Article Text

Abstract
Background Pancreatic cancer is characterised by the accumulation of a fibro-inflammatory stroma. Within this stromal reaction, myeloid cells are a predominant population. Distinct myeloid subsets have been correlated with tumour promotion and unmasking of anti-tumour immunity.
Objective The goal of this study was to determine the effect of myeloid cell depletion on the onset and progression of pancreatic cancer and to understand the relationship between myeloid cells and T cell-mediated immunity within the pancreatic cancer microenvironment.
Methods Primary mouse pancreatic cancer cells were transplanted into CD11b-diphtheria toxin receptor (DTR) mice. Alternatively, the iKras* mouse model of pancreatic cancer was crossed into CD11b-DTR mice. CD11b+ cells (mostly myeloid cell population) were depleted by diphtheria toxin treatment during tumour initiation or in established tumours.
Results Depletion of myeloid cells prevented KrasG12D-driven pancreatic cancer initiation. In pre-established tumours, myeloid cell depletion arrested tumour growth and in some cases, induced tumour regressions that were dependent on CD8+ T cells. We found that myeloid cells inhibited CD8+ T-cell anti-tumour activity by inducing the expression of programmed cell death-ligand 1 (PD-L1) in tumour cells in an epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MAPK)-dependent manner.
Conclusion Our results show that myeloid cells support immune evasion in pancreatic cancer through EGFR/MAPK-dependent regulation of PD-L1 expression on tumour cells. Derailing this crosstalk between myeloid cells and tumour cells is sufficient to restore anti-tumour immunity mediated by CD8+ T cells, a finding with implications for the design of immune therapies for pancreatic cancer.
- PANCREATIC CANCER
- MACROPHAGES
- GENE REGULATION
- SIGNAL TRANSDUCTION
- IMMUNE RESPONSE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Myeloid cells, including macrophages and immature myeloid cells/myeloid-derived suppressor cells (MDSCs), accumulate during the progression of pancreatic cancer.
Macrophages promote pancreatic cancer initiation, growth and metastasis.
Depletion of MDSC or macrophage subsets results in increased infiltration and activation of CD8+ T cells.
The programmed cell death 1 (PD-1)/PD-ligand 1 (PD-L1) immune checkpoint inhibits CD8+ T-cell anti-tumour responses in pancreatic cancer.
What are the new findings?
Myeloid cells are required for sustained mitogen-activated protein kinases (MAPK) signalling in pancreatic epithelial cells during the onset of carcinogenesis, notwithstanding the expression of oncogenic Kras.
Myeloid cells regulate expression of PD-L1 in tumour cells which then suppress anti-tumour immune responses mediated by CD8+ cells.
Expression of PD-L1 in pancreatic cancer cells is regulated by the MAPK signalling cascade and can be blocked by MAPK kinase (MEK) inhibitors.
Treatment with MEK inhibitors lowers the intratumoural expression of PD-L1 and renders the tumour susceptible to PD-1 blockade.
How might it impact on clinical practice in the foreseeable future?
Our data show that multiple pathways can converge to promote immune evasion in pancreatic cancer. These findings support a role for combined therapeutic modalities that disrupt distinct immunosuppressive pathways orchestrated by both malignant and non-malignant cells. To this end, our data show that inhibition of the MAPK signalling cascade with MEK inhibitors can reduce the expression of PD-L1 by tumour cells and allow for an anti-tumour immune response to develop. Together, our findings support the combination of MEK inhibition and immune therapy as a novel therapeutic strategy for pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma (PDA) and its precursor lesion, pancreatic intraepithelial neoplasia (PanIN), are characterised by the accumulation of a desmoplastic stroma rich in inflammatory cell infiltrates.1 While immune cells are abundant within the stroma, they mostly belong to immunosuppressive subsets, such as regulatory T cells (Tregs), T-helper (Th)17 cells, tumour-associated macrophages (TAMs) and multiple subsets of immature myeloid cells/myeloid-derived suppressor cells (MDSCs).2–4 Conversely, CD8+ T cells are infrequent and when present they express high levels of immune checkpoint receptors such as programmed cell death-1 (PD-1), possibly indicating an exhausted status.5 We have previously shown that depletion of CD4+ T cells in a genetically engineered mouse model of pancreatic cancer leads to increased CD8+ T-cell infiltration and activation, thus preventing oncogenic Kras-driven PanIN formation.3 Depletion of CD4+ T cells leads to decreased infiltration of several myeloid cell types, including macrophages and immature myeloid cells (MDSCs), indicating cross-regulation across different immune subsets. Macrophages and MDSCs are prevalent within the pancreatic cancer microenvironment2 and tumour-promoting capabilities have been ascribed to both cell types.6–8 CD11b-diphtheria toxin receptor (DTR) mice allow inducible depletion of CD11b+ cells, including macrophages and MDSCs, on administration of diphtheria toxin (DT).9 Here, by crossing CD11b-DTR mice with a mouse model of pancreatic cancer, the iKras* mouse,10 we set out to determine the contribution of myeloid cells to the initiation, progression and maintenance of pancreatic cancer.
Our results indicate that myeloid cells are required at multiple stages of pancreatic carcinogenesis. Depletion of myeloid cells early during pancreatic cancer development prevents PanIN formation in the setting of acute pancreatitis. At later stages, myeloid cells are critical for tumour growth. In accordance with the previous body of literature, we show that myeloid cells are instrumental in maintaining an immune suppressive network. We identify myeloid cells as the drivers of the immune checkpoint ligand PD-ligand 1 (PD-L1) expression in tumour cells through activation of epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MAPK) signalling. Accordingly, MAPK kinase (MEK) inhibition increases tumour vulnerability to PD-1/PD-L1 blockade, thus potentially exposing new therapeutic strategies.
Materials and methods
Mouse strains
By crossing CD11b-DTR mice (B6.FVB-Tg(ITGAM-DTR/EGFP)34Lan/J, Jackson Laboratory) with iKras* (p48Cre;TetO-KrasG12D;R26rtTa-IRES-EGFP)10 or iKras*p53* (p48Cre;TetO-KrasG12D;R26rtTa-IRES-EGFP;p53R172H/+) mice11 we generated iKras*;CD11b-DTR and iKras*;p53*;CD11b-DTR mice. To induce expression of oncogenic Kras, doxycycline (doxy) (Sigma-Aldrich) was administered in the drinking water, at a concentration of 0.2 g/L in a solution of 5% sucrose.12 Animals lacking the full complement of transgenes were used as control and treated in parallel with the experimental animals. CD11b-DTR mice were also backcrossed with friend virus B (FVB)/NJ or C57BL/6J mice (Jackson Laboratory) for syngeneic subcutaneous experiments. All mice were housed in specific pathogen-free facilities of the University of Michigan Comprehensive Cancer Center. All animal studies were approved by the University Committee on Use and Care of Animals.
In vivo experiments
To induce pancreatitis, mice aged 4–6 weeks were treated with 8-hourly intraperitoneal (i.p.) injections of caerulein (Sigma-Aldrich) at a dosage of 75 µg/kg body weight over 2 consecutive days. DT (Enzo Life Science) was administered i.p. every 4 days at a concentration of 25 ng/g. To establish the subcutaneous tumour model, 2×106 of primary mouse pancreatic cancer cell lines iKras*1, iKras*2 and iKras*3 derived from iKras*p53* tumours,11 ,13 4×105 of 65 671 cells (FVB/NJ strain)14 or 7940B cells (C57BL/6J strain)15 derived from KPC tumour (P48-Cre; loxP-stop-loxP (LSL)-KrasG12D; p53flox/+) were injected into CD11d-DTR mice of compatible genetic background. Tumour diameters were measured with digital callipers and the tumour volume was calculated by the formula: length×width2/2. For CD8+ T-cell depletion, anti-CD8 monoclonal antibody (BioXcell #BE0061; clone 2.43; 200 µg/mouse) was injected i.p. twice per week. Purified anti-mPD-1 antibody (BioXcell #BE0033-2; clone J43) was used for in vivo PD-1 blockade at a dose of 200 μg/i.p. injection, repeated every 3 days if needed. MEK inhibitor (MEKi) GSK1120212 (Selleckchem) was administered daily (1 mg/kg, i.p.) in a 10% (2-Hydroxypropyl)-β-cyclodextrin solution.
Cell culture
Primary human pancreatic cancer cell lines 1319, UM2, UM5, UM18 and UM19 were derived from patients with confirmed diagnosis of PDA at University of Michigan.14 ,16 For co-culture experiments, 1–2×105 cells were plated in 6-well trans-well dishes (0.4 µm pore size; Corning). Bone marrow (BM) cells were collected from mouse tibias and femurs. Ammonium-chloride-potassium (ACK) Lysing Buffer (Lonza) was used to eliminate erythrocytes. Anti-mIL6 and recombinant human interleukin (IL)-6 were purchased from R&D Systems. MEKi inhibitor GSK1120212 and EGFR inhibitor erlotinib were purchased from Selleckchem.
Statistical analysis
Graphpad Prism 6 software was used for all statistical analysis. All data were presented as mean±SE (SEM). Intergroup comparisons were performed using unpaired two-tailed Student’s t-test or two-way repeated measure analysis of variance and p<0.05 was considered statistically significant.
Detailed protocol and standard procedures are included in the Supplementary Methods.
Supplementary methods
Results
CD11b+ myeloid cells are required for KrasG12D-driven PanIN formation
To investigate the role of CD11b+ myeloid cells in pancreatic cancer, we generated p48Cre;TetO-KrasG12D;Rosa26rtTa-IRES-EGFP;CD11b-DTR (iKras*;CD11b-DTR) mice (figure 1A). Oncogenic Kras expression was induced in adult mice by doxycycline administration. To promote PanIN formation, we induced acute pancreatitis as previously described.10 ,17 ,18 We also depleted myeloid cells using administration of DT 1 day prior to pancreatitis induction (figure 1B; 4–8 mice/cohort; pancreata were harvested 1 day or 1 week later). Depletion of CD11b+ cells was confirmed by flow cytometry (see online supplementary figure S1A). Using this experimental approach, we detected acinar-ductal metaplasia (ADM) at day 1 after acute pancreatitis induction, as determined histologically and by the presence of cells co-expressing the acinar marker Amylase and the ductal marker cytokeratin 19 (CK19) (see figure 1C and online supplementary figure S1B, top row). One week later, we found clear differences in the histology of pancreas from iKras* mice compared with the other groups. While control and CD11b-DTR mice had undergone tissue repair, iKras* mice demonstrated extensive fibrosis and ADM (see figure 1C and online supplementary figure S1B). Occasional periodic acid-Schiff (PAS) staining, indicating low-grade PanINs was also observed (see online supplementary figure S1C). In contrast, iKras*;CD11b-DTR mice had also undergone tissue repair notwithstanding the expression of oncogenic Kras (see figure 1C and online supplementary figure S1B). Together, these findings suggest a critical role for CD11b+ myeloid cells in suppressing tissue repair after acute pancreatic inflammation in the setting of mutant Kras activation.

CD11b+ myeloid cells are required for oncogenic Kras-driven pancreatic intraepithelial neoplasia (PanIN) formation. (A) Genetic makeup of the iKras*;CD11b- diphtheria toxin receptor (DTR) (p48Cre;TetO-KrasG12D; Rosa26rtTa-IRES-EGFP;CD11b-DTR) and iKras*;p53*;CD11b-DTR (p48Cre;TetO-KrasG12D; Rosa26rtTa-IRES-EGFP;p53R172H/+;CD11b-DTR) mouse models is shown. (B) Experimental design is shown. Doxycycline water was given to iKras* and iKras*;CD11b-DTR mice to induce Kras* expression for 3 days before 2 consecutive days of intraperitoneal caerulein injections to induce pancreatitis. Mice also received diphtheria toxin (DT) injection to deplete CD11b+ myeloid cells 1 day prior to pancreatitis induction; n=4–8 mice per time point. (C) H&E and (D) phosph-ERK1/2 staining of wild-type control, CD11b-DTR, iKras* and iKras*;CD11b-DTR pancreas 1 day and 1 week post-pancreatitis induction. Scale bar 50 μm.
supplementary figures
The MAPK signalling pathway is a downstream effector of Kras (for review, see refs. 19 and 20). Elevated MAPK activity, detected as phosphorylated extracellular signal-regulated kinase1/2 (p-ERK1/2) staining, occurs during pancreatitis and pancreatic cancer.21–23 Moreover, MAPK activation is necessary for PanIN formation.24 We detected elevated p-ERK1/2 in all tissues at day 1 after induction of acute pancreatitis. One week later, p-ERK1/2 levels were still elevated in iKras* pancreas, but had become low to undetectable in control and CD11b-DTR pancreas. Similarly, in iKras*;CD11b-DTR pancreas, p-ERK1/2 levels were low (see figure 1D and online supplementary figure S1D). Thus, sustained MAPK activation and formation of pre-neoplastic lesions requires the presence of myeloid cells, notwithstanding expression of oncogenic Kras.
CD11b+ myeloid cells are required for pancreatic cancer growth
To further investigate the role of myeloid cells in pancreatic cancer, we elected to use a panel of primary mouse pancreatic cancer cell lines. Cell lines were derived from KPC25 tumours bred to pure FVB/NJ (line 65 671) or C57BL/6J background (line 7940B), to allow syngeneic transplantation. In addition, iKras*p53*-derived tumour lines (iKras*1–3) from our highly inbred colony were used for littermate transplantation (figure 2A).11 ,13 To maintain expression of mutant Kras in iKras*p53* cells transplanted in vivo, doxycycline was continuously administered. In a first set of experiments, strain-matched CD11b-DTR or wild-type (WT) mice received DT 1 day prior to subcutaneous injection of PDA cells (figure 2A). Tumour volumes were then measured daily in each group (n=4–6). Compared with WT mice treated with DT or control-treated CD11b-DTR mice, CD11b-DTR treated with DT showed significant inhibition of tumour growth (figure 2B) across different strains. Histological analysis highlighted the presence of well-differentiated tumours and large necrotic cores in the tumours from DT-treated CD11b-DTR mice (compared with small or absent areas of necrosis in the control groups) (figure 2C). Thus, CD11b+ myeloid cells are important for the implantation and outgrowth of transplanted PDA tumours.

Myeloid cells are required for pancreatic cancer growth and maintenance. (A) Experimental design showing subcutaneous tumour growth. Diphtheria toxin (DT) was given 1 day prior to subcutaneous tumour plantation. (B) Tumour growth curve of subcutaneous tumours is shown. Data represent mean±SEM, n=4–6. Statistical difference was analysed by two-way analysis of variance (ANOVA). (C) H&E staining of subcutaneous tumours from control and DT-treated CD11b-DT receptor (DTR) mice is shown. Scale bar 50 µm. (E) Experimental design showing subcutaneous tumour progression and maintenance. DT was given to deplete myeloid cells after the tumours were measurable. (F) Changes in tumour size (%) post-DT treatment are shown. Data represent mean±SEM, n=6–8. Statistical difference was analysed by two-way ANOVA between two groups. (H) H&E staining of subcutaneous 65 671 tumours is shown. Scale bar 50 µm. (G) Percentage of CD45+CD11b+ myeloid cells, CD45+CD11b+F4/80+ macrophages and CD45+CD11b+Gr1+ MDSCs in subcutaneous 65 671 tumours measured by flow cytometry. Data represent mean±SEM; each point indicates one tumour (n=5–6). The statistical difference was determined by two-tailed Student’s t-tests. MDSCs, myeloid-derived suppressor cells.
We then performed a second set of experiments where DT treatment was administered 6–8 days following the implantation of tumour cells, when tumour diameters were 5–8 mm (see figure 2D and online supplementary figure S2A). CD11b+ cell depletion resulted in growth arrest or tumour regression in a cell-line-dependent manner (figure 2E). While histological analysis revealed no change in tumour architecture in some cell lines (figure 2F), CD11b+ cells were depleted from tumours as seen by flow cytometry (figure 2G) and F4/80 immunostaining (see online supplementary figure S2B). Although the number of proliferative tumour cells positive for Ki67 was not altered by CD11b+ cell depletion (see online supplementary figure S2C), we did observe a significant increase in apoptotic cells, as measured by cleaved caspase 3 and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (see figure 3A–D and online supplementary figure S2D). This observation led us to investigate the mechanism underlying tumour cell death and consequently tumour regression that occurred with CD11b+ myeloid cell depletion.

Myeloid cell depletion induces tumour cell death and enhances tumour-infiltrating CD8+ T cells. (A) Experimental design is shown. (B) Immunohistochemistry for cleaved caspase 3 and (D) co-immunofluorescent staining for TUNEL (red) and cytokeratin 19 (CK19) (green) in control and diphtheria toxin (DT)-treated subcutaneous tumours are shown. Scale bar 50 µm. Quantification of cleaved caspase 3+ areas per slide (%) and TUNEL+ cell number per high power field (HPF, 400×) are shown in (C). Data represent mean±SEM, n=3. The statistical difference was determined by two-tailed Student's t-test. (E) Co-immunofluorescent staining for CD8 (red), GFP (green), CD3 (grey) and DAPI (blue) in control and DT-treated iKras*1 and iKras*2 subcutaneous tumours is shown. Scale bar 25 µm. Yellow arrows indicate CD3+CD8+ T cells. (F) Quantification of CD3+CD8+ T-cell number per HPF. Data represent mean±SEM, n=3–5. The statistical difference was determined by two-tailed Student’s t-test. (G) Quantitative real time-PCR showing interferon γ (Ifnγ), Ifnβ1 and perforin-1 expressions in control and DT-treated subcutaneous tumours. Data represent mean±SEM; each point indicates one sample (n=4–7). The statistical difference was determined by two-tailed Student’s t-test. DAPI, 4',6-diamidino-2-phenolindole.
Increase in intratumoural CD8+ T-cell infiltration and activation following CD11b+ cell depletion
The increase in tumour cell death seen with depletion of CD11b+ myeloid cells could be due to several distinct mechanisms, including a reduction in factors released by myeloid cells that support tumour cell survival or, alternatively, activation of an anti-tumour immune response. We found that depletion of myeloid cells in CD11b-DTR mice resulted in a decrease in expression of several macrophage markers including Arginase 1 (Arg1), Macrophage scavenger receptor 1 (Msr1) and Mannose receptor C type 1 (Mrc1) (online supplementary figure S3A), which is consistent with myeloid cell depletion. Each of these markers has also been associated with macrophages that can foster immunosuppression in the tumour microenvironment (for review, see refs. 26–28). Thus, we considered the possibility that myeloid cell depletion might reduce immunosuppression in PDA and thereby, restore productive immunosurveillance. On analysis, we found that CD8+ T-cell infiltration within tumours was increased in both iKras*1 and iKras*2 tumours (figure 3E,F) when myeloid cells were depleted, but decreased in iKras*3 tumours by flow cytometry (figure 4B). Since the decrease in T-cell infiltration in iKras*3 tumours was unexpected, we then evaluated the spatial localisation of CD8+ T cells by co-immunostaining for CD8+ and green fluorescent protein (GFP) which was used to mark PDA tumour cells. In control tumours we observed that CD8+ T cells were mainly confined to the tumour periphery. In contrast, in DT-treated CD11b-DTR mice, CD8+ T cells were found amidst the malignant cells (online supplementary figure S3B compares the localisation of CD8+ T cells, indicated by red arrows, at the boundary of the tumour, with few cells within the tumour mass on the top panel with the intermingling of GFP+ tumour cells and CD8+ cells on the bottom). Thus, while the overall number of CD8+ T cells was decreased in iKras*3 PDA tumours, this change mainly reflected a reduction at the periphery of the tumour and belied an actual increase in CD8+ T cells within the tumour mass. Consistent with the concept of increased CD8+ T-cell activation, we also observed an increase in the expression of T-cell activation markers including interferon γ (Ifnγ), Ifnβ1 and perforin-1 (Prf-1) following CD11b+ cell depletion (figure 3G). The increase in CD8+ T-cell infiltration and activation was accompanied by a decrease in the Treg population (online supplementary figure S3C). Thus, myeloid cell depletion has a complex effect on the immune microenvironment of pancreatic cancer, shifting the balance between immunosuppression and activation.

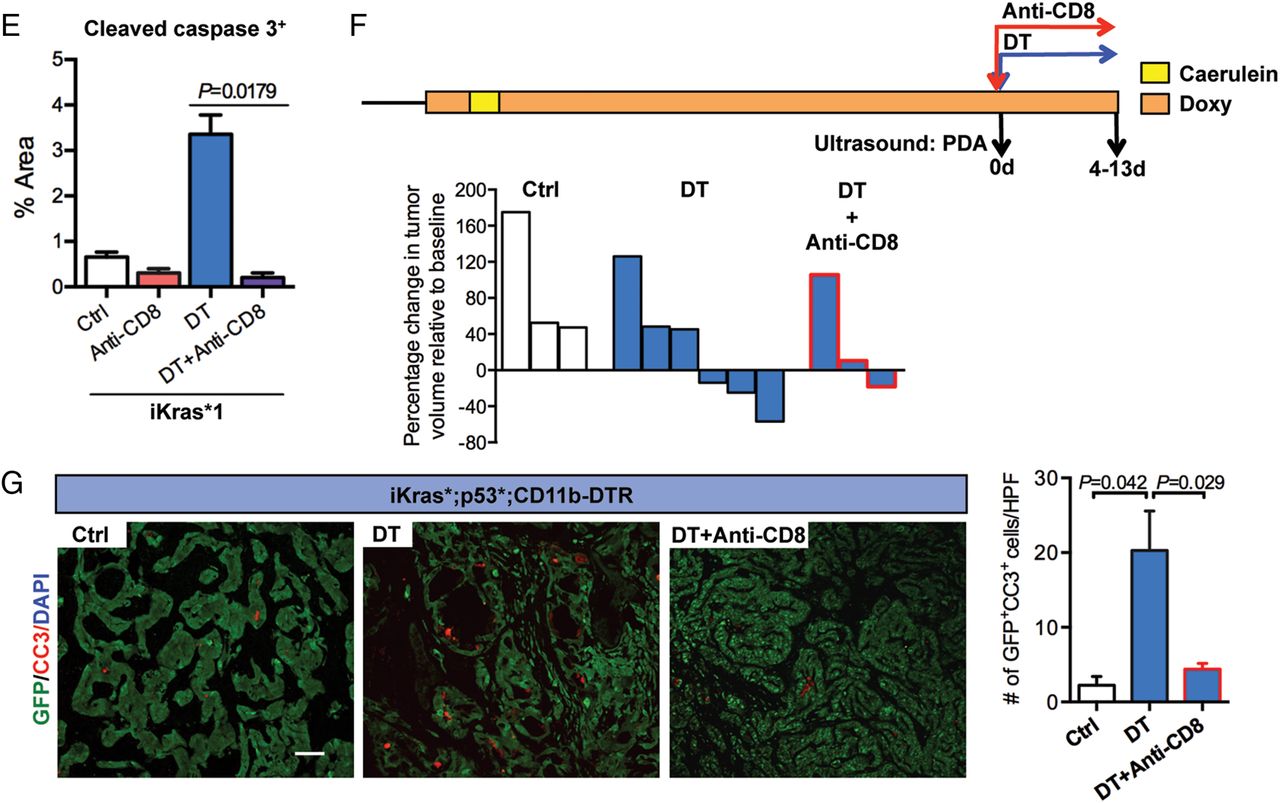
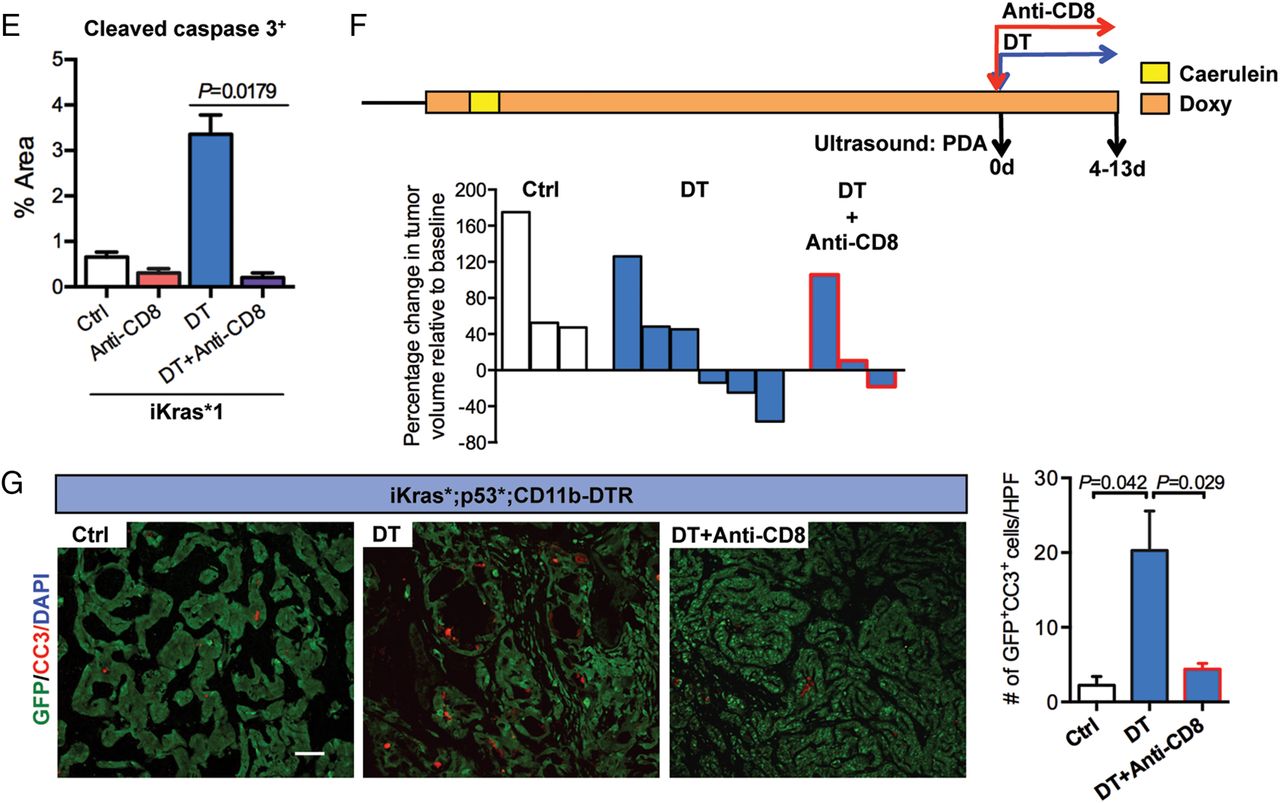
CD8+ T cells depletion rescues tumour progression in myeloid cell-depleted mice. (A) Experimental design is shown. (B) Percentage of CD3+CD8+ T cells in control, diphtheria toxin (DT) treated or both DT and anti-CD8-treated iKras*3 subcutaneous tumours measured by flow cytometry is shown. Data represent mean±SEM; each point indicates one tumour (n=4). The statistical difference was determined by two-tailed Student’s t-test. (C) Tumour size change (%) of iKras*1 and iKras*3 subcutaneous tumours extracted from the control, DT and/or anti-CD8 treated CD11b-DT receptor (DTR) mice is shown. Data represent mean±SEM, n=6. The statistical difference between groups was analysed by two-way analysis of variance. (D) H&E staining and immunohistochemistry for cytokeratin 19 (CK19) and cleaved caspase 3 in control, DT and/or anti-CD8-treated iKras*1 subcutaneous tumours are shown. Scale bar 50 µm. (E) Quantification of cleaved caspase 3+ areas per slide (%) is shown. Data represent mean±SEM, n=3. The statistical difference was determined by two-tailed Student’s t-test. (F) Experimental design: iKras*;p53*;CD11b-DTR mice were maintained on doxy water after pancreatitis induction and monitored for tumour formation. After pancreatic tumours identified by ultrasound, mice were given DT or in combination of anti-CD8 treatment. Tumour size was determined by ultrasound imaging. (G) Co-immunofluorescent staining for cleaved caspase 3 (CC3, red), GFP (green) and DAPI (blue) in tumours from control, DT with or without anti-CD8-treated mice is shown. Scale bar 50 µm. Graph depicts the quantification of apoptotic tumour cells per high power field (HPF, 200X). Data represent mean±SEM, three HPF for each mouse. Statistical difference was determined by two-tailed Student’s t-tests. DAPI, 4',6-diamidino-2-phenolindole.
Myeloid cells regulate CD8+ T-cell immunity against pancreatic cancer
We then tested whether myeloid cells promote tumour growth by inhibiting T-cell-mediated immune responses in PDA. For this purpose, animals with transplanted tumours were subdivided into cohorts and treated with anti-mCD8, DT or a combination of both (figure 4A). Depletion of CD8+ T cells in the tumours was verified by flow cytometry (figure 4B). We found that CD8+ T-cell depletion by itself had no effect on tumour growth (figure 4C). In contrast, DT treatment blocked tumour growth but this effect was dependent on CD8+ T cells as tumour growth and tumour cell viability were rescued when CD8+ T cells were depleted in mice treated with DT (figure 4C–E).
To determine whether the findings obtained in the transplantation model could be extended to spontaneous tumours, iKras;p53* or iKras*;p53*;CD11b-DTR mice were monitored for cancer formation by high-resolution ultrasound. Once tumours were detected, tumour volume was estimated by high-resolution ultrasound and the animals were either treated with DT or left untreated and changes in tumour volume were measured over time (figure 4F). We found that 50% of iKras*;p53*;CD11b-DTR mice treated with DT underwent tumour regression (figure 4F). Immunostaining of the tumour tissue revealed a 10-fold increase in apoptosis in the epithelial compartment (defined using the lineage tracing marker GFP present in the iKras* system) on myeloid cell depletion (figure 4G). We considered whether this increase might be explained by a lack of clearing of dead cells by macrophages or by active anti-tumour CD8+ T-cell-mediated responses. To distinguish among these possibilities, we performed CD8+ T-cell depletion together with CD11b+ cell depletion and observed an almost complete rescue of epithelial cell apoptosis (figure 4G). Thus, our data support a role for CD11b+ myeloid cells in ‘protecting’ tumour cells from T-cell responses.
We next examined the mechanism by which myeloid cells regulate CD8+ T-cell immunosurveillance in PDA. One mechanism that has been reported in many solid malignancies is activation of the PD-1/PD-L1 immune checkpoint.29 ,30 Myeloid cells are known to express PD-L1—a ligand of PD-1 which is expressed on activated T cells and acts to limit sustained activation within tumours. Thus, we first examined for PD-L1 expression on myeloid cells within PDA tumours. Using implanted iKras*p53* tumour lines in genetically compatible CD11b-DTR mice, we sorted CD45+CD11b+Gr-1+ MDSCs, CD45+CD11b+F4/80+ macrophages, CD45-GFP− stromal cells, CD3+CD8+ T cells and GFP+ CD45− tumour cells. Quantitative PCR analysis revealed that Pdcdlg1, encoding for PD-L1, was expressed by MDSCs, stromal cells, macrophages and, at lower levels, by tumour cells (see figure 5A and online supplementary figure S3D). Pdcdlg2, another immune checkpoint ligand for PD-1, was also detected in macrophages, but not in tumour cells (see online supplementary figure S3D and data not shown). DT treatment resulted in loss of Pdcdlg1 expression in tumour cells but not in the residual myeloid cells or in stromal cells (figure 5A). Immunohistochemistry similarly showed a decrease in PD-L1 protein on tumour cells on myeloid cell depletion (figure 5B). In these tumours, DT treatment caused an increase in IL-2 and Ifnγ expression and a decrease in Pdcd1 expression, suggestive of increased activation and proliferation of CD8+ T cells (see online supplementary figure S3E). We then measured PD-L1 expression by flow cytometry in KPC tumours. Myeloid cell depletion again resulted in a reduction in PD-L1 expression in epithelial cells of 65 671 tumours (figure 5C), although not in epithelial cells of 7940B tumours (data not shown), indicating variability across mouse tumour lines.

Myeloid cells regulate programmed cell death-ligand 1 (PD-L1) expression in pancreatic cancer cell through epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MAPK) signalling. (A) Quantitative real time (qRT)-PCR for Pdcdlg1 expression in flow sorted CD45−GFP− stromal cells and CD45+CD11b+Gr-1+ MDSCs; Pdcdlg1 expression in pancreatic intraepithelial neoplasia (PanIN) cells, iKras*3 in vitro cells and flow sorted GFP+CD45− subcutaneous iKras*3 tumour cells. Data represent mean±SEM, n=3. The statistical difference was determined by two-tailed Student’s t-test. (B) Immunohistochemistry for PD-L1 in control and diphtheria toxin (DT)-treated subcutaneous tumours is shown. Scale bar 25 µm. (C) Percentage of CD45−EpCAM+ tumour cells and CD45−EpCAM+PD-L1+ tumour cells in subcutaneous 65 671 tumours extracted form DT-treated wild-type mice or CD11b-DT receptor (DTR) mice measured by flow cytometry is shown. Data represent mean±SEM; each point indicates one tumour (n=6). The statistical difference was determined by two-tailed Student’s t-test. (D) Experimental design of the co-culture experiments is shown. (E) Western blotting showing EGFR, MAPK, AKT, STAT3 signalling pathway components in UM2 cultured alone, co-cultured with bone marrow (BM) cells and followed by vehicle, erlotinib or MAPK kinase (MEK) inhibitor (MEKi) treatment. (F) qRT-PCR showing PDCDLG1 expression and western blotting for PD-L1 levels in primary human pancreatic ductal adenocarcinoma (PDA) cell line UM2 cultured alone, co-cultured with BM cells and followed by vehicle, erlotinib or MEKi treatment. Data represent mean±SEM, n=3. The statistical difference was determined by two-tailed Student’s t-tests. (G) qRT-PCR for PDCDLG1 expression and western blotting for PD-L1 levels in primary human PDA cell line UM5 cultured alone, co-cultured with BM cells and followed by vehicle or MEKi treatment are shown. Data represent mean±SEM, n=3. The statistical difference was determined by two-tailed Student’s t-tests. AKT, protein kinase B (PKB), also known as Akt; MDSCs, myeloid-derived suppressor cells; qRT, quantative real time.
We then sought to investigate whether inhibition of the PD-1/PD-L1 immune checkpoint pathway was sufficient to recapitulate the effects of myeloid cell depletion. Thus, we treated tumour-bearing mice with anti-mPD-1 antibody (clone J43) or isotype control and harvested them at day 3 of treatment (see online supplementary figure S4A). PD-1 blockade failed to repress tumour growth in iKras*2 cells (see online supplementary figure S4A), in accordance with current clinical data.31 The fact that we did observe a response in the iKras*1 and 3 cell lines (see online supplementary figure S4A) possibly indicated that these lines are slightly more immunogenic. Accordingly, we observed an increase in intratumoural CD8+ T cells in iKras*1 and 3 cells (see online supplementary figure S4B), but not in iKras*2 cells (see online supplementary figure S4A and data not shown). Moreover, the levels of Ifnγ, Ifnβ1, Prf-1 and Gzmb—markers of CD8+ T-cell activation—increased in iKras*3 tumours, but not in iKras*2, on PD-1 blockade (see online supplementary figure S4C). Even long-term treatment with anti-mPD-1 had no effect on tumour growth over time in both syngeneic models (7940B and 65 671) (see online supplementary figure S4D and figure 6G). Moreover, we observed that anti-PD-1 treatment resulted in a compensatory increase in cytotoxic T-lymphocyte-associated protein 4 (Ctla4) expression, thus activating another immune checkpoint and eventually bypassing the PD-1 blockade (see online supplementary figure S4E).

Mitogen-activated protein kinase(MAPK) inactivation in vivo downregulates programmed cell death-ligand 1 (PD-L1) expression on tumour cells. (A) Experimental design is shown. (B) Changes in tumour size (%) post-MAPK kinase (MEK) inhibitor (MEKi) treatment. Data represent mean±SEM, n=4–6. Statistical difference between two groups was analysed by two-way analysis of variance (ANOVA). (C) By flow cytometry, the percentages of CD45−EpCAM+PD-L1+ tumour cells and CD45+CD11b+PD-L1+ myeloid cells in subcutaneous 65 671 tumours and iKras*3 tumours extracted from vehicle or MEKi-treated mice were measured. Data represent mean±SEM; each point indicates one tumour (n=4–6). The statistical difference was determined by two-tailed Student’s t-test. (D) Western blotting showing PD-L1 and phosph-ERK1/2 levels in subcutaneous iKras*3 tumours post-vehicle or MEKi treatment. Data represent mean±SEM, n=3. (E) Co-immunofluorescent staining showing E-cadherin (green), PD-L1 (red), phosph-ERK1/2 (magenta) and DAPI (blue) in vehicle or MEKi-treated subcutaneous 65 671 tumours. Scale bar 50 µm. (F) Co-immunofluorescent staining showing GFP (green), PD-L1 (red), phosph-ERK1/2 (magenta) and DAPI (blue) in vehicle or MEKi-treated subcutaneous iKras*3 tumours. Magenta arrows indicate GFP+PD-L1+phosph-ERK1/2+ tumour cells; green arrows indicate GFP+PD-L1−phosph-ERK1/2− tumour cells. Scale bar 50 µm. (G) Experimental design and graph showing the changes in tumour size (%) post-MEKi and/or anti-PD-1 treatment. Data represent mean±SEM, n=4–6. Statistical difference between two groups was analysed by two-way ANOVA. DAPI, 4',6-diamidino-2-phenolindole.
Myeloid cells induce expression of PD-L1 in tumour cells through EGFR/MAPK signalling
Although iKras* tumour cells expressed elevated levels of PD-L1 in vivo, culturing the cells in vitro led to rapid downregulation of Pdcdlg1 expression (see online supplementary figure S4F). This finding was consistent with our observation that PD-L1 expression was downregulated in tumours depleted of CD11b+ myeloid cells. To understand this biology, we conducted a series of co-culture experiments. First, we isolated CD11b+F4/80+ TAMs, a major subset of the CD11b+ cells infiltrating the tumours. We also isolated T cells from the tumours. We then co-cultured TAMs and T cells, individually or in combination, with the iKras* tumour cells, using trans-well plates. While tumour cells alone displayed very low expression of Pdcdlg1, we found that co-culture with macrophages, T cells or with a combination of the two, induced Pdcdlg1 expression in tumour cells (see online supplementary figure S5A).
We next investigated whether these findings were relevant to human tumours using four primary pancreatic cancer cell lines 1319, UM2, UM18 and UM19. First, we cultured these cells alone or with BM cells in trans-wells. We found that BM cells induced upregulation of expression of both Pdcdlg1 and Pdcdlg2 in three of the human tumour cell lines (see online supplementary figure S5B). Moreover, reciprocal effects on the co-cultured BM cells were also observed, with three of the lines inducing both macrophage differentiation and alternative polarisation (see online supplementary figure S5C).
In our studies investigating the mechanism by which myeloid cells regulate PD-L1 expression in PDA cells, we found that co-culture of human PDA cell lines with BM-derived cells led to an increase in EGFR phosphorylation in tumour cells, indicating activation of EGFR signalling (figure 5E). EGFR signalling has been reported to induce PD-L1 expression in non-small-cell lung cancer cell;32 however its role in PDA has not been previously reported. Thus, we investigated whether a similar effect is present in pancreatic cancer cells. To do this, human primary pancreatic cancer cells (UM2, UM5, 1319 and UM18) were co-cultured with BM-derived cells using the trans-well system. Then, the BM cells were removed and the tumour cells were treated with vehicle control or with erlotinib, an EGFR inhibitor (figure 5D). Although the majority of pancreatic cancer cells express a mutant form of Kras, activation of MAPK signalling in these cells is still partially dependent on EGFR signalling, a pathway that plays a key role in pancreatic carcinogenesis.22 ,33 Treatment with erlotinib led to a slight decrease in the levels of p-EGFR and of the downstream signalling component p-ERK1/2 and reduced the expression of Pdcdlg1 RNA and protein following co-culture with BM-derived cells (figure 5E, F). To determine whether MAPK signalling activation downstream of EGFR22 was responsible for induction of PD-L1, we used the MEKi GSK1120212 in a similar set of experiments. MEKi treatment led to a drastic reduction of p-ERK1/2 levels, as expected (figure 5E), and reduced PD-L1 expression in all the cell lines, independent of their basal levels of PD-L1 expression (see figure 5F, G and online supplementary figure S6B, S6C). Intriguingly, although a feedback mechanism between MAPK/ERK and PI3K/AKT (protein kinase B, also known as AKT) has been previously described in breast and colorectal cancer,34 ,35 treatment with MEKi only resulted in a modest upregulation of p-AKT but a surprisingly strong upregulation of p-Stat3 (see figure 5E and online supplementary S6B), through an increase in the expression of IL-6 (see online supplementary figure S6D).
In addition to EGFR ligands, MAPK signalling can be activated by other secreted factors. We have previously shown that IL-6 activates MAPK signalling in PanINs,36 and myeloid cells are a source of this cytokine.37 However, treatment with recombinant human IL-6 had no discernible effect on MAPK signalling (measured as p-ERK) in the UM2 line, although it led to increased p-Stat3, a direct downstream signalling effector. Conversely, when BM cells and tumour cells were co-cultured (see online supplementary figure S5A), the use of an anti-IL6 antibody had only a modest effect on p-ERK levels, while it did reduce p-Stat3 levels. In both cases, we did not observe any change in Pdcdlg1 expression (see online supplementary figures S6B, S6C, S6F). Thus, at least in the in vitro situation, IL-6 does not play a key role in the regulation of MAPK signalling or of Pdcdlg1 expression.
PD-L1 expression is regulated by MAPK signalling in vivo
To validate the mechanistic regulation of PD-L1 expression by MAPK signalling in pancreatic cancer cells in vivo, we treated mice bearing subcutaneous tumours with MEKi or vehicle (figure 6A). MEK inhibition led to reduced tumour growth in 65 671 and tumour regression in iKras3* (figure 6B). Flow cytometry analysis showed PD-L1-expressing tumour cells decreased in 65 671 (the percentage of CD45−EpCAM+PD-L1+ cells) and iKras*3 lines (the percentage of CD45−GFP+PD-L1+ cells). PD-L1-expressing myeloid cells also decreased in 65 671 (the percentage of CD45+CD11b+PD-L1+ cells) on MEKi treatment, but remained the same in iKras*3 (figure 6C). Western blot analysis in iKras*3-derived tumours confirmed a decrease in PD-L1 on MEKi treatment (figure 6D); however, since this was performed on bulk tumour tissue, it was not informative as to which cells were affected. For this reason, we then proceeded to perform co-immunostaining both in 65 671 and in iKras*3-derived tumours. In both sets of samples, we observed that tumour epithelial cells (as defined by co-expression with E-cadherin for the 65 671 cells and by expression of the lineage tracer EGFP for the iKras*3 line) expressed high levels of p-ERK and membrane PD-L1 (figure 6E, F). On MEKi treatment, p-ERK and PD-L1 levels were reduced. Interestingly, a subset of tumour cells in the iKras*3 sample were resistant to MEKi and the levels of p-ERK in those cells did not change on treatment. In addition, those individual cells also retained PD-L1 expression (figure 6F). Moreover, to test whether reduced PD-L1 expression on tumour cell may make them more responsive to anti-PD-1 blockade, we treated a cohort of 65 671-derived tumours with both MEKi and anti-mPD-1. The combination treatment showed a greater inhibitory effect on tumour growth and possibly tumour regression, in comparison with no effect of PD-1 inhibition alone and a modest decrease in growth with the MEKi by itself (figure 6G). Taken together, our data reveal a novel interaction between MAPK signalling and the PD-1/PD-L1 immune checkpoint that could be exploited therapeutically.
Discussion
The onset and progression of pancreatic carcinogenesis is accompanied by the accumulation of an inflammatory microenvironment, which includes a prevalence of immunosuppressive lymphoid and myeloid cell types.2 ,6 The interaction between tumour cells and immune cells is complex and is not fully understood. An understanding of this biology will be necessary if immune therapy is to be successfully applied to pancreatic cancer—a disease that has demonstrated remarkable resistance to current immunotherapeutic strategies.38 In the current study, we have investigated the function of a broad group of myeloid cells, characterised by expression of the CD11b surface marker, in regulating PDA progression and growth.
The relative abundance of different myeloid cell subsets changes during pancreatic carcinogenesis, with an accumulation of TAMs at early stages followed by an increasing accumulation of granulocytes and immature myeloid cells.6 ,39 Tumour-derived granulocyte-macrophage colony-stimulating (GM-CSF) factor is required for MDSC recruitment during the development of pancreatic cancer; in turn, MDSCs inhibit CD8+ T-cell dependent responses.40 ,41 Similarly, Gr-MDSCs (CD11b+ Gr-1high Ly6Cint) promote pancreatic carcinogenesis by inhibiting T-cell proliferation and inducing T-cell death after activation.6 Macrophages induce ADM at the onset of carcinogenesis and promote tumour growth.7 ,42 ,43 Macrophages are equally required in advanced tumours, where they are required to promote metastasis.44
Prevention of macrophage recruitment with an anti-CSF1 receptor (CSF1R) antibody reduces the overall number of TAMs and their immunosuppressive capacity. Interestingly, blockade of CSF1R signalling causes upregulation of the immune checkpoint molecules CTLA4 and PD-L1 on T cells and tumour cells, respectively, as a result of enhanced immune stimulation within the tumour microenvironment. This ability of PDA to rapidly adapt to immune pressure suggests the need for combination therapy in pancreatic cancer and, in fact, CSF1R inhibition combined with anti-PD-1 therapy led to tumour regression.45 While TAMs largely act in a pro-tumour manner, their biological activity can be redirected. Targeting the surface antigen CD40 in macrophages with an agonist antibody results in tumour-infiltrating macrophages becoming tumouricidal and causing tumour regression.46 Systemic release of Chemokine (C–C Motif) Ligand 2 (CCL2) and IFNγ on CD40 agonist treatment was recently shown to redirect macrophages from a pro-tumour status into a status favouring degradation of the fibrotic reaction and remodelling of the pancreas.15
In light of this extensive body of work, we endeavoured to investigate the effect of depleting a large subset of myeloid cells, including macrophages as well as MDSCs, in a genetically engineered mouse model of pancreatic cancer as well as in syngeneic transplantation model. We used a genetic approach, based on the use of the CD11b-DTR mouse that allows depletion of all CD11b+ cell subsets on treatment with DT. During the initiation of carcinogenesis, we observed that myeloid cell depletion was sufficient to prevent PanIN formation. At later stages of carcinogenesis, myeloid cells were required for tumour growth. Moreover, our in vivo findings show that expression of the PD-L1 immune checkpoint ligand on the surface of tumour cells depends on the presence of myeloid cells and that myeloid cell depletion restores CD8+ T-cell-mediated immune responses.
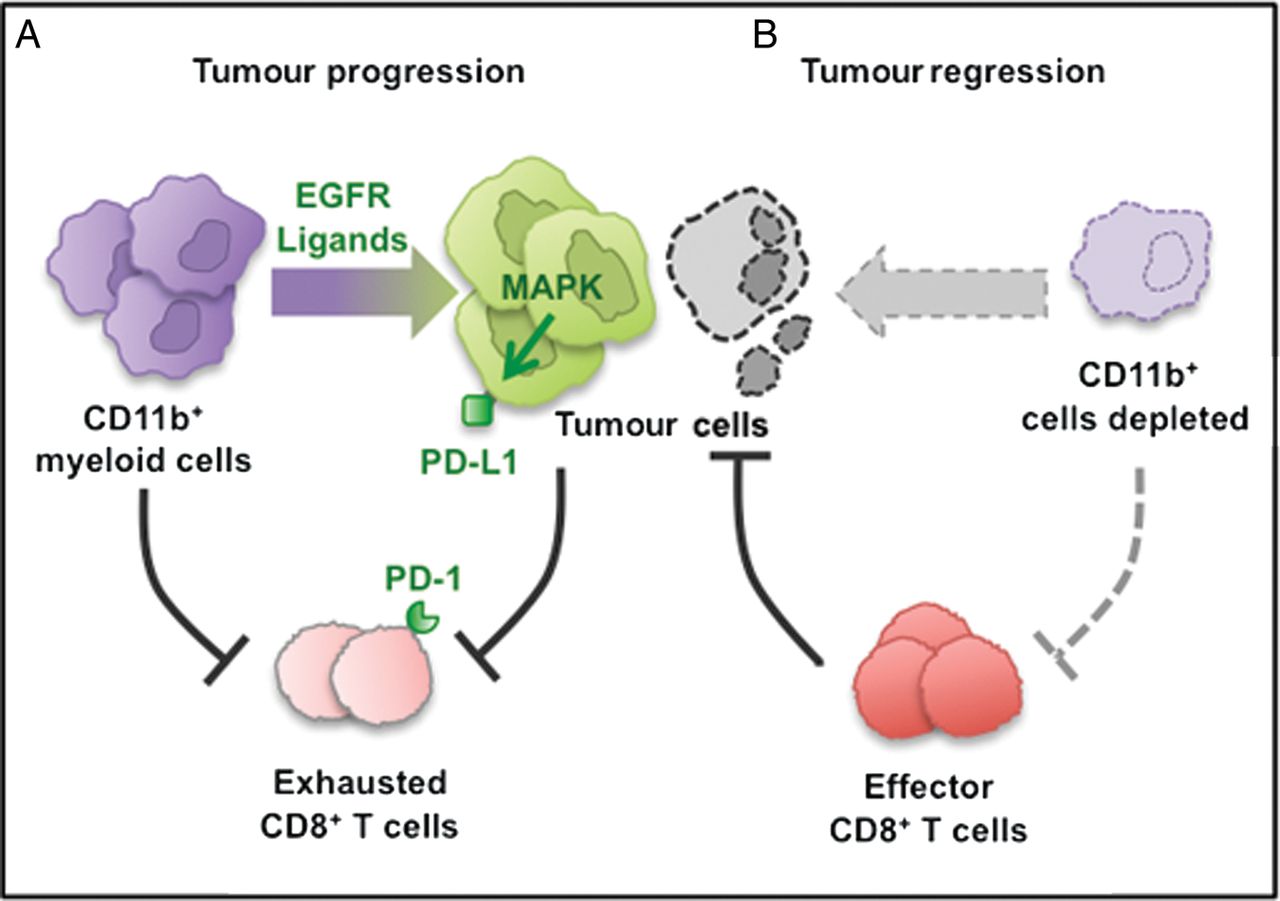
PD-L1 expression can be activated by multiple cytokines.47–50 In vivo, CD8+ T cells can drive its expression in melanoma tumour cells.51 Similarly, we find that T cells drive expression of PD-L1 in pancreatic cancer cells in an in vitro co-culture system. We show that myeloid cells can also promote PD-L1 expression in pancreatic cancer cells in vitro. Moreover, in vivo, depletion of myeloid cells leads to loss of PD-L1 on the tumour cells (see scheme in figure 7). This finding does not necessarily reflect a direct effect of myeloid cells on tumour cells, as myeloid cell depletion causes other changes within the tumour microenvironment such as a decrease in Tregs. Intriguingly, myeloid cell depletion had no effect on PD-L1 expression in other cell types such as stromal cells. We further show that EGFR/MAPK signalling is a key regulator of PD-L1 expression in the tumour cells. This finding parallels the observation that PD-L1 is expressed in EGFR-mutant non-small-cell lung cancer.32 In our system, myeloid cells are required for MAPK activation in the tumour cells. The mechanism underlying this finding is not fully established, but it is likely to involve activation of EGFR through its ligands. In fact, infiltrating macrophages have been shown to be a source of EGFR ligands in breast cancer52 and specifically of HB-EGF in the neoplastic pancreas.53 Based on these observations, the question arose whether MEK inhibition could condition the tumours by lowering the expression of PD-L1 and making them more responsive to anti-PD-1 treatment. In fact, combined MEK and PD-1 inhibition was more effective than either treatment alone in a syngeneic transplantation model of pancreatic cancer.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagram depicting our working model. The CD11b+ myeloid cells protect tumour cell viability by blocking CD8+ T-cell-mediated anti-tumour responses in pancreatic cancer. (A) CD11b+ myeloid cells block anti-tumour CD8+ T cells immune responses partially by activating the programmed cell death-1 (PD-1)/PD-ligand 1 (PD-L1) checkpoint. (B) CD11b+ myeloid cell depletion reverses immune suppression and enables CD8+ T-cell effector function, thus blocking tumour growth.
These findings might have important therapeutic implications. The observation that PD-L1 is expressed in pancreatic cancer cells5 led to the possibility of exploiting this molecule therapeutically. However, while PD-1/PD-L1 targeting in cancer has shown beneficial responses in other cancer types, it is by itself not effective in pancreatic cancer.31 There is more promise in the use of combination strategies. Combined PD-1 and CTLA4 targeting is more effective than either on its own.5 Our data indicate that PD-1 targeting should also be explored in combination with targeting the MAPK signalling pathway, one of the effectors of oncogenic Kras. Moreover, as the interaction between tumour cells and myeloid cell subsets is further explored, new potential therapeutic targets suitable for combined therapy might be identified.
An unexpected finding that will merit future investigation is the activation of Stat3 on MEK inhibition. The IL-6/Stat3 signalling pathway has been shown to promote pancreatic carcinogenesis17 ,37 and, thus, an eventual feedback mechanism linking MEK inhibition to Stat3 activation could potentially be the cause of concern but may also provide new opportunities for combined targeting of pancreatic cancer.
Acknowledgments
The authors thank Dr Jörg Zeller, Dr Wei Yan and Dr Bin Zheng for scientific discussion. They thank Kristen Long for helpful discussion and manuscript editing; Dawson Knoblock for technical assistance and helpful discussion and Kevin T Kane for lab support. The CK19 antibody was obtained from the Iowa Developmental Hybridoma Bank.
References
Footnotes
Contributors YZ, AV-D, EM, FMM, KF and DL performed the experiments and analysed the data; DMS established the primary human cell lines; ADR assisted ultrasound imaging; YZ, GLB and MPdM designed the study, interpreted the data and wrote the manuscript. MPdM directed the study.
Funding This project was supported by the University of Michigan Biological Scholar Program, the University of Michigan Cancer Center Support Grant (NCI P30CA046592), an American Cancer Society Scholar Grant, a grant by the Elsa U Pardee Foundation and NCI-1R01CA151588-01. EM was supported by a University of Michigan Program in Cellular and Molecular Biology training grant (NIH T32 GM007315) and by a University of Michigan Gastrointestinal Training Grant (NIH T32 DK094775). GLB was supported by a National Institutes of Health grant K08 CA138907.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Cell lines, tissue samples, reagents and original data and study design from this study will be freely shared with any investigators on request.