Article Text

Abstract
Objective The gut microbiota modulates host susceptibility to intestinal inflammation, but the cell types and the signalling pathways orchestrating this bacterial regulation of intestinal homeostasis remain poorly understood. Here, we investigated the function of intestinal epithelial toll-like receptor (TLR) responses in the dextran sodium sulfate (DSS)-induced mouse model of colitis.
Design We applied an in vivo genetic approach allowing intestinal epithelial cell (IEC)-specific deletion of the critical TLR signalling adaptors, MyD88 and/or TIR-domain-containing adapter-inducing interferon-β (TRIF), as well as the downstream ubiquitin ligase TRAF6 in order to reveal the IEC-intrinsic function of these TLR signalling molecules during DSS colitis.
Results Mice lacking TRAF6 in IECs showed exacerbated DSS-induced inflammatory responses that ensued in the development of chronic colon inflammation. Antibiotic pretreatment abolished the increased DSS susceptibility of these mice, showing that epithelial TRAF6 signalling pathways prevent the gut microbiota from driving excessive colitis. However, in contrast to epithelial TRAF6 deletion, blocking epithelial TLR signalling by simultaneous deletion of MyD88 and TRIF specifically in IECs did not affect DSS-induced colitis severity. This in vivo functional comparison between TRAF6 and MyD88/TRIF deletion in IECs shows that the colitis-protecting effects of epithelial TRAF6 signalling are not triggered by TLRs.
Conclusions Intestinal epithelial TRAF6-dependent but MyD88/TRIF-independent and, thus, TLR-independent signalling pathways are critical for preventing propagation of DSS-induced colon inflammation by the gut microbiota. Moreover, our experiments using mice with dual MyD88/TRIF deletion in IECs unequivocally show that the gut microbiota trigger non-epithelial TLRs rather than epithelial TLRs to restrict DSS colitis severity.
- EXPERIMENTAL COLITIS
- GUT INFLAMMATION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Studies in germfree mice showed that the gut microbiota has a protective role during dextran sodium sulfate (DSS) colitis in mice.
Gene targeting in mice showed that the toll-like receptor (TLR) signalling molecule, MyD88, mediates protective effects during DSS colitis.
Bone marrow transplant experiments showed that MyD88/TRIF-mediated TLR signalling acts in non-haematopoietic cells to protect mice from DSS colitis.
Distinct studies applying cell-type-specific genetic modulation of MyD88 showed that protective TLR signalling acts in intestinal epithelial cells (IEC), in B cells and in myeloid cells.
What are the new findings?
Mice with IEC-specific MyD88/TRIF deletion display unaltered DSS colitis severity, unequivocally showing that the microbiota modulate colon inflammation by triggering TLRs residing on non-epithelial cells rather than on IECs.
Mice with IEC-specific TRAF6 deletion display increased DSS colitis severity, showing that instead of TLR signalling, TLR-independent TRAF6 signalling in IECs limits acute DSS colitis.
Microbiota depletion abolished excessive colitis in mice lacking intestinal epithelial TRAF6, demonstrating that epithelial TRAF6 signalling limits DSS colitis by preventing the gut microbiota from exacerbating colon inflammation after DSS-induced epithelial damage.
These findings establish a model of intestinal-immune homeostasis in which epithelial TLR-independent TRAF6 signalling prevents the microbiota from propagating colitis, while bacteria that invade the mucosa trigger TLRs on non-epithelial cells to restrain colitis severity.
Introduction
The maintenance of intestinal homeostasis depends on a tightly regulated cross-talk between the gut microbiota, intestinal epithelial cells (IEC) and mucosal immune and stromal cells. Even though deregulated immune responses to the gut microbiota are thought to contribute to the development of IBDs,1 germfree mice were shown to be more susceptible to dextran sodium sulfate (DSS)-induced colitis,2 suggesting a beneficial role for the microbiota in this model of colon inflammation. Consistent with this observation, mice lacking individual toll-like receptors (TLR), or the critical TLR-signalling molecule, MyD88, showed increased sensitivity to DSS colitis, suggesting that microbiota-induced MyD88-dependent TLR responses protect from DSS-induced colon inflammation.3–7 Together with genetic mouse models showing that epithelial NF-κB activation prevents spontaneous and DSS-induced colon inflammation,8–11 the studies above collectively raised the possibility that bacteria might directly trigger epithelial TLR/MyD88-dependent NF-κB activation for controlling intestinal inflammation.12
This plausible hypothesis was questioned by an elegant study showing that mice lacking MyD88 specifically in B cells, but not mice lacking MyD88 in IECs, display increased lethality in response to DSS treatment.13 Additionally, restricted expression of MyD88 in myeloid cells was shown to abolish the susceptibility of MyD88-deficient mice to DSS colon injury.14 These observations indicated that MyD88-mediated protective effects in DSS colitis originate from B cells and myeloid cells rather than epithelial cells. On the other hand, another study observed that IEC-specific ablation of MyD88 compromised epithelial barrier function, and as such, did sensitise mice to DSS colitis,15 arguing in favour of epithelial-intrinsic TLR/MyD88-driven protection from colon inflammation. Moreover, while the above studies focused on MyD88-dependent TLR signalling, also TRIF-dependent epithelial TLR signalling might contribute to the protective microbiota effects in DSS colitis. Indeed, reciprocal bone marrow transplant experiments using wild-type and MyD88/TRIF double deficient mice showed that such complete blockage of TLR signalling in non-haematopoietic cells was sufficient to sensitise mice to DSS colitis.16 Taken together, despite the abundance of the gut microbiota adjacent to the epithelium, the role of epithelial TLR triggering by the luminal microbiota during DSS colitis currently remains unclear.
In this study, we experimentally addressed the intestinal epithelial function of TLR signalling during DSS colitis by using three distinct mouse models allowing IEC-specific deletion of the critical TLR signalling adaptors MyD88 and TRIF, as well as the downstream ubiquitin ligase TRAF6. Surprisingly, simultaneous IEC-specific deletion of both MyD88 and TRIF did not sensitise mice to DSS-induced colitis. Instead, IEC-specific ablation of TRAF6 rendered mice more susceptible to DSS-induced acute and chronic colitis, revealing that epithelial TRAF6-dependent but TLR-independent signalling is essential for limiting inflammation after DSS-induced damage of the colonic epithelium.
Results
Epithelial-specific TRAF6 deficiency sensitises mice to DSS-induced colitis
In order to investigate the role of intestinal epithelial TRAF6, we crossed mice harbouring LoxP-flanked Traf6 alleles17 with Villin-Cre transgenics expressing Cre recombinase under the Villin promoter.18 These TRAF6IEC-KO mice showed efficient deletion of TRAF6 in IECs, but endoscopic and histological examination did not reveal any colon or small intestinal abnormalities (see online supplementary figure S1), demonstrating that epithelial TRAF6 does not control intestinal immune homeostasis under basal conditions.
To assess whether epithelial TRAF6 regulates colon homeostasis under conditions of inflammation, we treated TRAF6IEC-KO mice and their TRAF6FL control littermates for 7 days with 2% DSS in the drinking water followed by 2 days recovery on normal drinking water, after which we sacrificed the mice on day 9. Starting from day 5 of DSS treatment, TRAF6IEC-KO mice lost significantly more weight than control animals, indicating that epithelial TRAF6 deficiency sensitised mice to DSS-induced colitis (figure 1A). Consistent with the increased weight loss, TRAF6IEC-KO mice showed increased intestinal bleeding and suffered from more severe diarrhoea (figure 1B, C). Endoscopic analysis on day 6 revealed enhanced colon inflammation in TRAF6IEC-KO mice compared with their TRAF6FL littermates, as indicated by a less translucent and more granular colonic wall and signs of diarrhoea (figure 1D). Moreover, TRAF6IEC-KO mice sacrificed on day 9 revealed considerably shorter colons compared to their littermate controls (figure 1E). Histopathological examination of colon sections showed more severe epithelial erosion, loss of goblet cells and areas of mucosal ulceration, as well as increased numbers of infiltrating mucosal and submucosal leukocytes in TRAF6IEC-KO mice compared with their control littermates, resulting in higher histological scores for both tissue damage and inflammation (figure 1F, G). Immunostaining of colon sections with specific antibodies revealed elevated numbers of infiltrating neutrophils as well as macrophages in TRAF6IEC-KO colons compared to their TRAF6FL littermates (figure 1H, I), which was accompanied by enhanced expression of several inflammatory cytokines and chemokines including tumour necrosis factor (TNF), IL-1β, CCL3 and CXCL2 (figure 1J, K). Taken together, these results demonstrate that TRAF6IEC-KO mice were more susceptible to DSS-induced colitis, revealing a previously unidentified function of TRAF6-mediated signalling in IECs that protects the colon from DSS-induced inflammation.
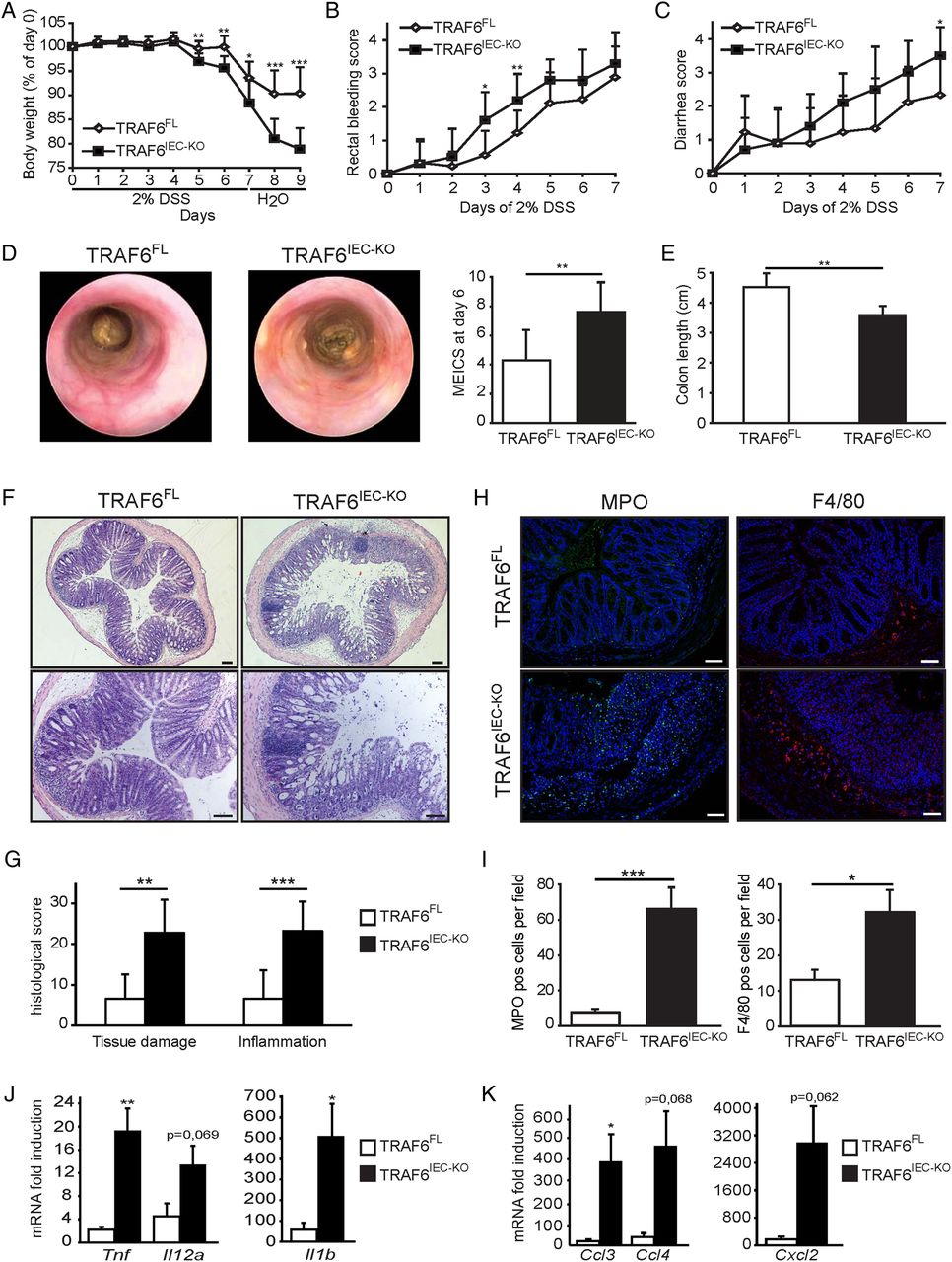
TRAF6IEC-KO mice are more susceptible to dextran sodium sulfate (DSS)-induced acute colitis. (A) Body weight change, (B) rectal bleeding score, and (C) diarrhoea score of TRAF6FL (n=9) and TRAF6IEC-KO (n=10) mice administered 2% DSS for 7 days followed by 2 days of normal drinking water. (D) Representative endoscopic pictures and mean endoscopic index of colitis severity of TRAF6FL and TRAF6IEC-KO mice after 6 days of DSS treatment. (E) Colon length and (F) representative H&E stained colon cross-sections of TRAF6FL and TRAF6IEC-KO mice at day 9 of the DSS colitis protocol. Bars, 100 μm. (G) Histological tissue damage and inflammation scoring at day 9 of the DSS protocol. (H) Representative pictures and (I) mean number±SEM per 200× magnification field of myeloperoxidase (MPO)-stained neutrophils and F4/80-stained macrophages in TRAF6FL and TRAF6IEC-KO mice at day 9 of the DSS colitis protocol. Bars, 100 μm. (J) Cytokine and (K) chemokine mRNA induction±SEM in colon of TRAF6FL and TRAF6IEC-KO mice at day 9 of the DSS colitis protocol compared to mRNA levels of untreated TRAF6FL and TRAF6IEC-KO mice. All data shown are representative of at least 2 independent experiments. All statistical analyses were performed with unpaired two-sided Student's t tests with unequal variance.
TRAF6IEC-KO mice develop chronic colitis after DSS treatment
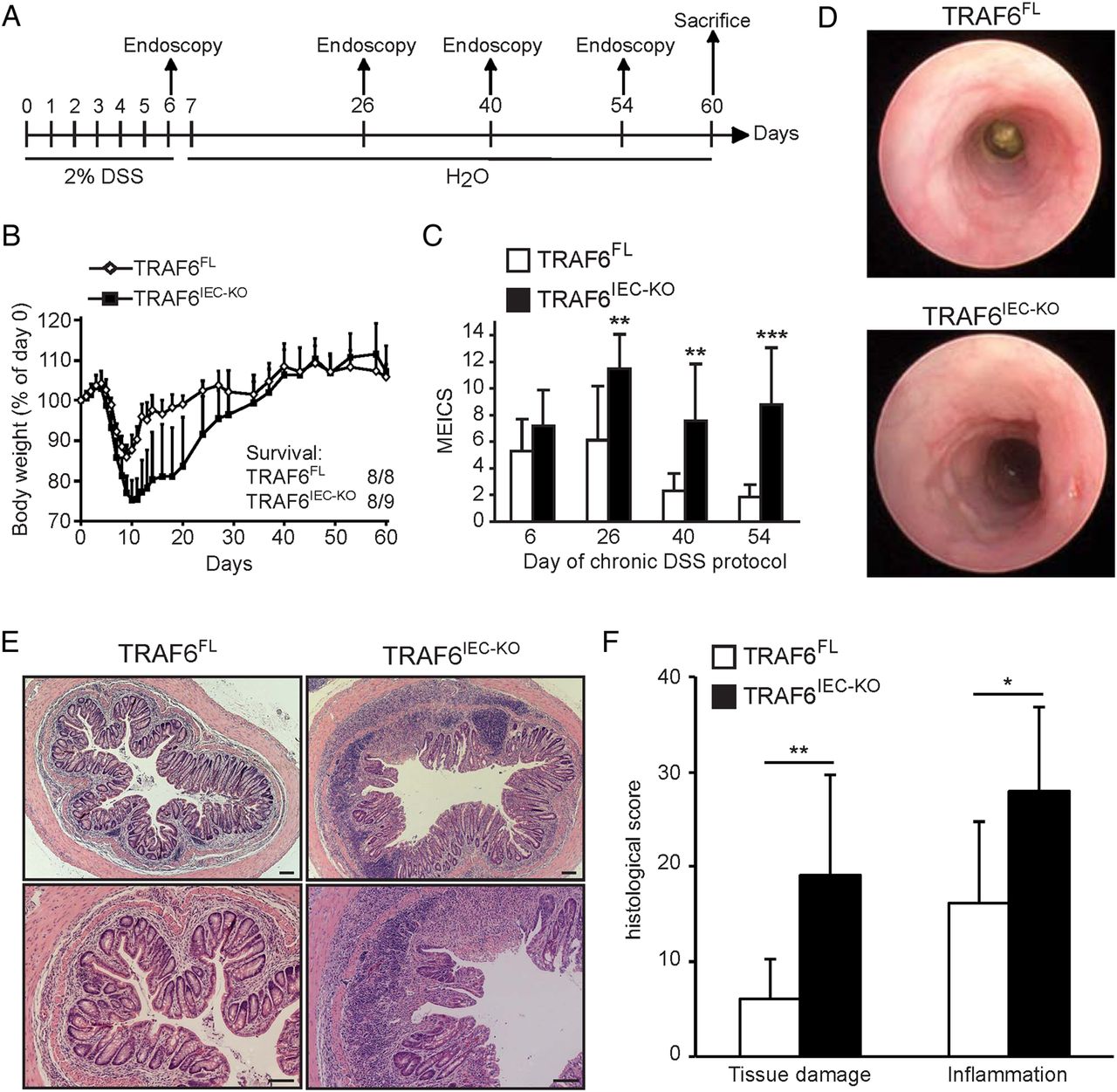
Although the exact mechanisms of how DSS evokes colitis are not entirely clear, DSS is thought to damage the colon epithelium by directly killing IECs, followed by a regenerative proliferation response that aims to restore the epithelial barrier, and as such, to limit propagation of colonic inflammatory responses. However, both epithelial cell death and regenerative proliferation upon DSS treatment were similar between TRAF6IEC-KO and littermate control mice (see online supplementary figure S2). These observations indicated that increased colitis severity in TRAF6IEC-KO mice could not be attributed to increased IEC death, and that even in a setting of ongoing severe inflammation, TRAF6-deficient IECs retain their capacity to elicit regenerative responses attempting to restore tissue homeostasis. Furthermore, this result suggests that rather than functioning in tissue maintenance or repair, anti-inflammatory effects might constitute the dominant factor by which epithelial TRAF6 protects from DSS colitis. This hypothesis prompted us to evaluate the role of epithelial TRAF6 in chronic DSS colitis by treating TRAF6IEC-KO and TRAF6FL littermates with 2% DSS for 6 days followed by a 7-week recovery period, during which colitis severity was assessed by endoscopy at regular time intervals (figure 2A). Consistent with the above observations showing sensitisation to DSS colitis yet preserved regenerative capacity of their IECs, TRAF6IEC-KO mice suffered from more severe acute colitis losing more than 20% of their original body weight by day 10, but fully recovered from this weight loss after about 35 days (figure 2B). Interestingly, despite the lack of body weight differences, endoscopic examination 40 and 54 days after initiation of DSS treatment revealed severe colon inflammation in TRAF6IEC-KO mice as opposed to an almost completely healed colonic wall in TRAF6FL animals (figure 2C, D). Histological examination of colon sections from animals sacrificed at day 60 revealed areas with severe mucosal and submucosal immune cell infiltrates accompanied by notable inflammation-associated tissue damage in TRAF6IEC-KO mice, while their littermate controls showed only mild signs of inflammation at this stage (figure 2E, F). Thus, while TRAF6-deficient IECs showed similar apoptotic and regenerative responses upon DSS treatment, they failed to limit ongoing inflammatory responses resulting in chronic colitis. These observations suggest that protective epithelial TRAF6 signalling in the colon is dispensable for IEC-intrinsic functions, but rather limits inflammation in a paracrine fashion after DSS treatment.

TRAF6IEC-KO mice develop chronic colon inflammation upon dextran sodium sulfate (DSS) treatment. (A) TRAF6FL (n=8) and TRAF6IEC-KO (n=9) mice were administered 2% DSS for 6 days followed by normal drinking water with endoscopies at the indicated time points until day 60 when they were sacrificed. (B) Body weight change and final survival at day 60. (C) Mean endoscopic index of colitis severity of TRAF6FL and TRAF6IEC-KO mice at indicated time points of the chronic DSS protocol. (D) Representative endoscopic pictures from TRAF6FL and TRAF6IEC-KO mice at day 54 of the chronic DSS colitis protocol. (E) Representative H&E stained colon cross-sections of TRAF6FL and TRAF6IEC-KO mice at day 60 of the chronic DSS colitis protocol. Bars, 100 μm. (F) Histological tissue damage and inflammation scoring at day 60 of the chronic DSS protocol. All data shown are representative of two independent experiments. All statistical analyses were performed with unpaired two-sided Student's t tests with unequal variance.
The gut microbiota drives increased DSS-induced colitis in TRAF6IEC-KO mice
Because TRAF6 mediates responses to bacterial TLR triggering as well as to host factors such as IL-17 and TGFβ,19 ,20 multiple stimuli could trigger the protective functions of epithelial TRAF6 during DSS colitis. In order to discriminate between bacterial and host factor-mediated TRAF6 signalling, we investigated whether epithelial TRAF6 deficiency also sensitised mice to DSS colitis after depletion of the gut microbiota. To do so, we treated both TRAF6FL and TRAF6IEC-KO mice with broad-spectrum antibiotics (AB) to reduce the gut luminal microbiota before administrating DSS. While AB-treated TRAF6IEC-KO mice lost more weight after DSS than their controls (figure 3A), parameters reflecting colon inflammation, such as stool consistency, rectal bleeding, colon length, as well as endoscopic and histological evaluation of colitis severity were comparable between microbiota-depleted TRAF6IEC-KO mice and their AB-treated controls (figure 3B, H). Indeed, whereas TRAF6IEC-KO mice harbouring normal gut microbiota showed more occult blood and diarrhoea, a shorter colon and more endoscopically visible colitis upon DSS treatment than their control TRAF6FL mice, these differences were not, or much less, apparent in microbiota-depleted TRAF6IEC-KO mice in comparison with their respective controls (figure 3B, E). Thus, in microbiota-depleted conditions, epithelial TRAF6-deficiency does not confer increased susceptibility to DSS-induced colitis, suggesting that intestinal bacteria themselves trigger protective epithelial TRAF6 signalling.

Excessive dextran sodium sulfate (DSS)-induced acute colitis in TRAF6IEC-KO mice is driven by the gut microbiota. TRAF6FL and TRAF6IEC-KO mice were treated with antibiotics (+AB) or were left untreated (−AB) for 4 weeks before administration of DSS. (A) Body weight change, (B) rectal bleeding score, and (C) diarrhoea score of TRAF6FL −AB (n=8), TRAF6IEC-KO −AB (n=10), TRAF6FL +AB (n=8), TRAF6IEC-KO +AB (n=11) mice that were administered 2% DSS for 7 days followed by 2 days of normal drinking water. For A–C, asterisks above the data points indicate statistically significant differences between TRAF6FL −AB and TRAF6IEC-KO −AB mice, while asterisks below the data points indicate statistically significant differences between TRAF6FL +AB and TRAF6IEC-KO +AB mice. (D) Colon length, (E) mean endoscopic index of colitis severity and (F) representative endoscopic pictures of indicated mice at day 9 of the DSS colitis protocol. (G) Representative H&E stained colon cross-sections of indicated mice, and (H) histological tissue damage and inflammation scoring at day 9 of the DSS colitis protocol. Bars, 100 μm. All data shown are representative of two independent experiments. All statistical analyses were performed with unpaired two-sided Student's t tests with unequal variance.
Anti-inflammatory effects of epithelial TRAF6 signalling during DSS colitis are independent of TLR signalling
The results obtained from AB-treated mice raised the possibility that microbiota may protect mice from DSS-induced colitis by triggering TLR-dependent protective responses in IECs. Alternatively, TLR-independent host-derived epithelial TRAF6 signalling may prevent the gut microbiota from driving inflammatory responses in non-epithelial cells during DSS colitis. Indeed, also in the latter case, the absence of gut microbiota would abolish sensitisation of TRAF6IEC-KO mice to DSS colitis. To discriminate between these two options, we generated mice lacking the upstream TLR signalling molecules MyD88 (see online supplementary figure S3) or TRIF21 in IECs, allowing directly assessing the role of epithelial TLR signalling in DSS colitis. Neither MyD88IEC-KO nor TRIFIEC-KO mice showed differences in DSS-induced colitis severity when compared with their respective controls (see online supplementary figures S4 and S5), demonstrating that neither MyD88-dependent nor TRIF-dependent TLR signalling in IECs is essential to protect the colon from DSS-induced inflammation.
We then investigated whether complete blockage of TLR signalling in IECs affects DSS-induced colon inflammation by comparing the DSS colitis response of mice lacking both MyD88 and TRIF specifically in IECs (MyD88IEC-KO/TRIFIEC-KO) with their MyD88FL/TRIFFL littermates. In order to directly compare this effect of epithelial TLR blockage with that of epithelial TRAF6 deficiency, we also included age-matched and sex-matched TRAF6FL and TRAF6IEC-KO mice housed in the same cages. Moreover, in order to more easily detect enhanced DSS susceptibility, in this experiment, we applied a mild colitis protocol by administrating only 1% DSS in the drinking water. Surprisingly, DSS-treated MyD88IEC-KO/TRIFIEC-KO mice showed similar weight loss, rectal bleeding and diarrhoea compared to their littermate controls (figure 4A, C). MyD88IEC-KO/TRIFIEC-KO and MyD88FL/TRIFFL mice also did not show considerable differences in colon length after being sacrificed at day 9 (figure 4D). Additionally, histopathological scoring of colon sections of mice sacrificed at day 9 failed to reveal considerable differences in tissue damage or colon inflammation between MyD88IEC-KO/TRIFIEC-KO and MyD88FL/TRIFFL mice (figure 4E, F). By contrast, TRAF6IEC-KO mice suffered from more severe DSS-induced colitis compared to their TRAF6FL littermates, but also to the MyD88FL/TRIFFL and MyD88IEC-KO/TRIFIEC-KO mice (figure 4A, F). Similar results were obtained when the mice were treated with 2% DSS (data not shown). Therefore, complete ablation of TLR signalling in IECs does not sensitise mice to DSS-induced colitis. Moreover, this result indicates that the observed protective effects of epithelial TRAF6 signalling in this model of colon inflammation are independent of MyD88/TRIF-mediated TLR signalling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
MyD88IEC-KO/TRIFIEC-KO mice are not more susceptible to dextran sodium sulfate (DSS)-induced acute colitis. (A) Body weight change, (B) rectal bleeding score, and (C) diarrhoea score of TRAF6FL (n=8), TRAF6IEC-KO (n=8), MyD88FL/TRIFFL (n=7) and MyD88IEC-KO/TRIFIEC-KO (n=6) mice administered 1% DSS for 7 days followed by 2 days of normal drinking water. For A–C, asterisks indicate statistically significant differences between TRAF6FL and TRAF6IEC-KO mice. (D) Colon length, (E) histological tissue damage and inflammation scoring and (F) representative H&E stained colon cross-sections of indicated mice at day 9 of the DSS colitis protocol. Bars, 100 μm. All data shown are representative of two independent experiments. All statistical analyses were performed with unpaired two-sided Student's t tests with unequal variance.
Discussion
Here, we show that IEC-specific deletion of both MyD88 and TRIF did not increase susceptibility of mice to DSS-induced colitis. This result is surprising, as several studies showed more severe DSS colitis in the absence of either the gut microbiota,2 TLR signalling,3–6 ,22 or epithelial NF-κB signalling,9–11 which collectively led to a widespread assumption that microbiota-induced TLR signalling in IECs protects mice from DSS-induced colitis.12 Even though a study showing an impaired epithelial barrier function and more severe DSS colitis in mice with epithelial MyD88 ablation reinforced this hypothesis,15 our findings provide unequivocal genetic evidence that the gut microbiota do not mediate protective effects in DSS-induced colitis by triggering epithelial TLR responses. Our results, thus, support studies showing that protective MyD88 signalling during DSS colitis acts in B cells to prevent systemic dissemination of intestinal bacteria, as well as in myeloid cells, to initiate a regenerative response of the epithelium that is necessary for tissue repair after DSS damage.13 ,14 Furthermore, while bone marrow chimaera experiments demonstrated that double MyD88/TRIF deficiency in radio-resistant cells was sufficient for sensitising mice to DSS colitis,16 our observations ruling out a role for epithelial MyD88/TRIF signalling suggest that this phenomenon may be mediated by TLR signalling in non-epithelial stromal cells. Thus, together with the above studies, our results support a model in which the microbiota invading the mucosa upon DSS-induced epithelial injury trigger TLR signalling in non-epithelial cells to coordinate a response that limits bacterial spreading, and that acts on epithelial cells in a paracrine fashion in order to preserve tissue integrity. By contrast, epithelial-intrinsic TLR signalling does not exert a crucial role in inflammatory or tissue-preserving responses during DSS colitis.
However, instead of epithelial TLR signalling, we found that TLR-independent epithelial TRAF6 signalling prevented development of DSS-induced excessive colon inflammation that was driven by the gut microbiota. Interestingly, TRAF6 was recently found to exert a very similar homeostatic function in dendritic cells (DC), as MyD88-independent TRAF6 signalling in DCs was essential to preserve immune tolerance to the small intestinal microbiota.23 However, the identity of the crucial TLR-independent innate immune receptors that control gut homeostasis via TRAF6 signalling in either DCs or IECs remains unclear. We observed that although epithelial TRAF6 deficiency did not affect IEC survival or proliferation, TRAF6IEC-KO mice developed chronic colon inflammation after DSS treatment. This suggests that the triggers initiating epithelial TRAF6 signalling do not limit DSS colitis by mediating epithelium-intrinsic functions involved in epithelial homeostasis, but rather, induce the release of epithelial factors that exert direct or indirect anti-inflammatory functions aiming to downregulate mucosal inflammatory reactions after DSS-induced damage. Numerous cytokines that signal in a TRAF6-dependent, but MyD88/TRIF-independent manner, might be responsible for eliciting such anti-inflammatory functions in IECs. For instance, inhibition of epithelial TGFβ signalling by expression of dominant negative TGFβ-RII specifically in IECs increased the sensitivity of mice to DSS colitis.24 Although TRAF6 is only involved in a subset of TGFβ-induced signalling pathways,19 it is plausible that the protective effect of epithelial TRAF6 in DSS colitis could, in part, be induced by TGFβ signalling. Additionally, several studies showed that mice deficient in IL-17 signalling fail to control DSS colon inflammation.25–27 Since TRAF6 mediates MyD88/TRIF-independent signalling in response to IL-17,20 a role for TRAF6 in protective effects induced by IL-17 is conceivable. However, future studies addressing the in vivo intestinal epithelial role of various receptors initiating TRAF6-dependent signalling will be required to reveal the identities of the crucial agents responsible for initiating protective epithelial TRAF6 signalling in colon inflammation.
Taken together, this study adds to our understanding of microbiota-host interactions controlling intestinal homeostasis. Our observation that epithelial MyD88/TRIF ablation does not sensitise to DSS colitis unequivocally shows that IEC-intrinsic TLR signalling is not needed to limit DSS-induced colon inflammation, and supports the notion that TLR/MyD88 signalling acts in non-epithelial cells, such as B cells, myeloid cells and stromal cells to promote recovery from DSS-induced injury. However, similar to their small intestinal homeostatic effect acting in DCs, TRAF6-dependent signalling pathways in IECs limit colon inflammation after DSS-induced injury. The comparison with the effects of MyD88/TRIF deficiency shows that the critical TRAF6-dependent epithelial pathways controlling DSS colitis are not initiated by TLR triggering. Together, our observations suggest that the gut microbiota triggers TLR-dependent and TLR-independent homeostatic signalling pathways in different cell types for preserving intestinal homeostasis. Further identification of these microbiota-induced pathways and cellular targets will be invaluable in understanding the mechanisms by which the microbiota affects the pathogenesis of chronic intestinal inflammation.
Materials and methods
Mice
TRAF6 and TRIF-floxed mice have been described.17 ,21 We generated MyD88-floxed mice using gene targeting by homologous recombination in Bruce-4 embryonic stem cells derived from C57Bl/6 mice28 as shown in online supplementary figure S3 using procedures as described.29 Germline transmitting chimaeras were generated using embryonic stem cells carrying the respective targeted gene. Mice were crossed to Flp-Deleter transgenics30 to excise the FRT-flanked neomycin selection cassette, and were then bred with villin-Cre mice18 to delete the respective genes in IECs. Flp-Deleter and villin-Cre mice were backcrossed into the C57Bl/6 genetic background for at least 10 generations. In all experiments, co-housed littermates carrying the LoxP-flanked alleles, but not expressing Cre recombinase, were used as controls. The mice used in this study were housed in individually ventilated cages in specific pathogen-free animal facilities at the Institute for Genetics of the University of Cologne. All animal procedures were conducted in accordance with European, national and institutional guidelines and protocols, and were approved by local government authorities.
Induction and clinical evaluation of DSS colitis
Seven-week-old to eight-week-old sex-matched co-housed littermates were administered 1% or 2% DSS (36–50 kDa; MP Biomedicals) in their drinking water ad libitum for 7 consecutive days, followed by 2 days of normal drinking water. Survival and clinical parameters, such as weight loss, rectal bleeding and diarrhoea were monitored daily. The appearance of blood in the stool was measured by haemoccult tests (Beckman Coulter), and was given a score from 0 to 4, defined as follows: 0 for no blood; 2 for positive haemoccult; and 4 for gross bleeding. The severity of diarrhoea was given a score from 0 to 4, defined as follows: 0 for well-formed pellets; 2 for pasty and semiformed stools; and 4 for liquid stools. All clinical scorings were performed in a blinded fashion. Postmortem, the entire colon was removed from caecum to anus, and the colon length was measured as a marker for inflammation.
For antibiotic treatment, 1 g/L ampicillin (ICN Biomedicals), 1 g/L neomycin (Sigma), 0.5 g/L meronem (AstraZeneca) and 0.5 g/L vancomycin (Sigma) were added to the drinking water starting from weaning for 28 days. AB-containing drinking water was refreshed every second day.
Endoscopic procedures
For monitoring colitis in vivo, mice were anaesthetised using intraperitoneal injection of ketamin (Ratiopharm)/Rompun (Bayer) and a high-resolution miniendoscope, denoted Coloview (Karl-Storz, Germany), was used as previously described.31 The mean endoscopic index of colitis severity (MEICS) was determined in a blinded fashion by evaluating stool consistency, fibrin deposition, vasculature, translucency and granularity of the colon wall. Each of these five different parameters of inflammation was given a score from 0 to 3, resulting in a total MEICS ranging from 0 to 15.
Histology and immunohistochemistry
Colon tissue was fixed overnight in 4% paraformaldehyde, embedded in paraffin, and 4 μm sections were made. Cell death was evaluated on paraffin sections by TUNEL staining (DeadEnd Fluorometric TUNEL System, Promega) according to the manufacturer's instructions. For quantification, the numbers of TUNEL-positive cells in at least three different 200× magnification fields from three different cross-sections each were counted per mouse. For assessing cell proliferation, mice were injected intraperitoneally with 100 mg/kg BrdU 2 h before sacrifice. Immunostaining was performed with anti-BrdU (Chemicon). For quantification, the numbers of BrdU-positive cells in all well-oriented and longitudinally sectioned crypts in at least three different 200× magnification fields from three different cross-sections each per mouse were counted. For staining of macrophages and neutrophils, primary antibodies for F4/80 (Serotec) and MPO (Dako), respectively, and secondary Alexa-coupled antibodies (Invitrogen) were used. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Vector Laboratories). All immunefluorescence pictures were taken with a fluorescence microscope (Leica) at the same exposure and intensity settings. All histological scorings and quantifications were performed in a blinded fashion.
Histopathological evaluation
For assessment of intestinal pathology, an expert blinded to the genotypes scored H&E-stained intestinal sections based on three parameters of tissue damage and four parameters of inflammation, and multiplied these scores with a factor accounting for the extent of the tissue being affected, as described previously.32 Briefly, for assessing tissue damage, three distinct scores were attributed to the degrees of crypt hyperplasia, epithelial injury/erosion and oedema (each from 0 to 3, with 0 absent, 1 slight, 2 moderate and 3 severe). For assessing inflammation, three distinct scores were attributed to the numbers of mononuclear cells, polymorphonuclear cells and lymphocytic cells (also from 0 to 3, with 0 absent, 1 slight, 2 moderate and 3 severe). A fourth inflammation parameter accounted for the location of the inflammatory infiltrate: 0 absent, 1 mucosal, 2 submucosal, 3 transmural extending into muscularis and serosa, and 4 diffuse. The sum of these tissue damage or inflammation scores was then multiplied by a factor according to the fraction of the tissue being affected: 1, <10%; 2, 10%–25%; 3, 25%–50%; and 4, >50%. This semiquantitative composite histological pathology scoring system results in tissue damage and inflammation scores ranging from 0 to 36 and 0 to 48, respectively.
Southern blotting
Ten micrograms of genomic DNA was digested with the indicated restriction enzymes, separated on agarose gels and transferred to nitrocellulose. Hybridisation was performed with the indicated 32P-labelled probes.
IEC isolation and western blotting
IECs were isolated by sequential incubation of intestinal tissue in 1 mM dithiothreitol (DTT) and 1.5 mM EDTA solutions, and protein extracts were prepared from IECs as described previously.33 Protein extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (Millipore). Membranes were probed with primary antibodies anti-TRAF6 (MBL International Corporation), anti-α-tubulin (Sigma), anti-MyD88 (ProSci Incorporated), anti-green fluorescent protein (GFP) (Biozol). Membranes were incubated with secondary HRP-coupled antibodies (GE Healthcare, Jackson ImmuneResearch) and developed with chemiluminescent detection substrate (Thermo Scientific).
Quantitative real-time PCR
Total RNA from colon was isolated using Trizol (Invitrogen), cDNA was prepared with Superscript (Invitrogen) and analysed by real-time PCR with TaqMan gene expression assays (Applied Biosystems). Values were normalised to the level of the reference gene Tbp.
Statistics
All data shown represent the means ± SD unless otherwise indicated. Statistical analyses were performed with unpaired two-sided Student's t tests with unequal variance using Microsoft Excel software, and statistically significant differences are indicated as follows: *p<0.05; **p<0.01; ***p<0.001.
Acknowledgments
We thank C Uthoff-Hachenberg, J von Rhein, E Mahlberg, D Beier, J Buchholz, B Hülser and B Kühnel for excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
KV and AP authors share first authorship.
MP and AW authors share senior authorship.
Correction notice This article has been corrected since it published Online First. The affiliations for Manolis Pasparakis have been updated.
Contributors The study was designed by AW, KV and MP. Experiments were performed by AW, KV and P-SW. AP generated MyD88 and TRIF-floxed mice, GvL generated TRAF6-floxed mice. Results were analysed by AW, KV and MP. MP coordinated the project. AW wrote the paper with input from all other authors.
Funding MP acknowledges funding from the ERC (2012-ADG_20120314), the DFG (SFB670, SFB829, SPP1656), the European Commission (Grants 223404 (Masterswitch) and 223151 (InflaCare)), the Deutsche Krebshilfe (Grant 110302), the Else Kröner-Fresenius-Stiftung and the Helmholtz Alliance (PCCC). AW was supported in part by a fellowship from the Humboldt Foundation, P-SW was supported by a fellowship from the International Graduate School in Genetics and Functional Genomics at the University of Cologne. AW holds a postdoctoral fellowship from the Research Foundation-Flanders (FWO) and is supported by the FWO Odysseus return grant number G.0C49.13N.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.