Article Text

Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 genome engineering has revolutionised biomedical science and we are standing on the cusp of medical transformation. The therapeutic potential of this technology is tremendous, however, its translation to the clinic will be challenging. In this article, we review recent progress using this genome editing technology and explore its potential uses in studying and treating diseases of the liver. We discuss the development of new research tools and animal models as well as potential clinical applications, strategies and challenges.
- GENE THERAPY
- GENE TARGETING
- GENE TRANSFER
- HEPATOCYTE
Statistics from Altmetric.com
Introduction
Clinical application of gene therapy—the transfer of a wild-type allele to the human patient to replace a mutated gene1—has proven very challenging for several reasons, including inefficient delivery into the target cell, the need for sustained expression of the transgene, and deleterious patient immune response. The first gene therapy trial2 sought to correct adenosine deaminase deficiency by transferring a copy of the wild-type gene into haematopoietic stem cells ex vivo and re-infusing the cells into the patient. This trial demonstrated the feasibility of gene therapy, which prompted a flurry of trials in the biomedical community. A few years later, a fatal systemic inflammatory response occurred with a liver-directed adenoviral vector3 and it became clear that retroviral integration could result in unexpected neoplasias.4 While many of these initial setbacks have been overcome by improvements in vector design and cell-based therapy procedures, there is still considerable room for improvement. Recent advances in genome editing are driving a fundamental paradigm shift from overexpression of defective gene products to precisely modifying a patient's own DNA. The notion of treating disease by removing or repairing harmful mutations is a tantalising one, and may be a solution to the many disorders not amenable to pharmacological treatment.
Genome editing has been attempted for some time, but the complexity of zinc finger nucleases, combined with the secrecy of proprietary technology, delayed further development. Later, Transcription Activator-Like Effector Nuclease technology became available, and genome editing started to gain momentum. Both technologies had one major drawback: the nucleases used to cut DNA were inefficient. This changed with the development of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 genome editing: this technology is more efficient than previous generations of designer nucleases, and it has the added benefit of being simple to use, from design to execution. Many physicians and scientists are now searching for the best clinical applications for this promising technology.
Glossary
Cas9: CRISPR-associated protein 9, an endonuclease from bacteria that forms a ribonucleoprotein with the sgRNA, which can be directed to cause a double-strand break at most variable ∼20 base pair (bp) DNA sequences via sgRNA target sequence.
sgRNA: single guide RNA. An artificial chimaera of crRNA and tracrRNA, the two bacterial RNA components that direct Cas9 to DNA sequences for cleavage. The first ∼20 bp of sgRNA (or crRNA) are variable and complementary to the target site.
PAM: protospacer adjacent motif. The sequence required immediately downstream of the target sequence. The PAM varies depending on the bacterial origin of the Cas9 protein.
DSB: double-strand break. CRISPR/Cas9 introduces a blunt DSB in the target DNA three bps upstream of the PAM.
NHEJ: non-homologous end joining. A method of DSB repair that does not use a template strand, and which can result in the introduction of insertions or deletions of variable length at the cut site.
HDR: homology-directed repair. A repair mechanism using a DNA template to repair double-stranded DNA breaks via homologous recombination.
CRE-loxP technology: a method of inducing site-specific recombination between two sites (LoxP sites), which is catalysed by the Cre recombinase protein. This phage recombination system is the ‘gold standard’ of creating conditional knockout alleles in mouse transgenesis.
Zygote: a fertilised egg, which is still at the single-cell stage. The genome of the zygote is amenable to editing by injected CRISPR/Cas9 molecules. Modifications in zygotes are then carried on in all subsequent cell divisions during embryonic development.
The liver has several advantages over other organs for somatic genome editing for both hepatic disorders and for systemic metabolic conditions triggered by a mutated or dysregulated gene expressed in the liver. First, the liver is an immune-privileged organ and favours immune tolerance over induction of immunogenicity.5 Second, many gene therapy vectors, including nanoparticles, have a natural tropism towards the liver, which should help to reduce the risk of a severe immune response (see below). Third, the ‘exit strategy’ in the liver is more favourable than in other organ systems such as the brain or heart, so if CRISPR/Cas9-mediated genome editing leads to deleterious complications such as neoplastic growth, the problematic area could be more readily resected. Such an outcome is of course not desirable, but must be carefully weighed against the potential benefits to patients when introducing CRISPR/Cas9 into the clinic.
Here, we will discuss how CRISPR/Cas9 is used in research as well as its potential clinical applications. We will explain the benefits of this technology as well as discuss the major hurdles involved in translating it to the clinic.
CRISPR/Cas9 genome editing
The CRISPR/Cas9 genome editing system is derived from a naturally occurring antiviral immune system found in many species of bacteria. The first discovery came in 1987, when Ishino et al 6 noticed a cluster of repeat sequences, interrupted by variable spacer sequences, later referred to as CRISPR.7 However, it was not until 2005 that these spacer sequences were recognised as foreign in origin8–10 and postulated to play a role in host adaptive immunity.8 This defence mechanism relies on a family of CRISPR-associated (cas) genes.7 ,11 ,12 The Cas9 gene encodes an RNA-guided nuclease that normally protects the host from phage infection through sequence-specific destruction of foreign DNA.13 ,14 Years of work by several groups finally culminated in the identification of all key components of a recombinant CRISPR/Cas9 system (box 1) and the demonstration of its functional capability in mammalian cells.15–18
The mechanism of CRISPR/Cas9 activity consists of a few essential components (figure 1A): a processed spacer/repeat sequence that is transcribed to generate the ‘crRNA’,19 which binds to a separately transcribed and partially complementary ‘tracrRNA’.14 Together, these RNAs are able to guide the Cas9 protein to a target site, defined by complementarity to the spacer sequence, in combination with a species-specific protospacer adjacent motif (PAM), which must be present immediately following the target site for DNA cleavage to occur.20 ,21 This mechanism was further simplified in 2012, when Jinek et al 22 showed that a chimaeric crRNA:tracrRNA transcript, termed ‘single guide RNA’ (sgRNA), is equally capable of directing Cas9 to its target sequence to cut DNA. The Cas9/sgRNA complex surveys the genome for complementary sites based on Watson-Crick base pairing between the guide sequence and DNA. On successful recognition of the target, Cas9 undergoes a conformational change that engages its two nuclease domains.23–25 The nuclease domains cleave both strands of the target DNA at approximately –3 nucleotides before the PAM, generating a double-strand break (DSB).26 Single amino acid mutations in either nuclease domain of Cas9 result in a ‘nickase’,27 ,28 which produces single-stranded DNA nicks rather than DSBs. Likewise, mutations of both nuclease domains result in a version of Cas9 that can survey and bind to DNA, but that is incapable of cutting. This catalytically inactive or ‘dead’ Cas9 (dCas9) can be modified with fusion proteins to activate or repress transcription28–31 or change the epigenetic status of the local chromatin.32–35 However, it is the ability of Cas9 to efficiently and easily generate DSBs in DNA that has opened the door for a multitude of basic research and therapeutic applications.

CRISPR/Cas9 structure and simplified repair mechanism. (A) The molecular elements of CRISPR/Cas9. The bacterial Cas9 protein that binds to two different single-stranded RNAs (crRNA and tracrRNA), which are partially complementary in bacteria but have been combined as sgRNA for research purposes. The species-dependent protospacer adjacent motif (PAM) must be present immediately following the guide sequence for DNA cleavage to occur. (B) Repair mechanism in mammalian cells induced by CRISPR/Cas9-mediated DNA cleavage (simplified). It is important to note that perfectly repaired DNA will be repeatedly cut until the targeting sequence is modified. See text for more details on non-homologous end joining (NHEJ) and homology-directed repair (HDR). Red arrow is the location within the target sequence where the double-strand break takes place.
The CRISPR/Cas9 system is the most efficient and flexible genome editing system to date, requiring only customisation of the sgRNA to generate DSB at a user-designated site. The PAM for the most commonly used Streptococcus pyogenes Cas9 (SpCas9) is ‘NGG’,20 which occurs frequently in the genome and generally imposes only minimal constraints on target site selection and sgRNA design. The second most commonly used Cas9 ortholog from Staphylococcus aureus has a more restrictive ‘NNGRRT’ PAM,36 although recent efforts have been successful in partially re-engineering Cas9 PAM specificities through rational design.37 ,38
Once the Cas9 has bound and cleaved the DNA, the host's DNA repair machinery repairs the DSB by either homologous recombination (homology-directed repair (HDR)) or non-homologous end joining (NHEJ) (figure 1B). HDR uses a template strand to precisely repair a DSB, such that no mutations are introduced at the cleavage site. While this occurs naturally during the cell cycle using the sister chromatid as repair template,39–41 exogenous template DNA can also be introduced for integration based on homology to the target site. This process is dramatically more efficient for insertion of foreign genetic material at sites when Cas9 is used to introduce targeted DSBs. While HDR has been achieved for years with conventional gene targeting in mouse embryonic stem cells, CRISPR/Cas9 makes this feasible in most dividing cells.
NHEJ is the dominant DSB repair pathway in most mammalian cells and organisms, and is active in all phases of the cell cycle. As the name suggests, this pathway does not require a homologous template, and can repair irregular breaks that have overhanging strands such as those that often result from exogenous DNA-damaging agents. In contrast to HDR, NHEJ can result in genomic insertions and/or deletions (referred to as ‘indels’) during processing of the ends of the DSB42 (figure 2A). These indels are of varying size and, when targeted within protein coding sequences, can create inactivating missense or nonsense mutations. Most indels introduced by Cas9 cleavage are small frameshift mutations (usually ±1 or 2 bases). Frameshift mutations that occur before the penultimate exon in a protein-coding gene usually result in nonsense-mediated decay (NMD) of the mutant transcript, effectively ‘knocking out’ the gene. When multiple sgRNA are used in combination, the NHEJ pathway can also be used to delete intervening DNA sequences.
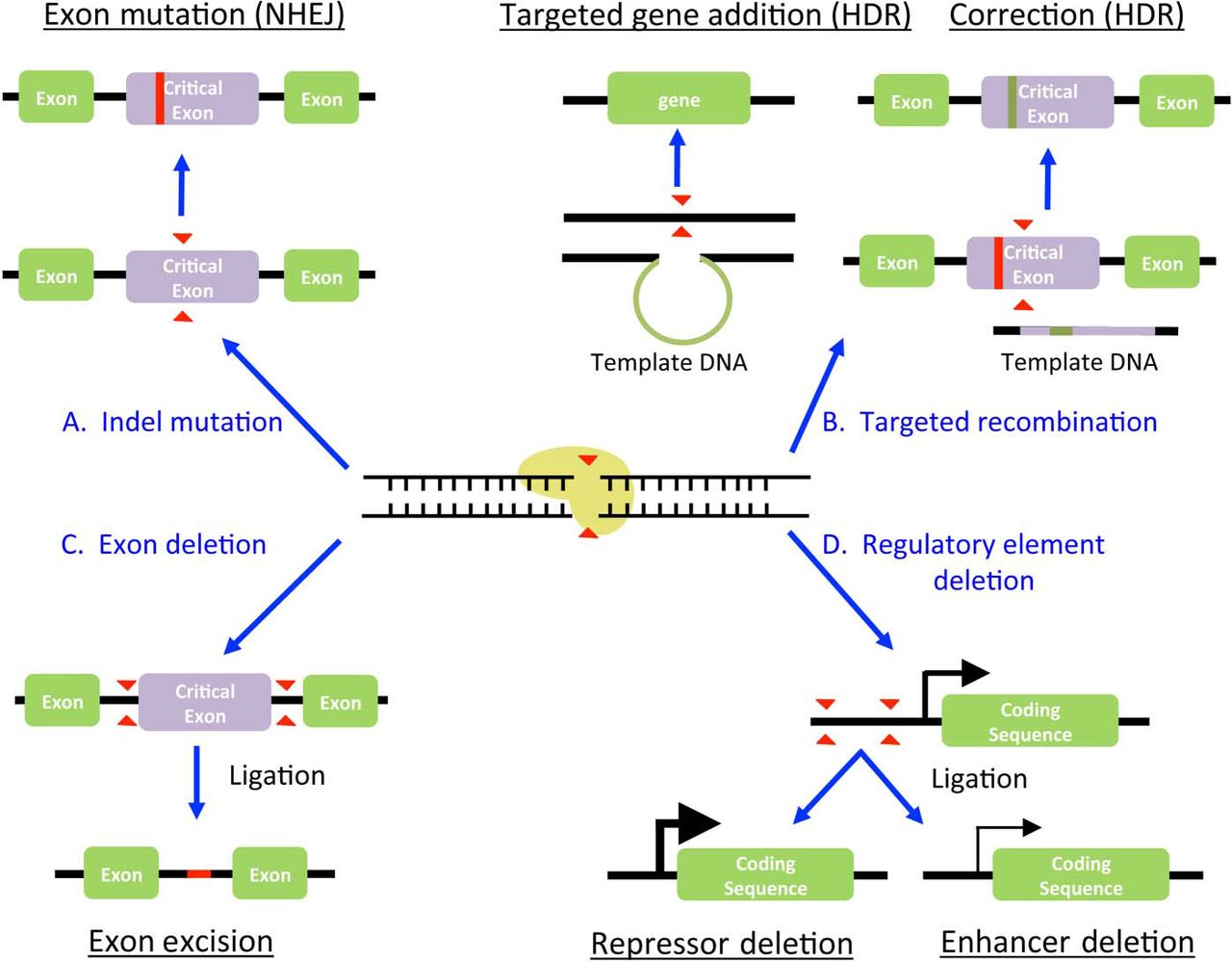
Genome editing approaches for biomedical applications. Examples of genetic modifications are given. For details, see ‘CRISPR/Cas9 preclinical and research applications’ or ‘Medical applications for CRISPR/Cas9 in the liver’ section. HDR, homology-directed repair; NHEJ, non-homologous end joining.
Both DSB repair pathways are used in parallel by a single cell and have their own potential utilities and pitfalls. HDR is primarily active during the S and G2/M phases, while cells in G0/G1 almost exclusively repair DSB by NHEJ.39–41 In addition, there is evidence that even during S phase, when HDR is most likely to occur, NHEJ is still the favoured repair mechanism in human cells.39 The mechanism of DSB repair is important to consider when designing CRISPR/Cas9 strategies in quiescent cells such as hepatocytes.
In the following sections, we will discuss how CRISPR/Cas9 has facilitated research in the field of hepatology, primarily as it pertains to human liver disease. We will also discuss potential clinical applications of CRISPR/Cas9 for liver disease and disorders, which might benefit from hepatic genome editing strategies.
CRISPR/Cas9 preclinical and research applications
In vitro applications
A major milestone in the development of CRISPR/Cas9 was its adaptation for use in mammalian cells,17 ,18 which provided researchers with a powerful tool to study genetic perturbations in tissue homeostasis and disease. Since then, a significant body of work has used CRISPR/Cas9 in the tissue culture setting to explore various aspects of liver biology. While these studies are diverse and involve many genetic targets, there are common underlying themes and strategies.
So far, most efforts have been focused on either deleting a gene by NHEJ or introducing a transgene by homologous recombination. When knocking out a gene, there are several potential strategies. The most common method is to use a single sgRNA targeted to a critical exon in order to introduce indels, likely causing a frameshift mutation and/or NMD (figure 2A). This approach has been used to validate, in vitro, potential oncogenes discovered in human liver cancers, such as acid sensing ion channel 1a43 and eukaryotic elongation factor 2 kinase.44 Similarly, isocitrate dehydrogenase 1, commonly mutated in intrahepatic cholangiocarcinoma,45 has been deleted in the HepG2 cell lines to dissect its biological and metabolic functions.46
To increase the chances of gene deletion, several groups have used multiple sgRNAs targeting different regions of the gene of interest simultaneously. For instance, the aspartate β-hydroxylase gene has been targeted by three separate sgRNAs to generate a HepG2 knockout line.47 CRISPR/Cas9-induced NHEJ can also be used for the deletion of entire genomic regions; when two DSBs are induced at the same time in close proximity to each other, it is likely that the intervening region will be lost during NHEJ repair27 ,48 ,49 (figure 2C). There are several advantages to this research design. For instance, Pankowicz et al 50 has shown that whole exons can be removed without the risk of unpredictable indels in the reading frame of the hydroxyphenylpyruvate dehydrogenase (HPD) gene. This deletion strategy can also be used to delete a specific enhancer region, as has been done in the cytochrome P450 (CYP) 2D6 (CYP2D6) promoter of HepG2 cells51 (figure 2D). Similarly, Hep3B cells have been modified using CRISPR/ Cas9 to delete the zic family member 2 binding region in the octamer binding protein 4 promoter.52
CRISPR/Cas9-induced DSB can also be used to increase the rate of HDR, which is particularly useful for introducing transgenes at virtually any desired locus (figure 2B). This approach is being broadly used in biomedical science, particularly to generate reporter cell lines for screens.17 ,53 ,54 The strategy has also been used for in vitro lineage tracing during bile duct development. In this case, the fluorescent reporter mCherry, and the selection marker puromycin N-acetyl-transferase, were knocked into the cytokeratin 7 locus in human induced pluripotent stem cells.55 CRISPR/Cas9-assisted HDR has also been used in a potential therapeutic setting for the urea cycle disorder arginase 1 deficiency.56
Finally, the nuclease domain of Cas9 can be inactivated (dCas9), and other functional domains can be attached.28–35 This technology has been used to characterise the metformin response in the liver. In one study, metformin responsive enhancers were identified, and targeted with a dCas9-VP64 fusion protein to measure the transcriptional response in the Huh7 cell line.57 Activation of genes in this region via the CRISPR-guided VP64 transactivator showed significantly increased levels of ataxia telangiectasia mutated (ATM), exophilin 5, DEAD-Box Helicase 10 mRNA, suggesting a possible role for these proteins in the mechanism of action of metformin.
In vivo applications
Somatic genome editing using CRISPR/Cas9
As scientists move from cell culture to in vivo systems, delivery of macromolecules, such as the CRISPR/Cas9 machinery, becomes a limiting factor. However, the field of hepatology has greatly benefited from the development of an in vivo transfection method called hydrodynamic tail vein injection (HTVI), which is a simple and efficient way to deliver DNA into murine hepatocytes.58 ,59 The exact nature of the uptake is not known, but up to 40% of hepatocytes can be transfected with this method. Hydrodynamic tail vein injection has already proven very useful for a variety of CRISPR/Cas9 applications.
Several groups have used CRISPR/Cas9 editing to promote tumour growth or study tumour suppressor candidate genes in the liver.60–62 For instance, Xue et al 60 targeted the tumour suppressors Pten and p53 alone and in combination using HTVI. Each sgRNA was able to create indels and disrupt its respective gene, although with relatively low frequencies (2%–7%). While the individual gene mutations did not generate tumours within the study's 3-month timeframe, combining Pten and p53 sgRNAs was capable of generating liver tumours similar to those of transgenic animals with CRE-loxP-deleted Pten and p53. In addition, they generated a gain-of-function mutation in β-catenin by targeting the Ctnnb1 gene with sgRNAs and providing a single-stranded DNA template for HDR. This template was successfully integrated in only ∼0.5% of hepatocytes, as measured by nuclear localisation of β-catenin. This study highlights several points to consider when using CRISPR/Cas9 for somatic genome engineering in the liver. First, it demonstrates the ease of bypassing the lengthy process of embryonic stem cell targeting with CRE-loxP technology. This is tempered by the low rate of indel formation, but this is not limiting in a positive selection setting such as expansion of liver cancer. Furthermore, it demonstrates the considerably lower rates of HDR compared with NHEJ in the liver. Another study using multiple sgRNA in a sleeping beauty transposon cassette demonstrated development of hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma by deleting several tumour suppressor genes in parallel.61 This study revealed a high (79%) incidence of biallelic mutations within the multiple sgRNA target sites, confirming the high efficiency of NHEJ in hepatocytes. Another recent study used a genome-wide sgRNA library to identify new liver tumour suppressors in transgenic p53−/− and myc overexpressing embryonic liver progenitors injected subcutaneously.62 In a second step, they validated the tumour suppressor neurofibromatosis type 1 using somatic genome editing in the murine liver.
Somatic editing with CRISPR/Cas9 has also been successfully used to explore experimental therapies for liver disease.50 ,63–65 Hereditary tyrosinemia type I (HT-I) is caused by a defect in the fumarylacetoacetate hydrolase (fah) gene, which leads to a toxic accumulation of tyrosine catabolites in the liver. Yin et al used HTVI to transfect livers of fah−/− mice with sgRNA, Cas9 and single-stranded DNA templates.65 They observed an initial HDR editing efficiency of 0.4% of hepatocytes. However, these edited hepatocytes displayed a growth advantage over non-edited hepatocytes and eventually expanded up to 33% of the liver, improving the weight loss phenotype over the 30-day observation period. In a follow-up study using nanoparticles to deliver Cas9 and adeno-associated virus (AAV) for the sgRNA and template DNA, they found a higher initial editing rate of up to 6%.64 Yang et al explored a dual AAV CRISPR/Cas9 approach in the spfash mice, a murine model for ornithine transcarbamylase deficiency. Mice injected as neonates showed correction in 10% of hepatocytes, which improved survival when challenged with a high-protein diet, but the same therapeutic approach in adult mice was much less successful (<1.7% correction). Furthermore, deep sequencing revealed very high frequencies (up to 50% adult, 35% neonate) of indels in the sgRNA target site, consistently exceeding the correction rate in all mice analysed. These studies indicate that HDR can be achieved in the settings of growth during early development, as well as in metabolic disease models with regenerating hepatocytes. However, it should be noted that alleles that do not undergo HDR are very likely to get NHEJ-induced mutations. This phenomenon is especially important to consider for compound heterozygotes, where unintended NHEJ editing of the other allele could generate more deleterious mutations.
Some disorders will be amenable to an approach called ‘metabolic pathway reprogramming’, which avoids generating potentially dangerous mutations in the diseased gene (figure 3). This approach deletes a disease pathway-associated gene in order to reroute metabolic pathways and improve a metabolic condition. As a proof-of-principle, we applied this strategy to fah–/– mice and were able to rescue the lethal phenotype with a one-shot therapy targeting the hydroxyphenylpyruvate dioxygenase (hpd) gene, which acts upstream of fah. By deleting critical hpd exons with two separate sgRNAs, we safely inactivated hpd in ∼8% of hepatocytes following hydrodynamic delivery. Double mutated hepatocytes (fah−/−/hpd−/− ) had a growth advantage and almost completely repopulated the liver over the course of 2 months. In a similar approach, Liang et al 66 deleted the Fas receptor and were able to demonstrate resistance to concanavalin A-induced fulminant hepatic failure.

{kind=link}
{kind=link}
{kind=link}
Schematic representation of metabolic pathway reprogramming within a single pathway. In a healthy individual, the metabolic pathway proceeds as normal, with functional clearance of intermediate metabolites. Toxic accumulations in a diseased state occurs due to a non-functional or hypomorphic downstream enzyme (E2), which can be alleviated by using CRISPR/Cas9 to delete a wild-type enzyme (E1) in a disease-associated pathway leading to a more benign phenotype.
Finally, Jarrett et al 67 recently demonstrated that somatic genome editing with CRISPR/Cas9 can be used for disease modelling. Using AAV to deliver sgRNA targeting the low-density lipoprotein receptor (ldlr) to Cas9 transgenic animals, a novel model for familial hypercholesterolaemia was established. The nearly complete removal of hepatic Ldlr protein produced a severe hypercholesterolaemia and atherosclerotic plaque. They further showed that dual AAV vectors could be used for concomitant disruption of apolipoprotein B (apoB), which dramatically reduced plasma cholesterol levels and offered complete protection from atherosclerosis. Besides gaining new insight into familial hypercholesterolaemia and apoB as a therapeutic target, this model establishes a new method to efficiently knockout genes in the liver without the need for laborious germline targeting.
Zygote injection of CRISPR/Cas9
CRISPR/Cas9 has revolutionised the generation of genetically modified organisms. Previously, gene targeting was accomplished by first modifying embryonic stem cells, followed by blastocyst injection and screening for germline transmission in offspring.68 This process was inefficient, time-consuming and expensive, limiting the rate at which animal research could be performed. In 2013, several groups reported that zygote injection with CRISPR/Cas9 could lead to efficient gene knockout at several loci in zebrafish,69–71 mice,72 ,73 rats74 ,75 and rabbits,76 completely bypassing the need for targeting in embryonic stem cells. Since then, several groups have used CRISPR/Cas9 to generate knockout models of animals in order to study hepatic gene function. In zebrafish, this method has been used to study hepatic glucose metabolism on deletion of both the leptin77 and insulin receptor.78
In mice, zygote injection of CRISPR/Cas9 has been used to generate models for the study of lipid disorders. For example, the E167K mutation of the transmembrane 6 superfamily member 2 gene (TM6SF2) had been previously correlated with lower total plasma cholesterol and low-density lipoprotein (LDL) cholesterol, but increased susceptibility to non-alcoholic fatty liver disease.79 ,80 Fan et al 81 investigated the long-term consequences of TM6SF2 deletion in mice, using a CRISPR/Cas9-generated knockout model. In a separate study, Nakagawa et al 82 used CRISPR/Cas9-injected zygotes to generate a conditional knockout (CRE-loxP technology) model for cAMP responsive element binding protein 3-like 3 (creb3l3). Generation of these mice was an impressive use of HDR in zygotes through simultaneous insertion of loxP sites with donor oligos, however, with a very low efficiency (1 mouse of 277 injected zygotes). In order to elucidate the role of the protein in the liver and intestine, mice were crossed with either albumin-Cre (liver) or villin-Cre (intestine) transgenic mice. Only hepatic deletion of the CREB3L3 resulted in increased plasma lipid and cholesterol content. Interestingly, this is a deviation from the global CREB3L3 knockout, which results in hypocholesterolaemia.
Rats have also been modified using CRISPR/Cas9 and have notable differences from their mouse counterparts. For example, a rat model of HT-I generated by zygote injection of CRISPR/Cas9 displayed all of the hallmarks of the disease, including hypertyrosinemia, liver failure and renal tubular damage. Importantly, these rats also developed cirrhosis and fibrosis, which occur in affected humans but no other animal models of the disease.83 Recently, two other rat models for cytochrome metabolism have been generated using a CRISPR/Cas9 zygote injection strategy. Both CYP2E184 and CYP3A1/285 knockout rats have been successfully generated by targeting critical exons of the according genes. In both cases, rats were demonstrated to be viable and fertile, with drug metabolism profiles consistent with successful gene knockout.
To study human drug metabolism in mice, Barzi et al recently generated a novel mouse model that used zygote injections to simultaneously knockout three genes: fah, common gamma chain of IL-2 receptor and the recombination activating 2 gene. The injected zygotes were homozygous for a conditional knockout (CRE-loxP) allele of the P450 oxidoreductase (por).86 Por is the only electron donor for all P450 cytochromes and a deletion results in a functional inactivation of the whole cytochrome superfamily. The resulting mice can be repopulated with human hepatocytes, and on deletion of the murine por, were found to demonstrate human-like drug metabolism. This next generation humanised mouse shows the power of CRISPR/Cas9 for simultaneously targeting multiple genes on existing complex genetic backgrounds.
In addition, CRISPR/Cas9-generated knockout models generally avoid the need for selection cassettes, which have been shown to interfere with cellular gene regulation and local gene expression.87–89 Although murine zygote injections are believed to have a lower CRISPR/Cas9-induced off-target mutation rates than human cell lines,48 ,90 ,91 it is advisable to backcross offspring with wild-type mice to eliminate potential confounding off-target mutations. For generation of knockout models, CRISPR/Cas9 zygote injection has essentially replaced classical gene targeting in mouse embryonic stem cells, and might ultimately supplant targeting in embryonic stem cells for conditional alleles and targeted knockins. Due to the rapid success of this technology, we are likely to see many more applications for hepatic research soon.
Medical applications for CRISPR/Cas9 in the liver
CRISPR/Cas9 technology has revolutionised science and has the potential to transform medicine too. This has also been recognised outside of academia by commercial entities developing CRISPR/Cas9 products. However, as far as we know, there are currently no clinical trials ongoing for liver disease using CRISPR/Cas9 technology. The following section outlines some strategies for medical applications in the field of hepatology, while the major translational challenges are addressed in a separate section. We will only describe somatic editing approaches in the liver, since germline manipulations have important ethical, regulatory and societal implications that require further discussion.
Deletion of dominant disease-causing alleles
Current CRISPR/Cas9 technology is more effective at disrupting or removing target genes than correcting mutations. Therefore, mutated genes of the liver that have a detrimental effect in the liver or other organs are likely the lowest hanging fruit for therapeutic application of CRISPR/Cas9. For instance, familial transthyretin (TTR) amyloidosis is an autosomal dominant disorder presenting with varying degrees of neuropathy, cardiopathy, GI impairment and ocular depositions. The extracellular deposition of TTR-derived fibres in all affected organ systems is responsible for this heterogeneous disorder. In theory, this condition could be treated by the removal of the mutated allele. The allele could be deleted by several CRISPR-mediated strategies, each of which has unique advantages and caveats (figure 2).
The successful clinical application of a somatic genome editing approach using CRISPR/Cas9 technology will depend on several general and disease-specific factors. For instance, it is essential to know where the mutated protein is expressed and to what extent hepatic production contributes to disease pathology. It is also important to know what percentage of edited alleles will alleviate symptoms (ie, the threshold of correction). Clinical studies or case reports from orthotopic liver transplantations (OLT) can help to guide the selection of targets for diseases where patients are most likely to benefit.
Patients with TTR amyloidosis and severe polyneuropathy undergo therapeutic OLT92; however, patients with cardiomyopathy may experience accelerated disease progression after OLT due to deposition of wild-type TTR on a template of amyloid derived from the mutated TTR.93 ,94 Thus, in theory more patients could benefit from somatic genome editing in the liver than OLT. A gene editing approach might also be applied to fusion proteins resulting from translocations, such as the DNAJB1-PRKACA chimaeric transcript in fibrolamellar HCC.95 In this case, careful design of the CRISPR/Cas9 vectors would be needed to target only the precise breakpoint, while avoiding other alleles of the genes involved in the translocation.
Deletion of foreign DNA and/or associated host factors
CRISPR/Cas9 has recently been explored for its therapeutic potential in targeting human pathogens, particularly viral infections with double-stranded DNA intermediates such as papillomavirus96 ,97 and Epstein-Barr virus.98 ,99
Notably, HBV has emerged as an attractive target for CRISPR/Cas9 in the laboratory and, potentially, the clinic. HBV is a partially double-stranded DNA virus that infects primate and human hepatocytes at least partially by using the sodium-taurocholate cotransporting polypeptide (NTCP) as a receptor.100 HBV establishes covalently closed circular DNA (cccDNA) within hepatocytes, which is maintained episomally in the nucleus and mediates chronic infection. This DNA replicative intermediate is an attractive target for the clinical use of CRISPR/Cas9.
Several groups have demonstrated targeted mutation of HBV in both the cell culture and animal model settings. Lin et al 101 were the first group to use CRISPR/Cas9 to target HBV. Using cotransfection of Huh7 cells with sgRNA pairs and an HBV expressing vector, they were able to achieve up to 96% suppression of intracellular hepatitis B surface and core antigens. Using a HTVI model of HBV replication in mice, they were also able to show in vivo editing of HBV in about 5% of the DNA copies. A variety of groups have since reported similar findings, targeting various regions of the HBV genome.102–109 In addition, Kennedy et al 102 also showed a synergistic effect between targeting HBV with CRISPR/Cas9 and using traditional antiviral therapies. It is important to note that HBV infects only humans and chimpanzees. Mouse models for HBV infection do not recapitulate the formation of cccDNA in hepatocytes, which is a target for therapy in humans. Dong et al 103 used CRISPR/Cas9 targeting in an in vivo model for cccDNA expression110 to show a modest decrease in cccDNA concentration within the liver and a corresponding decrease in serum levels of the viral hepatitis B surface antigen and hepatitis B e antigen. Human liver chimaeric mice replicate HBV111 ,112 and may be the best model to demonstrate the effectiveness of a CRISPR/Cas9 antiviral approach.
In addition to DNA viruses, it has recently been proposed that RNA viruses might be targeted with CRISPR/Cas9 as well. The Cas9 variant from Francisella novicida (FnCas9) is reported to be capable of targeting mRNA from HCV.113 Price et al developed FnCas9 to target both the positive and negative strands of HCV, and were able to inhibit HCV protein expression in a cell culture model of HCV infection. However, the same effect was observed when the nuclease domain was inactivated, suggesting that simply binding to the RNA was sufficient to interfere with translation. Interestingly, FnCas9 is also able to target DNA for double-strand cleavage similar to SpCas9, indicating the possibility for a dual targeting strategy in the future.
Aside from targeting hepatotropic viruses, CRISPR/Cas9 could be used to target host factors that are essential for viral replication but do not result in a severe hepatic phenotype. For instance, targeting the NTCP receptor to inhibit HBV entry would be a possibility, especially considering that the liver specific-knockout model has a surprisingly moderate phenotype in mice.114
Substrate reduction approach
A CRISPR-mediated approach to substrate reduction may be therapeutic when essential liver-produced proteins and metabolites lead to harmful effects even in the absence of a specific monogenic disorder, for instance, in hyperglycaemia and hyperlipidaemia. The liver has not been fully exploited in regard to antidiabetic therapies115 and somatic genome engineering opens many new therapeutic approaches towards lowering blood glucose levels.
The liver is also a key organ in lipid and lipoprotein metabolism.116 It is the site of synthesis and secretion of very low-density lipoprotein (VLDL) particles that carry triglyceride, cholesterol and fat-soluble vitamins to peripheral tissues. VLDL contain the apoB-100 (APOB) protein as their primary structural component, and are catabolised in the circulation to produce LDL. VLDL and LDL particles are both cleared primarily by the liver through the action of the LDLR.117 A substrate reduction approach using CRISPR/Cas9 could be pursued for APOB, which is currently targeted by antisense oligonucleotides in severe cases of homozygous familial hypercholesterolaemia.118 However, careful design would be needed to avoid mechanism-related hepatic fat accumulation with APOB disruption.
Another attractive target for somatic editing in the liver is proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein secreted by the liver that binds to apoB-lipoprotein particle and marks the LDLR for degradation on endocytosis.119 Adenoviral delivery of CRISPR/Cas9 into mice was very efficient in reducing PCSK9 and cholesterol in the blood, when ∼50% editing was achieved in the liver.120 Equally impressive results have been seen with AAV-mediated delivery with the S. aureus CRISPR system.36 Successful editing has also been achieved in human liver chimaeric mice,121 demonstrating the feasibility of targeting the human PCSK9 gene in hepatocytes. Although the benefits of a ‘one-time treatment’ are obvious and substantial in terms of sustained LDL lowering and patient compliance, it seems unlikely that these therapies will replace statins and PCSK9 inhibitors, which can be withdrawn at will if an adverse event should arise. Still many other proteins that are not currently targetable with small molecule inhibitors may also be candidates for substrate reduction.
Metabolic pathway reprogramming
Our group recently introduced a novel concept called metabolic pathway reprogramming50 (figure 3). This therapeutic approach can be applied to inborn errors of metabolism to avoid accumulation of toxic metabolites. Using this strategy, metabolic substrates can be rerouted to non-toxic avenues by inhibiting disease-associated genes instead of editing the mutated gene. As mentioned above, in a proof-of-concept study we converted HT-I (fah−/− deficiency) into type III (HT-III) by deleting the hpd gene. HT-III is a much more benign condition and in mice, we showed that double mutated hepatocytes (fah−/−/hpd−/− ) have a growth advantage and eventually repopulate the whole liver. The reprogramming rescued both viability in the mice and all related symptoms associated with HT-I.
Tyrosinemia is currently treated by nitisinone,122 a small molecule inhibitor of HPD. However, the pharmacological block is incomplete, and although nitisinone reduces the risk of HT-I patients developing HCC, the incidence of this cancer is still significantly greater in this patient group.123 ,124 fah−/− mice treated with nitisinone also suffer an increased risk of HCC,125 but this risk disappears when the mice are crossed with hpd−/− (HT-III) mice.126 If metabolic pathway reprogramming for HT-I can eliminate the elevated risk of HCC, it might become a realistic alternative to nitisinone that could offer lasting correction. Metabolic pathway reprogramming should be applicable to many other inborn errors of metabolism, where there are currently no therapeutic options. These experimental therapies will take time to develop, and require a detailed knowledge of the pathway to be targeted.
CRISPR/Cas9-mediated correction of mutated genes
The most obvious therapeutic use for CRISPR/Cas9 is in correcting a mutated gene. Correcting a mutation by HDR necessitates a template with the wild-type sequence. The sister chromatid can serve as such a template and intragenic recombination has been described in many species. In humans, however, the evidence for intragenic recombination in somatic cells is poor127 and clonal expansion in the liver of HT-I patients cannot be explained by this phenomenon.128 ,129
The requirement of an additional element, whether single-stranded oligonucleotide or double-stranded DNA, complicates the approach from both a design and delivery standpoint. As mentioned previously, the more efficient NHEJ will compete with HDR in every cell. The efficiency of HDR is dependent on cell type and cell cycle,39 but varies considerably between different mammalian species, which is strikingly evident when comparing HDR in mouse and human embryonic stem cells.130 Therefore, we will have to await studies in primary human hepatocytes to reasonably assess the chances of success with CRISPR/Cas9-mediated HDR.
The first few studies demonstrating the feasibility of CRISPR/Cas9-mediated gene corrections in the murine liver were encouraging.63–65 Nevertheless, the efficacy of gene correction in these studies was low. Additionally, all three studies were performed in the setting of increased mitotic rates, either in the expanding neonatal liver in the spfash mice63 or in adult fah−/− mice with constantly proliferating hepatocytes.64 ,65 Therefore, genetic mutations detected shortly after birth or inborn errors of metabolism with liver regeneration such as HT-I are the best candidates for such an approach. Conceptually, however, almost all genetic liver diseases could be cured using this approach if it was possible to increase the rate of HDR by proliferation or other means (see section ‘Translational challenges of CRISPR/Cas9 genome editing’). Even if we were able to increase the rate of HDR significantly, this approach to gene correction would still likely introduce deleterious new mutations in the uncorrected alleles.
Although our focus here is on hepatocytes, the above clinical strategies could potentially be applied to disorders of non-parenchymal liver cells such as inhibiting cirroghenic factors of myofibroblasts or correcting factor VIII (haemophilia A) in liver endothelial cells.
Translational challenges of CRISPR/Cas9 genome editing
Delivery
Delivery is a challenge for virtually every macromolecular therapy. Efficient hepatic genome editing will also depend on efficient and safe delivery of CRISPR/Cas9 components into the liver. While nanoparticles have shown promising results for transient expression of Cas9 or sgRNA in the liver,64 ,131 ,132 these agents lag behind viral vectors in their clinical development. AAV vectors have been used successfully in over 120 clinical trials to date133 ,134 (http://www.clinicaltrials.gov), with only little vector-related toxicity or adverse events. Liver-directed AAV vectors are in late stage clinical trials to treat haemophilia B (factor IX deficiency), and considerable success has already been achieved with AAV2,135 and AAV8-based vectors.136 ,137 However, these clinical trials report a moderate dose-dependent immune response against the viral capsid. Therefore, reducing the dose with a more efficient AAV would be desirable.
The tropism and transduction efficiency of AAV is capsid (serotype)-dependent.138 Many AAV serotypes have been isolated from adenoviral stocks, non-human primates, or humans, and many of them evaluated in mice, dogs and non-human primates for liver-directed gene transfer.139–141 However, before extrapolating results from animal studies to humans for gene therapy, potential differences in uptake, delivery to the nucleus, uncoating, second strand synthesis of the recombinant genome and expression of the transgene must be considered. Humanised FRG mice111 ,142 ,143 provide a unique in vivo platform to further evaluate candidate AAV serotypes for transduction efficiency of human hepatocytes. Several groups have recently published their first results using this animal model,144–147 but not all groups used the same set of serotypes. Additional experimental differences make it difficult to compare between these studies. One fundamental challenge with these studies is accounting for the effects of residual murine hepatocytes, which are more easily transduced by numerous serotypes than human cells. Better model systems and approaches are needed to find the best AAV serotypes for delivery to the human liver.
There are several remaining clinical hurdles for AAV gene therapy, including avoiding pre-existing neutralising antibodies to AAV,135 ,148 managing the immune responses to the capsid and transgene product,149 and the possibility of loss of episomal AAV genomes to cell division. While these challenges are more significant for classical gene therapy using a gene addition approach, CRISPR/Cas9 genome editing does not require sustained expression. Moreover, as we and others have shown in mouse models, successful genome editing in the liver can be achieved after only a few days.50 ,63–65
Despite significant early setbacks, adenovirus may still be the best alternative to AAV for liver-directed gene therapy. Adenovirus is known to elicit a strong immune response in both mice and humans3 ,150 and most humans have immunological memory from previous exposure.151 Helper-dependent adenovirus, so-called ‘gutless’ adenoviruses, have a better safety profile and retain excellent transduction efficiency of hepatocytes in experimental animal models,152 but have not been clinically developed for use in human livers. Herpes and retroviral vectors have a poor tropism for hepatocytes and the latter requires cell division. Similarly, lentiviral vectors, even when pseudotyped with vesicular stomatitis virus glycoprotein, are usually absorbed by non-parenchymal cells in the liver.153
In summary, choosing a safe and efficient delivery method for CRISPR/Cas9 applications is challenging, particularly when strategies with different macromolecular components (protein, DNA or RNA) are combined.
Genotoxicity
The potential genotoxicity of CRISPR/Cas9 and the vectors used for its delivery is a key issue that must be addressed prior to clinical translation. Cas9 cuts DNA only at sites that are recognised by its cognate sgRNA, and has no inherent nuclease activity in the absence of a sgRNA.154 However, base pairing between the sgRNA and the target site need not be perfect, and cleavage can still occur at sites with multiple mismatches.91 ,155 Considerable computational efforts have been undertaken to improve sgRNA design, as well as predict the most likely off-target sites in the genome.156 ,157 While there are positions in the sgRNA/DNA duplex that are unfavourable to Cas9 activity, there are no universal rules that can be applied to ensure complete specificity. Validation and testing of sgRNA must ultimately be performed empirically.
Thus far, CRISPR/Cas9 seems to have a relatively low frequency of off-target mutagenesis in the liver with the S. pyogenes system. Viral delivery of SpCas9 with adenovirus produced no detectable mutagenesis of top predicted off-target sites in normal mice120 or human liver chimaeric animals.121 We detected off-target mutagenesis of 2.8% in one of the 30 predicted sites using CRISPR/Cas9 to correct HT-I in the liver.50 Most recently, we used AAV vectors for sustained expression of sgRNAs in an SpCas9 transgenic mouse model and observed an off-target 5% mutagenesis rate with an sgRNA that had an on-target efficiency of 54%.67 Ran et al identified a Cas9 ortholog from S. aureus that is small enough to be delivered within AAV vectors. This ortholog of Cas9 appears to be highly efficient in the liver, and more specific than the commonly used S. pyogenes version.36 Two studies have used the S. aureus Cas9 system in the murine liver, and neither observed detectable off-target mutagenesis at predicted sites;36 ,63 however, more extensive genomic analyses were not performed on the livers.158 ,159
While the overall specificity of CRISPR/Cas9 for liver-directed genome editing looks promising, it should be noted that more robust genome wide and unbiased methods to assess specificity in human hepatocytes are needed. As shown in murine CRISPR/Cas9 cancer models, even rare events disrupting tumour suppressor genes can trigger hepatocellular cancers.60–62
In addition to CRISPR/Cas9 itself, genotoxicity due to insertional mutagenesis of viral vectors should be considered. Adenovirus would seem to pose little risk since genomic integration in the liver is believed to be rare,160 while the significance of insertional mutagenesis of AAV is controversial. Several groups have demonstrated that delivery of AAV to neonatal mice can cause HCC161 due to insertion at the murine Rian locus,162 ,163 which is permissive to integration only early in development.162 In contrast, high-dose delivery of AAV vectors to adult mice generally does not increase the incidence of HCC,164 ,165 where integration events are estimated to occur in ∼1/588 hepatocytes.165
We know far less about the risks of AAV integration in the human setting. In 2015, Nault et al published a study reporting integrations of wild-type AAV2 genomes in liver biopsies from patients with HCC.166 The frequency of integration events was paradoxically lower in the adjacent normal tissue relative to the tumour. Nonetheless, AAV integrations were found to expand clonally in several well-established proto-oncogenes known to underlie HCC driven by HBV infection (CCNA2, CCNE1, KMT2B, TERT). There has been only one study investigating rAAV integration in subjects receiving liver-directed gene therapy, where three human liver biopsies were examined.167 Integration events were rare (<0.1%), randomly distributed and not detected at any known HCC hotspots. Given the lack of any epidemiological relationship between AAV infection and HCC, it seems unlikely that AAV will pose an inherent cancer risk, but this risk may also depend on the nature of the genetic cargo and activity of the transgene.
Most recently, we discovered integration of short Inverted Terminal Repeats (ITR) sequences at a DSB generated by CRISPR/Cas9 with AAV vectors in mouse liver.67 DSBs are known to promote AAV integration into the genome in cultured cells,168 and in the context of genome editing nucleases this is an even more important consideration. Advances in sequencing technology will continue to improve our ability to detect rare and potentially genotoxic events, and it will be important for the field to adopt uniform procedures to assess the risk of off-target genome editing and vector integration. These risks must be weighed against the tremendous potential benefit to patients.
Immunogenicity
The majority of current clinical AAV applications seek to correct genes using constitutive promoters that may be more or less active than the endogenous promoter for that particular protein. This strategy makes protein expression difficult to regulate, and risks adverse events due to immune responses or unfavourable biological effects of excess transgene. Most importantly, gene addition relies on sustained expression of the transgene to achieve the therapeutic effect, while genome engineering only requires a small window of expression to accomplish the desired genomic alteration. From then on, the changes are genetically encoded and passed down to daughter cells.
One challenge in using CRISPR/Cas9 is the possibility that bacterial Cas9 will cause an unfavourable or prohibitive immune response in humans. Recent work using S. pyogenes Cas9 to delete the tumour suppressor Pten in the liver described the development of Cas9-specific antibodies and IL-2 secretion from Cas9-primed splenocytes.169 Of note, the Cas9 was delivered by adenoviral vectors, which are known to elicit an immune response and might have served as adjuvant.170 Another similar study, using adenovirus and CRISPR/Cas9 in the liver observed that Cas9 and the viral genome were massively reduced (1%) 90 days after injection.171 In contrast to previous study, the immune response was not analysed. Interestingly, both studies report long-term persistence of high-level genome editing on their target genes, in the case of Pten with the expected hepatomegaly and nonalcoholic steatohepatitis-like symptoms. Although it could be argued that Pten deletion might lead to a selective advantage and bias the results, these studies demonstrate that an immune response may not necessarily pose a problem for genome engineering.
The question remains how this immune response can be reduced and managed in the patient setting. A less immunogenic delivery method could help, but considering the fairly quick process of genome editing and the immunogenicity of bacterial proteins, it might be worth considering short-term immune suppression on injection. The road to clinical application is long and the bacterial kingdom is enormous, so there is plenty of time to find new and hopefully less immunogenic variants of the Cas9 protein or additional programmable nucleases from other species.172 ,173 Potentially, gene therapy vectors could also be designed as self-deleting vectors harbouring one or several sgRNA target sites.
In the case of gene correction (see section ‘Medical applications for CRISPR/Cas9 in the liver’), additional and likely long-term immune response might be triggered. However, we speculate that gene correction with proper regulation may be less immunogenic than strong transgenic expression from a gene therapy vector.
Promoting HDR
One major hurdle for gene correction strategies (see section ‘Medical applications for CRISPR/Cas9 in the liver’) for the liver is that active cell division is required for HDR. As the liver is largely quiescent organ under normal and even pathological circumstances, improved efficiency for HDR events must be achieved in order for CRISPR/Cas9-mediated HDR to be a viable possibility for liver repair studies.
Several groups have used small molecule inhibitors to block components of the NHEJ pathway in vitro and in vivo. 174–178 Inhibiting DSB repair is a common strategy used in chemotherapy. However, excessive inhibition of DSB repair would impair normal cell repair processes, resulting in cell cycle arrest or apoptosis. Recently, it has also been shown that regions of heterochromatin in the DNA are difficult for Cas9 to target, and that these regions are edited with much lower efficiency than regions of euchromatin.179 This decreased efficiency could negatively impact the already low-efficiency HDR, and could hinder our ability to target genes that have a fluid chromatin state.
A potential alternative strategy to increase HDR is to use a novel CRISPR-Cas effector molecule Cpf1.172 Cpf1 is an enzyme that generates staggered 5′ prime overhang DSB rather than the blunt DSB created by Cas9. Cpf1 also cuts distal to the PAM, potentially allowing for more cycles of cutting and opportunities for HDR before the sgRNA-binding site is lost to NHEJ mutations. Using this enzyme could increase the rate of HDR, although currently there is not enough experience with Cpf1 to support its theoretical utility.
For now, the most efficient strategy, as demonstrated in mice,179 is correct genes in the immature, ideally neonatal liver, where there is more proliferation. An alternative is to choose therapeutic targets with a proliferation phenotype such as HT-I. Partial hepatectomy is also a theoretical possibility to make hepatocytes proliferate in animal models, but this strategy is unlikely to be beneficial in a clinical setting.
Summary
More than 40 years ago, the first restriction enzyme was isolated and enabled development of modern molecular biology. Now CRISPR/Cas9 genome engineering takes the next step and enables even more precise genetic manipulation in living cells and organisms. Although it has the advantage that there is no need for sustained gene expression—a major limitation of traditional gene therapy (gene addition)—there is still concern about specificity and off-target cutting, which could result in genetic alterations that lead to cancer or other problems.
The CRISPR/Cas9 technology has rapidly conquered laboratories across the world and has generated a huge body of research, including the first preclinical therapeutic studies. Progress has already been made in animal models of ornithine transcarbamylase deficiency and HT, and work treating other inborn errors of metabolism is not far behind. We believe that liver disorders are particularly amenable to CRISPR/Cas9 genome editing and that the first clinical applications are close at hand.
Acknowledgments
We would like to thank Paul Overbeek, Vicky Brandt and Catherine Gillespie for careful review of the manuscript. The authors apologise to those authors whose work was not cited owing to space limitations. K-DB is supported by the National Heart Lung and Blood Institute (NHLBI) grant R01HL134510, the Texas Hepatocellular Carcinoma Consortium (THCCC) (CPRIT #RP150587) and the Diana Helis Henry and Adrienne Helis Malvin Medical Research WRL is supported by the NHLBI grant HL132840, FPP was supported by T32HL092332 and KEJ by T32HL07676.
References
Footnotes
Contributors All authors have contributed, written and accepted final version of the manuscript.
Funding National Heart and Lung Institute (HL132840, R01HL134510, T32HL07676 and T32HL092332) and Cancer Prevention and Research Institute of Texas (RP150587).
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.