Article Text

Abstract
Objective Congenital sodium diarrhoea (CSD) refers to a form of secretory diarrhoea with intrauterine onset and high faecal losses of sodium without congenital malformations. The molecular basis for CSD remains unknown. We clinically characterised a cohort of infants with CSD and set out to identify disease-causing mutations by genome-wide genetic testing.
Design We performed whole-exome sequencing and chromosomal microarray analyses in 4 unrelated patients, followed by confirmatory Sanger sequencing of the likely disease-causing mutations in patients and in their family members, followed by functional studies.
Results We identified novel de novo missense mutations in GUCY2C, the gene encoding receptor guanylate cyclase C (GC-C) in 4 patients with CSD. One patient developed severe, early-onset IBD and chronic arthritis at 4 years of age. GC-C is an intestinal brush border membrane-bound guanylate cyclase, which functions as receptor for guanylin, uroguanylin and Escherichia coli heat-stable enterotoxin. Mutations in GUCY2C were present in different intracellular domains of GC-C, and were activating mutations that enhanced intracellular cyclic guanosine monophosphate accumulation in a ligand-independent and ligand-stimulated manner, following heterologous expression in HEK293T cells.
Conclusions Dominant gain-of-function GUCY2C mutations lead to elevated intracellular cyclic guanosine monophosphate levels and could explain the chronic diarrhoea as a result of decreased intestinal sodium and water absorption and increased chloride secretion. Thus, mutations in GUCY2C indicate a role for this receptor in the pathogenesis of sporadic CSD.
- CHRONIC DIARRHOEA
- GUANYLATE CYCLASE
- BACTERIAL ENTEROTOXINS
- PAEDIATRIC DIARRHOEA
- INTESTINAL ION TRANSPORT
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- CHRONIC DIARRHOEA
- GUANYLATE CYCLASE
- BACTERIAL ENTEROTOXINS
- PAEDIATRIC DIARRHOEA
- INTESTINAL ION TRANSPORT
Significance of this study
What is already known on this subject?
Congenital diarrhoeal diseases represent a group of rare inherited enteropathies with a disease-onset early in life. There is a broad range of different monogenic conditions, and a number of patients have not been classified so far.
A GUCY2C mutation, p.S840I, has been described in 32 affected members of a large Norwegian family with autosomal dominantly inherited diarrhoea of infantile onset, frequently associated with IBS, bowel obstruction and IBD. GUCY2C encodes guanylate cyclase C (GC-C), and ligand binding to GC-C results in production of cyclic guanosine monophosphate (cGMP). The mutation conferred a higher cGMP production upon ligand-binding to GC-C.
What are the new findings?
We identify the molecular basis of a clinical entity, congenital sodium diarrhoea (CSD) as four distinct de novo mutations of GUCY2C occurring in four patients. GUCY2C mutations identified in patients with CSD caused increases in basal and ligand-stimulated GC-C activities.
We report that patients with GUCY2C mutations presented with intrauterine onset of diarrhoea requiring ileostomata within the 1st month of age. One patient with a p.L775P mutation in GC-C developed IBD and arthritis at 4 years of age thereby implicating GUCY2C as an IBD gene.
How might it impact on clinical practice in the foreseeable future?
This study provides a rationale for including GUCY2C into a fast, targeted or panel-based routine testing of patients suspected with CSD or patients with diarrhoeal disease.
Studying patient-derived tissues or organoids with GUCY2C mutations might contribute to the understanding of cGMP-mediated stimulation of chloride secretion by cystic fibrosis transmembrane conductance regulator (CFTR) and inhibition of sodium absorption by the sodium-hydrogen exchanger 3 (NHE3).
Introduction
Congenital diarrhoeal disorders are rare and heterogeneous enteropathies, generally with severe clinical manifestations. These disorders can be caused by defects in absorption and transport of nutrients and electrolytes, in enterocyte differentiation and polarisation, in enteroendocrine cell differentiation, and by modulations of the intestinal immune response. Clinical and histological criteria and, increasingly, mutation detection in known disease genes are used to classify these disorders.1 Some types of congenital diarrhoeal disorders are amenable to dietary treatment, while others require chronic nutritional support or small bowel transplantation.
Congenital sodium diarrhoea (CSD) refers to a secretory diarrhoea presenting with maternal polyhydramnios, prominent abdominal distension due to dilated fluid-filled loops of the intestine, which may lead to intestinal obstruction at birth, and high faecal losses of sodium and chloride. CSD associated with choanal atresia, corneal erosions and tufting enteropathy (syndromic CSD) can be distinguished from the originally described classic form of CSD.2 Syndromic CSD is caused by SPINT2 loss-of-function mutations,3 whereas the molecular basis for classic CSD remains unknown.3 ,4
Known genetic entities of CCDs are most often autosomal recessively inherited; however, a single heterozygous mutation in GUCY2C, encoding guanylate cyclase C (GC-C) has been reported as the cause of a dominantly inherited form of secretory diarrhoea (Familial Diarrhoea Syndrome) in 32 individuals of a Norwegian family.5 The mutation in the Norwegian study was a hyperactivating mutation present in the catalytic domain of GC-C. This mutation resulted in increased sensitivity of GC-C to its endogenous ligands guanylin and uroguanylin and to the heat-stable enterotoxin (ST) of Escherichia coli, giving rise to increased levels of cyclic guanosine monophosphate (cGMP) in the epithelial cell, resulting in enhanced fluid and ion secretion, and diarrhoea. In contrast, inactivating mutations of GUCY2C in three Bedouin families resulted in meconium ileus6 ,7 as a consequence of diminished fluid and ion secretion that is normally maintained by the action of guanylin and uroguanylin on GC-C.
In the Norwegian kindred, the predominant phenotype of affected family members mimicked IBS, resulting in three to four loose stools daily, while 8 out of the 32 affected family members already presented in infancy with severe dehydration. Small-bowel obstruction and/or IBD were/was diagnosed in 20–25% of these patients. As opposed to this relatively mild type of diarrhoea, the four patients with CSD described here presented with a more severe phenotype uniformly beginning prenatally and in whom one patient developed early and severe IBD. All four patients harbour different heterozygous de novo mutations in GUCY2C. These mutations are novel and lie in the catalytic domain of the receptor and in regions of the receptor that allosterically regulate ligand-mediated activation of the guanylate cyclase domain.
Methods
Chromosomal microarray analysis, exome and Sanger sequencing
Genomic DNA was extracted from blood samples by standard methods. Genome-wide copy-number variant detection (chromosomal microarray analysis) following hybridisation of the patient's DNA sample to a HumanCytoSNP-12v2 BeadChip single-nucleotide polymorphism (SNP) array (Illumina, Little Chesterford, UK) interrogating 299 140 markers was performed according to the manufacturer's instructions. Raw SNP call data were processed with the Genotyping Analysis Module of GenomeStudio 1.6.3 (Illumina). Copy-number variants and segments of loss-of-heterozygosity were called and visualised using Nexus (BioDiscovery, Hawthorne, USA) software.
The exomes of the four patients were enriched from genomic DNA using the Illumina Truseq Exome Enrichment Kit and sequenced using the Illumina HiSeq 2000 according to the manufacturer’s protocol, producing 4.54–10.72 gigabytes of paired-end 100-bp sequence reads per sample by analysis with CASAVA V.1.8 (Illumina). Sequencing reads were aligned to the human genome (hg19) with Burrows-Wheeler transformation,8 sorted, converted to BAM format and indexed with SAMtools.9 PCR duplicates were removed with PICARD (http://picard.sourceforge.net) and SNPs and small indels were identified with the SAMtools mpileup software. All variants were submitted to SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation/) for annotation, categorisation into synonymous and non-synonymous SNPs or indels, and for filtering using the data from dbSNP138, the 1000 Genomes Project, the National Heart, Lung, and Blood Institute Exome Sequencing Project. The exome aggregation consortium server (exac.broadinstitute.org) was subsequently searched for known GUCY2C variants.
Single exons of GUCY2C were sequenced in the index cases and in available family members to test for the presence of mutations detected in index patients by whole-exome sequencing. PCR and sequencing primer sequences were based on the ENSEMBL reference entries for mRNA (NCBI reference sequence for mRNA NM_004963.3), Ensembl reference entry (ENST00000261170) and for genomic DNA (ENSG00000070019); sequences are available from the authors upon request. Codon numbering was based on the published online amino acid and mRNA sequences of these genes (http://www.ncbi.nlm.nih.gov/) using the first methionine as an initiation codon. GoTaq polymerase and buffer (Promega, Mannheim, Germany) were used to PCR-amplify 20–50 ng of genomic DNA in 35 cycles of 20 s at 95°C, 20 s at 60°C and 30 s at 72°C in addition to a 7 min final extension, applied to all sets of primers (table 2). The amplicons were cleaned using ExoSap-IT (USB, Vienna, Austria), and subsequently sequenced using an M13 universal primer on an ABI 3730s automated sequencer, with BigDye terminator mix (Applera, Vienna, Austria). Sequence chromatographs were analysed using the Sequence Pilot computer program (JSI medical systems, Kippenheim, Germany).
Site-directed mutagenesis and characterisation of mutant GC-C proteins
We generated mutant complementary DNA (cDNA) containing the GUCY2C point mutations identified in patients as described previously.5 Non-mutant and mutant GUCY2C cDNAs were cloned into the mammalian expression vector pcDNA3 (Invitrogen), and the respective proteins were transiently expressed in HEK293T cells.10 We measured ligand-stimulated GC-C activity in intact cells 72 h after transfection, after the addition of varying concentrations of ST or uroguanylin (for 15 min) or guanylin (for 60 min).
Results
Clinical characterisation of patients with CSD
Diagnosis of CSD was based on characteristic clinical findings as described in the introduction.2 Patients were examined by paediatric gastroenterologists and questionnaires regarding bowel symptoms and general health were completed. The clinical findings of four unrelated patients, in whom whole-exome sequencing (see Methods) revealed GUCY2C mutations (see Results) are summarised in table 1. There was no parental consanguinity in any family, and all patients were sporadic cases.
Molecular and clinical findings in four patients with congenital sodium diarrhoea with GUCY2C mutations
The patients with GUCY2 mutations were born at gestational ages between 32 weeks and 40 weeks and are now between 34 months and 86 months old. In three out of four patients polyhydramnios was noted. All birth weights were appropriate for gestational age. Postnatal growth parameters in three of the four subjects developed in the low-normal range while patient A1111 grew along the 90th centile. The clinical features and postnatal clinical courses were rather similar: in none of the patients meconium passing was documented. Secretory diarrhoea with high stoma fluid was reported in three of the four patients with high faecal sodium concentrations and alkaline faecal pH characteristic from the early postnatal period.
All patients presented with a large abdomen due to intestinal dilation after delivery (figure 1). No anatomical bowel obstruction was found in any of these patients but obstruction due to massive fluid-filled dilated intestinal loops led to installation of enterostomata in all four patients with GUCY2C mutations during their 1st week of life. Gut histology did not reveal any specific abnormality. Complications of the disease included sepsis, ileus, volvulus and severe colitis in patient B1933, as reported in detail previously.11 All four patients needed parenteral fluids for at least 2 years with two of them still being dependent on total parenteral nutrition. In three patients in whom sodium loss was calculated, it amounted up to 33 mmol/kg/day. Two patients could be weaned off total parenteral nutrition and, despite recurrent diarrhoeal episodes, could lead a normal life.
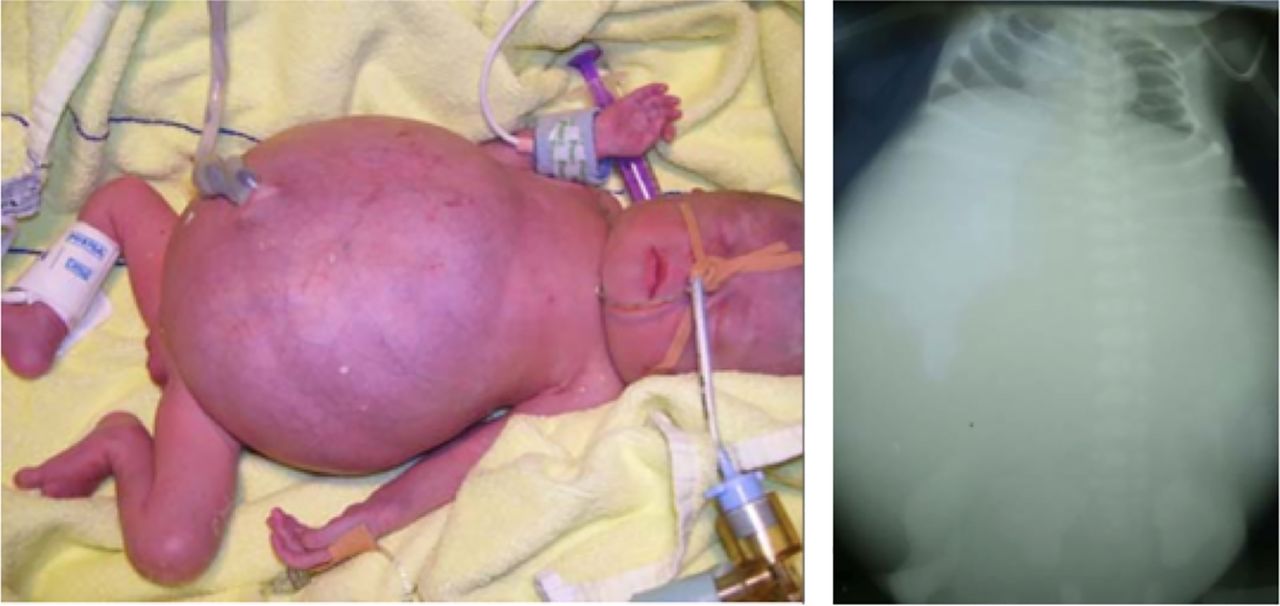
Characteristic clinical presentation of congenital sodium diarrhoea at birth. Note extensive distension of the abdomen due to fluid-filled intestinal loops due to an activating GUCY2C mutation in patient A1111.
Exome sequencing reveals missense mutations in GUCY2C in patients with CSD
To reveal the molecular basis of CSD, we performed whole-exome sequencing in genomic DNA samples from patients with CSD. The average sequencing coverage ranged between 45-fold and 90-fold with over 90% of the target covered by more than 8-fold in all samples. In the four patients, we identified 21451, 20667, 21025 and 21192 coding variants, of which 488, 597, 484 and 452 non-synonymous variants (missense, nonsense, cancel-start, read- through, splice-site and frameshift) were not present in dbSNP134. Among these rare and novel non-synonymous variants, no common gene in any two of the four patients with CSD was found under a recessive model with homozygosity or compound heterozygosity of mutations causing disease. However, we identified GUCY2C as the single gene that harboured heterozygous variants in all four patients, as confirmed by Sanger sequencing (table 1, figure 2). Neither of these different GUCY2C mutations, c.1519A>G (p.K507E), c.2324T>C (p.L775P), c.2376G>C (p.R792S), c.2548A>G (p.N850D) was present in parental samples, that is, all mutations had occurred de novo in patients, a first indication that they were causing disease. These GUCY2C mutations were not listed in public databases of gene variants listed in Methods. Chromosomal microarray analyses were normal in these patients.
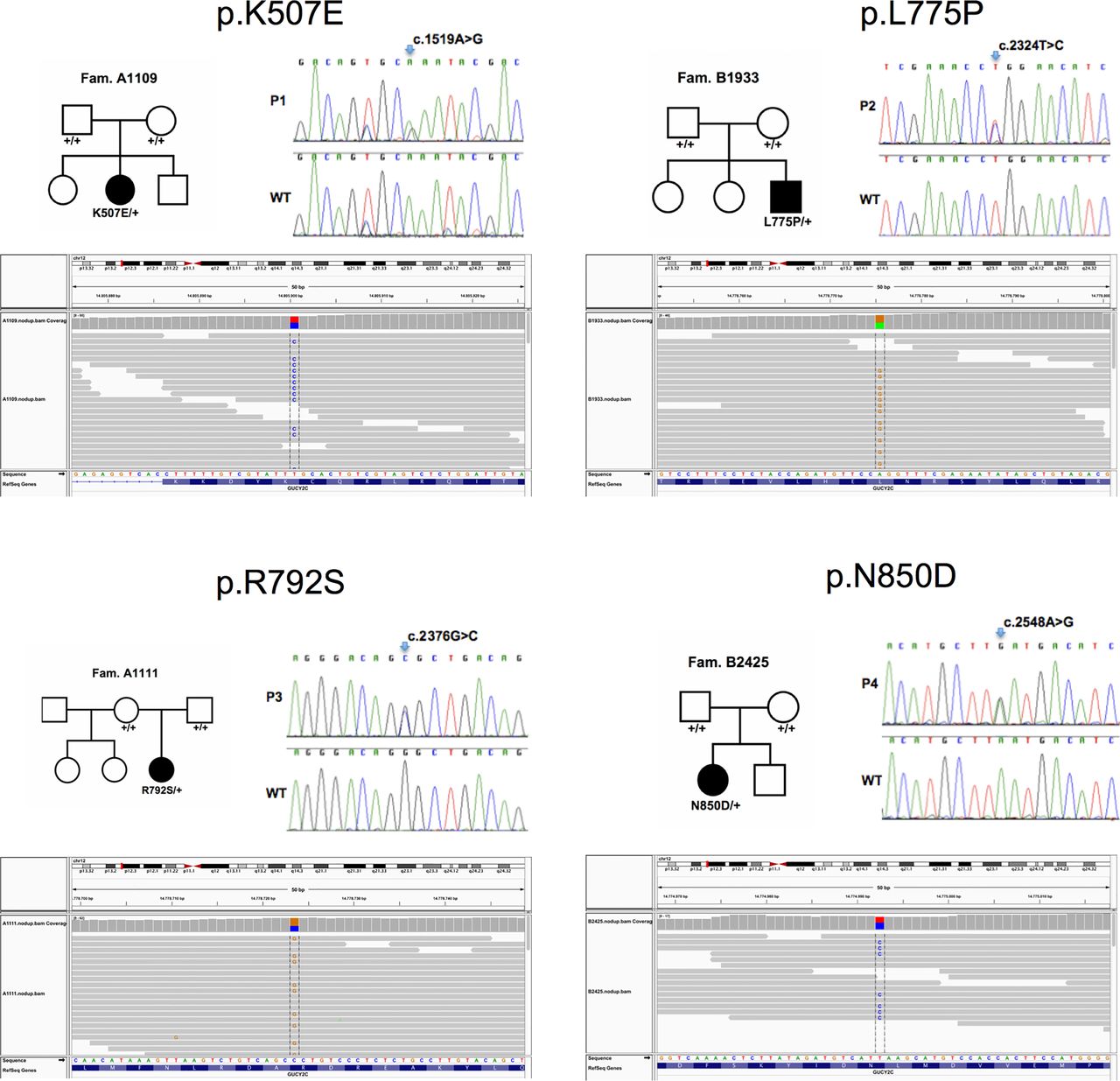
Identification of GUCY2C mutations in patients with congenital sodium diarrhoea. Upper panels: Pedigrees of patients’ families (left) and detection of heterozygous mutations in GUCY2C in four patients by Sanger sequencing (right). Lower panel: Corresponding aligned sequence reads spanning the GUCY2C mutations identified in the patients using the Integrated Genomics Viewer (http://www.broadinstitute.org/igv/).
Molecular characterisation of mutations in GC-C
The identified GUCY2C missense mutations were predicted to cause amino acid alterations in different cytoplasmic domains of the protein (figure 3A). All the residues for which mutations were observed in these patients are conserved in GC-C orthologs from fish to mammals (figure 3B).

Localisation and conservation of mutations in guanylate cyclase C (GC-C). (A) Schematic showing the domain organisation of GC-C, and mutations associated with disease thus far reported in GC-C. Shown on the top in magenta are earlier reported mutations leading to familial diarrhoea syndrome (S840I)5 or meconium ileus (1160A>G; and the insertion leading to truncation of the protein after Asn 757).6 Shown in red below are mutations reported in this study. Amino acids in black represent residues at predicted boundaries of the domains in GC-C. Residues 1–23 are predicted to be the signal sequence. The extracellular domain (residues 24–430) is the region where heat-stable enterotoxin, guanylin or uroguanylin bind. The receptor is a single transmembrane spanning domain protein, with the predicted transmembrane helix present from residues 431–453. Activity of GC-C is regulated allosterically by the kinase homology domain (residues 489–735). The linker region (residues 736–810) is important for suppressing basal activity of GC-C. The guanylate cyclase (catalytic) domain (residues 811–1010) is followed by a C-terminal domain (residues 1011–1073) that is important for apical localisation of GC-C. Note the presence of disease-associated mutations in almost all domains of GC-C. (B) Sequence conservation of the amino acids for which mutations in GC-C were identified in this study. Highlighted in blue is K507, pink is L775, green is R792 and yellow is N850. All residues are conserved from fish to mammals.
We measured the properties of mutant GC-C receptors by determining basal cGMP accumulation and ligand-stimulated activation of GC-C in transfected HEK293T cells. Equivalent expression of wild type and mutant receptors was confirmed by western blotting (figure 4A), with the two immunoreactive bands representing differentially glycosylated forms of the receptor.12 As seen in figure 4B, basal levels of cGMP were significantly elevated in cells expressing the p.L775P mutation, and no further stimulation in the presence of ST was observed (figure 4C), in agreement with our earlier results.10 Elevated basal cGMP levels were seen in all the other three mutations as well (figure 4A) with the p.R792S mutation showing almost 100-fold higher levels of basal intracellular cGMP as compared with wild type. The molecular phenotypes of these four novel mutations in GC-C are in contrast to the mutation (p.S840I) seen in the published Norwegian family, where no increase in basal activity of the receptor was observed.5

Functional properties of mutant guanylate cyclase C (GC-C). (A) Western blot of individual lysates prepared from cells transiently transfected with plasmids allowing expression of either wild type or mutant GC-C proteins. Blot shows almost equivalent expression of non-mutant and mutant receptors, and two differentially glycosylated forms of GC-C are seen. Data shown are representative of three independent transfections. (B) Intracellular cyclic guanosine monophosphate (cGMP) in cells transiently expressing either wild type or mutant GC-C proteins. Cells were lysed in 0.1N HCl and cGMP measured by radioimmunoassay. Note the elevated levels of cGMP, even in the absence of ligand, in cells expressing mutant receptors. Values shown are mean±SD of three independent determinations in a single experiment, with experiments repeated thrice. (C) Either heat-stable enterotoxin (10−7 M), guanylin (10−5 M) or uroguanylin (10−6 M) was applied to cells harbouring the indicated mutant GC-C receptors, and ligand-stimulated levels of cGMP in cells were measured. Transient overexpression of mutant GC-Cs showed enhanced ligand-mediated cGMP production of the R792S, K507E and N850D mutant receptors. Values shown are mean±SD of three independent determinations in a single experiment, with experiments repeated thrice.
Compared with wild type receptor, ligand-mediated activation by ST, uroguanylin and guanylin was significantly elevated in cells expressing the mutant receptors (figure 4C), except in cells expressing the p.L775P mutation, as seen earlier,10 where the abnormally high basal activity was not further increased by ligand binding. The maximum activation of GC-C was seen in p.K507E and p.R792S receptors.
Enhanced sensitivity of mutant receptors for ligands
We next asked if the increased responsiveness of p.K507E, p.R792S and p.N850D mutant receptors to ST and the endogenous ligands was a consequence of increased affinity of ligand binding to the receptors. We prepared membrane fractions from cells expressing wild type and mutant receptors, and monitored binding of ST and uroguanylin. As shown in table 2, the affinity of ST to all mutant receptors was similar to that of the wild type receptor. Moreover, the Kd for uroguanylin binding, as monitored by displacement assays, was also similar (table 2). Thus the enhanced activity of the mutant receptors was not a consequence of increased ligand-binding activity.
Binding affinity of ligands to wild type and mutant GC-C
In the case of the p.L775P mutant receptor, the non-responsiveness to further stimulation by cGMP implies that the disease phenotype is caused by the constitutively elevated levels of cGMP in the enterocyte, independent of the levels of guanylin or uroguanylin in the gut.
We next measured the EC50 for the ST peptide and uroguanylin for the three mutant receptors that were stimulated by the ligand (figure 5). All mutant receptors showed a significantly enhanced response to the ST peptide, and in the case of p.R792S, there was almost a 500-fold increase in efficacy for ST, as monitored by the dose required for half maximal cGMP production (figure 5A). It is intriguing that this enhanced response to ST peptide occurred in spite of similar affinities of ST binding to the receptor. Uroguanylin was also more potent in terms of enhancing cGMP production by mutant receptors, with the p.R792S mutant showing almost a 1000-fold increase in efficacy (figure 5B). Thus, normal concentrations of circulating uroguanylin (and presumably guanylin) would elicit supraphysiological accumulation of intracellular cGMP, leading to diarrhoea.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Enhanced sensitivity of mutant guanylate cyclase C (GC-C) receptors to heat-stable enterotoxin (ST) and uroguanylin. Varying amounts of ST peptide (A) or uroguanylin (B) were applied to cells expressing either wild type or mutant forms of GC-C, and cyclic guanosine monophosphate (cGMP) produced was measured by radioimmunoassay. Values shown are the mean±SD of three independent determinations across three experiments. EC50 values were calculated across all experiments and are shown in the table (inset) as mean±SD.
Membranes prepared from HEK293 cells expressing wild type or mutant GC-C were incubated with increasing concentrations of 125I-labelled STY72F peptide. Bound peptide was measured by filtering and data analysed to determine the Kd. For uroguanylin, a fixed concentration of 125I-labelled STY72F peptide (100 000 dpm) was incubated along with varying concentrations of uroguanylin and bound radioactivity measured. Data were analysed for binding to a single site. Values shown are the mean±SD for assays performed in duplicate with membranes prepared from three independent transfections.
Discussion
The classical form of CSD was first described in 1985,13 ,14 and less than 10 cases have been described in the literature.2 We identify activating de novo mutations in GUCY2C as the molecular basis of CSD in four patients. In these patients, secretory diarrhoea begins prenatally, as indicated by polyhydramnios and massive abdominal protrusion due to fluid-distended intestinal loops at birth. After birth, all cases of CSD require total parenteral nutrition for treatment of dehydration for at least several months. Sodium supplementation has been provided to all patients and is needed in the treatment of severe dehydration and to maintain normal body growth by preventing total body sodium depletion. A genotype-phenotype correlation might be inferred from our in vitro experiments where the highest cGMP levels are seen in two severely affected patients that still require parenteral nutrition, and one of whom has developed severe IBD at 4 years of age. The enhancement in activity of GC-C was far higher than that seen with the Norwegian p.S840I mutation, likely explaining the more severe phenotype of our patients. Thus, three of the four mutant receptors were hyperactivated by exogenous (ST) and endogenous ligands.
The consequences of increased cellular cGMP levels in downstream targets relevant for intestinal ion transport, sodium-hydrogen exchanger 3 (NHE3) and cystic fibrosis transmembrane conductance regulator (CFTR), are known. While CFTR is stimulated by cGMP with resultant increased chloride and water secretion into the intestine, NHE3 is inhibited by cGMP via protein kinase G-II phosphorylation leading to reduced sodium absorption. Since regulation of NHE3 and CFTR may be closely orchestrated—cellular trafficking and surface expression show opposite regulation—both effects might contribute to secretory diarrhoea.15 ,16 Neither the Norwegian study nor our study has actually investigated intestinal electrolyte absorption/secretion. Thus, firm conclusions about which one of these mechanisms is most important in this disease cannot be drawn, although probably both are contributing to the increased loss of sodium and chloride in the faeces in our patients with activating GC-C mutations. Studying intestinal biopsies and organoids from patients with different mutations in GUCY2C might elucidate the interplay of cGMP-mediated chloride secretion and sodium absorption via CFTR and NHE3, respectively.
Evidence for dominant GUCY2C mutations predisposing to IBD was inferred from the Norwegian family with familial diarrhoea syndrome.5 In addition, the observation of increased expression of GUCA2B, encoding uroguanylin, in colonic mucosa from patients with diarrhoea-predominant IBS,17 may aggravate the situation in patients with mutations in GUCY2C. The roles of GC-C in gut inflammation are supported by animal studies. GC-C−/− mice are more susceptible to dextran sodium sulfate-induced colitis relative to wild type controls.18 However, the observation that GC-C−/− mice appear more prone to lipopolysaccharide-mediated proinflammatory gene expression as compared with GC-C+/+ mice19 indicates that the exact relationship between GC-C expression and IBD is still unclear.20 Nhe3−/− mice exhibit diarrhoea,21 and develop spontaneous distal chronic colitis and are highly susceptible to dextran sulfate (DSS)-induced mucosal injury.22 Alterations in sodium and chloride transport were associated with inflammation and may represent a mechanism leading to changes in the intestinal microbial composition. The chloride/bicarbonate exchanger down regulated in adenoma (DRA), and NHE3 were observed to be downregulated in mucosal biopsies of patients with IBD,23–26 and in two different animal models of colitis.27 A significant decrease in the phylogenetic diversity of the luminal and mucosal microbiota of Nhe3−/− mice compared with the littermates was observed.28 NHE3 was thus considered to play a critical role in the composition of the gut microbiota and its deficiency may thus contribute to dysbiosis observed in patients with IBD.28 Very recently, it has been demonstrated in vivo, that acute Clostridium difficile infection resulted in decreased mucosal NHE3 expression and elevated stool sodium, which created an altered microbiota composition compared with healthy subjects and that was favoured by C. difficile.29 GUCY2C mutations represent monogenetic variants that provide a high susceptibility to develop early onset and late onset IBD, and provide support for the role of the micromilieu at the brush border for disease pathogenesis.
In conclusion, our study demonstrates that dominant de novo mutations in GUCY2C cause CSD, thereby stimulating further search for mutations in this gene in unexplained causes of diarrhoeal diseases, as well as future research on drugs and peptides to regulate GC-C activity and restore normal sodium homoeostasis.
References
Footnotes
TM, IR and PH-E contributed equally. ARJ and SSV contributed equally.
Correction notice This article has been corrected since it published Online First. The Open Access licence has now been added.
Contributors TM, B-SP, AF, IF, HW, HZ and ARJ performed genetic analyses. IR and SSV performed functional studies. PH-E, EM, CH, AM, LM, BGPK, AB, JV and SR contributed to the clinical and histopathological characterisation of the patients. TM, ARJ and SSV designed the study and contributed to the coordination of studies and wrote the manuscript with input from coauthors. All authors discussed the results and commented on the manuscript.
Funding This work was supported by grants from Jubiläumsfonds der Österreichischen National bank (grant no. 14496), the Tiroler Wissenschaftsfonds (grant no. UNI-0404/1286) and the Department of Biotechnology and Science and Technology, Government of India. IR was supported by a fellowship from the Indian Institute of Science and SSV is a JC Bose National Fellow supported by the Department of Science and Technology, Government of India.
Competing interests None declared.
Patient consent Parental consent obtained.
Ethics approval Ethics committee of the Medical University of Innsbruck.
Provenance and peer review Not commissioned; externally peer reviewed.