in non-tumour (NT) and tumour (T) sections from wild-type (Wt) (n=35) and FAAHko (n=26) livers (A) and from wild-type (n=20) and CB1ko (n=11) livers (B). (C and D) Proliferation markers Mki67 and Ccnb2 were determined by qPCR in non-tumour and tumour tissue from wild-type and FAAHko livers (C), and in non-tumour and tumour tissue from wild-type and CB1ko livers (D). (E and F) Proliferation was determined in non-tumour and tumour sections from FAAHko (n=11) and FAAH/CB1dko (n=6) mice by Ki-67 staining and morphometric quantification (E) as well as qPCR for proliferation markers Mki67 and Ccnb2 (F). (G and H) Expression of pErk was determined by immunohistochemistry in non-tumour and tumour tissue from wild-type (n=10) and FAAHko (n=10) livers (G), and from wild-type (n=11) and CB1ko (n=9) livers (H). *p<0.05, **p<0.01.

Fatty acid amide hydrolase (FAAH) and CB1 promote fibrogenesis in hepatocarcinogenesis. (A and B) Liver fibrosis was assessed by Sirius red staining and morphometric quantification in non-tumour (NT) and tumour (T) sections from wild-type (Wt) and FAAHko livers (A), and non-tumour and tumour sections from wild-type (n=20) and CB1ko (n=11) livers (B). (C and D) Fibrosis markers Acta2 and Col1a1 were determined by qPCR in non-tumour and tumour tissue from wild-type and FAAHko livers (C), and in non-tumour and tumour tissue from wild-type and CB1ko livers (D). (E and F) Liver fibrosis was assessed by Sirius red staining and morphometric quantification in tumour and non-tumour sections from FAAHko (n=11) and FAAH/CB1dko (n=6) mice (E) as well as qPCR for fibrogenic genes Acta2, and Col1a1 (F). *p<0.05, **p<0.01.
CB2 promotes T cell accumulation within HCC lesions
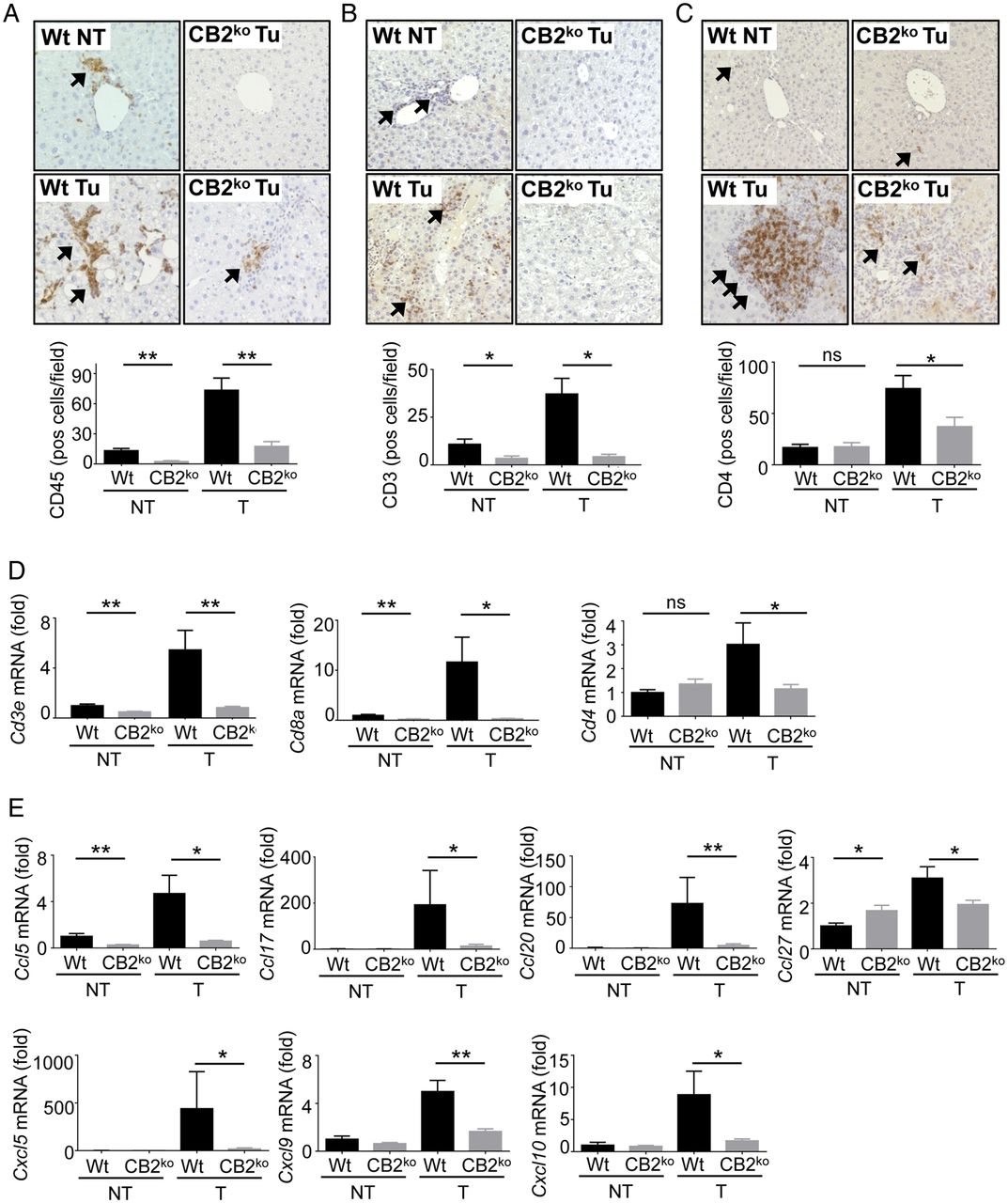
In contrast to CB1, we did not observe an effect of CB2 deficiency on proliferation, as determined by Ki-67 staining and qPCR for mKi67 and Ccnb2 mRNA (see online supplementary figure S7A, B). Moreover, there was also no significant difference in apoptosis (see online supplementary figure S7C), fibrogenesis (see online supplementary figure S7D) or inflammation (see online supplementary figure S7E) between wild-type and CB2 knockout mice. Consistent with previous publications,22 we observed high expression of CB2 within tumours in CD45-positive cells, in particular in F4/80-positive macrophages (see online supplementary figure S8). Accordingly, the highest CB2 expression in isolated cells was in macrophages, and we observed virtually no expression in a murine HCC line as well as four human HCC lines (see online supplementary figure S9). As CB2 has been implicated in the suppression of inflammation and immune responses,22 ,23 we next evaluated the infiltration of inflammatory cell subsets in wild-type and CB2-deficient livers. Immunohistochemistry for pan-leucocyte marker CD45 showed a strong infiltration of CD45-positive cells in tumours of wild-type mice but a significant reduction in tumours of CB2-deficient mice (figure 8A). While we found no significant differences in macrophage and B cell markers between wild-type and CB2-deficient mice (see online supplementary figure S10), there was a strong increase in T cell infiltration into HCC that was significantly suppressed in CB2-deficient mice, as determined by qPCR and immunohistochemistry for pan-T cell marker CD3 as well as suppression of Cd4 and Cd8a mRNA (figure 8C, D). Of note, recent publications have demonstrated a key role for CD4+ T cells in antitumour effector population.24–26 To further understand how CB2 status was linked to the recruitment of T cells, we next analysed expression of T-cell-recruiting chemokines in our HCC model as well as in acute liver injury models. We observed a strong reduction in the mRNA expression of chemokines with known roles in the recruitment of T cells,27 ,28 including Ccl5, Ccl17, Ccl20, Ccl27, Cxcl5, Cxcl9 and Cxcl10 in CB2-deficient mice (figure 8E). Accordingly, there was a trend towards reduced recruitment of CD3-positive T cells in CB2-deficient mice following acute liver injury by a single dose of CCl4 or concanavalin A and a significant reduction of Cd3a and Cd4 mRNA expression as well as the recruitment of CD4-positive cells following a single dose of CCl4 (see online supplementary figures S11 and S12). Moreover, mRNA levels of several chemokines were significantly reduced following a single dose of CCl4 (see online supplementary figure S12) as well as in macrophages isolated from CCl4-treated mice (see online supplementary figure S13), thus confirming that the reduction of T-cell-recruiting chemokines occurred indeed in this cell population. Together, our findings indicate that CB2 modulates the expression of T-cell-recruiting chemokines and the recruitment of T cells, in particular CD4+ T cells in acute and chronic settings. In summary, these data suggest that CB2 is likely to affect hepatocarcinogenesis through CB2-mediated modulation of T-cell-recruiting chemokines in macrophages and subsequent recruitment of CD4+ T cell that mediate antitumour responses—but not through effects of CB2 on proliferation or fibrosis. Hence, CB1 and CB2 exert opposite effects on hepatocarcinogenesis, mediating their effects through distinct cellular targets and mechanisms (figure 9).

Mechanisms by which CB2 contributes to hepatocellular carcinoma (HCC) development. (A) Livers from wild-type (Wt) (n=15) and CB2ko (n=15) mice were stained for CD45 and quantified. (B) Liver from wild-type (n=15) and CB2ko (n=15) mice were stained for CD3 and quantified. (C) Liver from wild-type (n=15) and CB2ko (n=15) mice were stained for CD4 and quantified. (D) Cd3e, Cd4 and Cd8a mRNA levels were determined in livers from wild-type and CB2ko mice by qPCR. (E) Expression of Ccl5, Ccl17, Ccl20, Ccl27, Cxcl5, Cxcl9 and Cxcl10 were compared between liver from wild-type (n=15) and CB2ko (n=15) mice by qPCR. *p<0.05, **p<0.01. NT, non-tumour; T, tumour.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic overview of mechanisms by which CB1 and CB2 affect hepatocarcinogenesis. Increased expression of CB1 within tumours results in the promotion of hepatocellular carcinoma (HCC) formation via increased proliferation and increased fibrogenesis. CB2 expression in macrophages limits HCC development by triggering the recruitment of T cells including CD4+ T cells, a population with a known key role in antitumour responses.
Discussion
Based on a large number of in vitro and in vivo studies employing cancer cell lines, activation of the ECS had been considered a novel antitumour strategy.12–15 ,29 In contrast to this long-held paradigm, several lines of evidence from our study now show a tumour-promoting role of the AEA-CB1 signalling axis: (i) activation of the endocannabinoid signalling by FAAH knockout increases hepatocarcinogenesis and (ii) inhibition of endocannabinoid signalling by CB1 in either wild-type or FAAH-deficient mice decreases hepatocarcinogenesis. These findings are consistent with a recently published study showing tumour-promoting effects of CB1 in hepatocarcinogenesis.30 In view of the moderate changes in endocannabinoids but strongly increased CB1 expression and profound effects of CB1 receptor knockout, the main mechanism by which the ECS becomes activated in hepatocarcinogenesis appears to be the upregulation of CB1. Together with the fact that AEA—the endocannabinoid that is upregulated in FAAH-deficient mice—predominantly activates CB1, these data demonstrate a cancer-promoting role of the AEA-CB1 signalling axis. One likely explanation for differences between the tumour-promoting effects of AEA in our in vivo studies and the antitumour effects of AEA in previous in vitro studies lies in different concentrations. Whereas AEA exist only in nanomolar concentration in vivo, AEA-induced growth arresting and/or cytotoxic effects in vitro could only be observed in micromolar range. In fact, we found that micromolar AEA concentrations kill a large number of murine and human hepatoma cell lines (data not shown). It is unlikely that these concentrations can be achieved in vivo. Moreover, it is likely that micromolar concentrations of endocannabinoids may trigger CB1-independent and CB2-independent cytotoxic effects as demonstrated by us and others.31 ,32
Our study provides several mechanisms by which the CB1-AEA axis promotes HCC development. Most notably, the AEA-CB1 axis had a major impact on proliferation as evidenced by reduced Ki-67 expression in FAAH-deficient and CB1-deficient mice. These findings are consistent with previous studies11 and our own data showing a key role for CB1 and FAAH in regulating liver regeneration, and a recently published study on promotion of hepatocarcinogenesis by CB1.30 Moreover, our findings of increased pERK in tumours from FAAH-deficient mice and decreased pERK in tumours from CB1-deficient or CB1-/FAAH-double-deficient mice are consistent with a previous study showing that AEA increases cancer cell proliferation via transactivation of EGFR.21 In addition, we found increased fibrosis in FAAH-deficient and reduced fibrosis in CB1-deficient mice, respectively, consistent with previous reports on fibrosis-promoting effects of CB1.6 Fibrosis is considered a risk factor for HCC development.33 Accordingly, a hepatic stellate cell signature was associated with outcomes after curative HCC resection.34 Our data showing alterations of fibrosis and proliferation in non-tumour tissue suggest that the AEA-CB1 axis may act predominantly on developing tumour rather than fully established HCC.
Our data differ from several previous in vivo studies, in which cannabinoids or endocannabinoids suppressed tumour formation. In one study, mice treated with the stable AEA analogue metanandamide displayed drastic reduction of tumour volume after transplantation of a transformed rat thyroid cell line.14 Likewise, treatment with mixed CB1/CB2 agonist WIN-55,212-2 resulted in inhibition of skin tumour growth.16 In view of these results, it will be important to determine whether the tumour-promoting effects of the AEA-CB1 axis may be restricted to the liver, for example, by studying the effect of FAAH and CB1 knockout on carcinogenesis in other organs, in particular in types where cannabinoids have been suggested to exert antitumour effects such as gliomas, breast, thyroid, skin and colon cancer.13–16 ,35 Alternatively, CB2-agonistic effects of specific cannabinoids or endocannabinoids might explain antitumour effects seen in previous studies.
Analogous to previous studies on the role of cannabinoid receptors in fibrosis,6 ,8 we observed opposite functions of CB1 and CB2, with exacerbation of hepatocarcinogenesis in CB2-deficient mice. In contrast to previous studies, we found no difference in fibrosis between wild-type and CB2-deficient mice. It is possible that fibrogenesis in the context of carcinogenesis differs from toxin-induced and bile duct ligation-induced fibrosis in regards to the involvement of CB2. In contrast to CB1, we also found no influence of CB2 deficiency on proliferation. Consistent with previous studies showing high expression and key functions of CB2 in the immune system,22 ,23 we found predominant expression of CB2 in F4/80-positive macrophages and a role for CB2 in the expression of T-cell-recruiting chemokines both in liver under different disease conditions and in macrophages isolated from injured liver. We found that CB2 deficiency significantly decreased the recruitment of CD4+ T cells in hepatocarcinogenesis and in acute liver injury. In light of the key role of CD4+T cells in antitumour immunity,24–26 it is likely that the increased tumour load in CB2-deficient mice is related to a failed tumour immunosurveillance. Future studies with conditional ablation of CB2 in macrophages are needed to confirm that CB2 in macrophages is relevant to trigger these antitumour responses. Moreover, we did not find alterations of CB2 expression in CB1-deficient mice (see online supplementary figure S14), thus excluding that reduction of HCC development by CB1 deficiency was mediated through increased CB2 expression. Interestingly, recent studies have shown a downregulation of CB2 by lipopolysaccharide (LPS),36 suggesting that the chronically increased LPS levels in patients with end-stage liver disease could contribute to a tumour-friendly environment via suppression of CB2-mediated antitumour immunity.
Besides CB1 and CB2, additional cannabinoid receptors such as TRPV1 and GPR55 might be involved in hepatocarcinogenesis. Our finding that TRPV1 deletion did not alter hepatocarcinogenesis in wild-type as well as FAAH-deficient mice excludes that TRPV1 acts as a relevant endocannabinoid receptor in hepatocarcinogenesis. This is consistent with the unaltered TRPV1 expression in murine and human hepatocarcinogenesis. As we have not compared FAAH/CB1dko mice with CB1ko mice, we cannot completely exclude that FAAH regulates additional tumour-promoting endocannabinoids that do not act through CB1, CB2 or TRPV1. As such, GPR55, whose expression was elevated in murine and human HCC, would be a potential candidate. Based on our data in CB1/FAAHdko mice, however, the main contribution appears to be through CB1 and additional receptors such as GPR55 are likely to make only minor contribution. We were not able to investigate potential contributions of GPR55 to hepatocarcinogenesis due to the lacking availability of GPR55 knockout mice.
Our study raises several issues with high relevance for patients with cancer. In view of ongoing efforts to develop cannabinoids for antitumour treatment, our data indicate an urgent need for more detailed studies that analyse specific contributions of CB1 and CB2 in cancer. As such, it is likely that receptor-specific interventions will be needed that suppresses tumour-promoting effects of the ECS without interfering with tumour-suppressive effects. Based on our findings, non-selective CB agonists or inhibitors of endocannabinoid degradation do not appear to be suitable for antitumour therapies. Our data suggest that CB1 antagonists or CB2 agonists may protect from HCC development. In this regard, CB1 antagonists could ‘kill several birds with one stone’ by decreasing HCC development as well as the development of liver fibrosis and/or hepatic steatosis. Consistent with our findings, tumour-suppressive effects of CB2 receptor agonists have been reported by Vara et al.37 However, we did not find expression of CB2 in HCC or HCC cell lines; thus, a direct effect of CB2 agonists on tumour cells, as proposed by Vara et al, is unlikely in our opinion.
Importantly, our findings emphasise the urgent need to investigate whether cannabis consumption or medical treatment with cannabinoids results in increased tumour development or acceleration of tumour growth, respectively. Daily cannabis consumption has been linked with progression of liver fibrosis and increased hepatic steatosis in patients with chronic hepatitis C.38 ,39 Based on our findings, it is conceivable that cannabis consumption may also promote the development of HCC or other cancers. With cannabis representing the most commonly used illicit drug worldwide,40 effects on carcinogenesis could be profound, in particular in patients with alcoholic liver disease or HCV infection who not only have a higher risk for HCC development but often also a higher prevalence of cannabis consumption.38
Acknowledgments
The authors thank Dr Benjamin Cravatt (Scripps Research Institute, La Jolla, USA) for providing FAAH-deficient mice, Dr Beat Lutz (University of Mainz, Germany) for providing CB1 floxed mice and Drs William Blaner and Serge Kramer (both Columbia University) for endocannabinoid measurements.
References
Footnotes
K-TS and IM contributed equally.
Contributors K-TS, IM, G-YG and AA designed and performed research. SWC provided human samples and supported their analysis. RF analysed microarray data. RFS, IM and K-TS drafted the manuscript. RFS designed and oversaw the study.
Funding This study was funded by 5R01DK075830 (to RFS), a German research foundation scholarship to IM (ME 3723/1-1), and Korea National Research Foundation to K-TS (NRF-2015R1C1A1A01053232).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics committee Chuncheon Sacred Heart Hospital (IRB no. 2015-30).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Are data will be freely shared.