Article Text

Abstract
The ‘IN’ of chronic inflammation—that is, the mechanisms of cell entry into the intestinal mucosa, bacterial and foreign antigen invasion, angiogenesis, and the control of gut inflammation through intestinal microvasculature—has received a great deal of attention in studies of the pathogenesis of inflammatory bowel disease (IBD). This has resulted in the validation of several targets for the treatment of experimental inflammation—both on immune and non-immune cells—some of which have translated into effective treatments for patients with IBD. An important aspect of this has been our growing understanding of the role the intestinal vascular microcirculation plays in the initiation and perpetuation of the inflammatory process, by regulating the migration of leucocytes into the interstitial space. However, it is becoming increasingly clear that it is also important to focus on the ‘OUT’ of chronic inflammation—that is, the lymphatics and their role in controlling tissue oedema, leucocyte exit, bacterial antigen and inflammatory chemokine clearance. As our understanding of the lymphatics and the role they play grows, another rich source of non-immune cell targets for therapeutic intervention is gradually being revealed. This article describes current knowledge of the roles played by the vascular and lymphatic endothelium throughout the gut in the pathogenesis of IBD, and how this differs from their role under physiological conditions, as well as discussing current and future therapeutic targets that have been identified.
- Endothelial cells
- inflammatory bowel disease
Statistics from Altmetric.com
Introduction
The study of inflammatory bowel diseases (IBD) has largely focused on the roles played by the classical components of the immune system, a unidirectional viewpoint that has dominated the study of Crohn disease (CD) and ulcerative colitis (UC), the two major forms of IBD. However, as the emphasis of studies has broadened to encompass environmental factors, intestinal microbial flora, the tissue response and the underlying genetics, an increasing number of reports have called attention to the significant contribution of non-immune cells such as epithelial, endothelial, mesenchymal, nerve and vascular cells, platelets and the extracellular matrix to the pathogenesis of IBD.1–3 These cell types are not only able to perform many of the functions traditionally attributed to classical immune cells,4 but they also secrete or express on their surface molecules involved in the immune response, particularly those of innate immunity.2 5–8 Indeed, it is becoming clear that there is a highly integrated network of immune and non-immune cells, involving many different types of interaction.1–9 This adds a further layer of complexity to our current understanding of the mechanisms underlying the pathogenesis of IBD.
An extensive body of research has demonstrated that the intestinal vascular microcirculation plays a central role in both the initiation and perpetuation of the inflammatory process. Its ability to regulate both the type and number of leucocytes that migrate into the interstitial space places it in a unique and key position to govern the infiltration of leucocytes into the gut.10–12 During inflammation, the phenotype of activated vascular endothelial cells (VECs) includes leakiness, leucocyte adhesiveness and procoagulant activity, and, further down the line, angiogenesis.13 14 However, while research has devoted a lot of attention to the investigation of the mechanisms of inflammation and how leucocytes arrive in the inflamed tissue, the mechanisms that mediate the resolution of inflammation, including the exit of leucocytes from the inflamed tissue into the lymphatics, have only recently begun to be elucidated.15
Although the role played by the lymphatic endothelium has not received the same level of attention as that of the vascular endothelium, the very recent development of techniques to isolate and culture lymphatic endothelial cells (LECs) in vitro has enabled significant progress in the study of the lymphatics by allowing the characterisation of LECs. Unique markers that differ from VECs have been identified, as well as mechanisms through which they are able to mediate post-inflammatory clearance of both leucocytes and chemokines. However, few data have been published thus far specifically describing LECs in the gut, either under physiological conditions or in the presence of inflammation.16 17
In this review, I describe the current knowledge of the roles played by the vascular and lymphatic endothelium throughout the gut in the pathogenesis of IBD, and how this differs from their role under physiological conditions. I will also discuss the potential of the two endothelial systems as distinct and disparate key targets for therapeutic intervention.
Microvascular endothelium
The highly specialised cellular system that forms the intestinal microvasculature performs a wide range of biological tasks that are the crucial underpinning of multiple physiological processes, such as blood flow, the flow of nutrients, tissue homoeostasis, and cell trafficking and distribution. In addition, the vascular endothelium has also been implicated in pathological processes such as inflammation. Studies of the functions performed by VECs have been significantly advanced by the availability of cultured human umbilical vein endothelial cells (HUVECs). However, it has also become clear that VECs have many tissue-specific characteristics, and stress and inflammatory stimuli can change the expression patterns of adhesion molecules, activate unique sets of genes, and trigger distinct chemokine secretory patterns.18 19 This tissue specificity of VECs limits the information that can be gained from the study of HUVECs.
For the study of VECs in the mucosa of the gut, this deficiency drove the development of protocols for routine isolation and long-term culture of pure populations of human intestinal microvascular endothelial cells (HIMECs).19 The importance of this development to the study of intestinal inflammation was exemplified by the demonstration of unique patterns of leucocyte adhesion and growth for HIMECs compared with HUVECs. This demonstration also supports the belief that tissue-specific mediators and transcription factors contribute to the induction or maintenance of a specific tissue VEC profile.11 19–21
Leucocyte recruitment
Recruitment of leucocytes from the vascular circulation into inflamed tissues is an important step in the inflammatory response which is entirely regulated by the microvascular endothelium. In response to activation by cytokines and other inflammatory mediators, the microvascular endothelium expresses cell adhesion molecules (CAMs) and chemokines that enhance interactions with leucocytes and their subsequent recruitment.12 22 23 The leucocyte extravasation cascade involves multiple steps, including tethering/rolling, activation, adhesion, spreading and transmigration, and has been extensively reviewed elsewhere.24–26 Many in vitro and in vivo studies have focused on the contribution of molecular elements at each step of the extravasation cascade to the pathogenesis of IBD.27
Leucocyte homing to the gut is mainly mediated by the CCR9 chemokine receptor, which binds to CCL25 on the surface of the ileal endothelium.28–30 Subsequent adhesion is then supported by the binding of integrins expressed on the surface of the leucocytes to the CAMs expressed on endothelial cells (figure 1). Indeed, adhesion is the crucial event that determines which leucocytes will ultimately migrate into the tissues. In the gut, this is mainly mediated by CD11a/CD18 on the leucocytes binding to intercellular adhesion molecule (ICAM)-1, or by binding of α4β1 or α4β7 to vascular cell adhesion molecule (VCAM)-1 and mucosal addressin cell adhesion molecule (MAdCAM)-1.23–27 The importance of this pathway is reflected in the fact that it has been a rich source of targets for therapeutic intervention. Indeed, one drug targeting this pathway has been approved by the Food and Drug Administration for the treatment of moderate-to-severe CD: natalizumab is a recombinant humanised IgG4 directed against a4 integrins. New drugs that target leucocyte–endothelial interactions are now under development, including vedolizumab (a humanised antibody to α4β7 integrin), PF-547659 (a monoclonal antibody to MAdCAM-1), rhuMab β7 (an anti β7 integrin) and CCX282-B (an oral CCR9 inhibitor) (figure 1).

Cell adhesion molecules involved in the multistep paradigm of leucocyte recruitment, and therapeutic targets that have been developed or are under development for the treatment of patients with inflammatory bowel disease. ICAM-1, inter cellular adhesion molecule-1; VCAM-1, vascular cellular adhesion molecule-1; MadCAM-1, mucosal addressin cellular adhesion molecule-1; VLA, very late antigens.
In patients with IBD, the homing of leucocytes to the microvasculature is profoundly altered. Indeed, HIMECs isolated from chronically inflamed areas of the intestine of patients with IBD have a significantly enhanced capacity to adhere leucocytes compared with HIMECs either from uninflamed areas from the same patients or from controls.19 31 In accordance with this, in the intestinal microvasculature of patients with IBD, there is enhanced expression of the gut-specific homing model MAdCAM-1, which plays a major role in the recruitment of the gut-specific α4β7 integrin-expressing leucocytes into the mucosal immune compartment.32 33 In addition, increased expression of ICAM-1 and VCAM-1 further ensures continuous recruitment of leucocytes to the gut.34
Characterisation of HIMECs has demonstrated that other molecules are also specifically upregulated in the endothelium of patients with IBD. Fractalkine is a unique CX3C chemokine that acts as an adhesion molecule and is upregulated in the endothelium of patients with IBD.35 Moreover, patients with IBD have greater numbers of circulatory T cells that express the CX3C chemokine receptor CX3CR1 than controls.35 Fractalkine could therefore be an important mediator of interactions between VECs and leucocytes under conditions of intestinal inflammation.
Finally, intestinal endothelial cells also express CD40, an important immune costimulatory molecule for T cells and platelets, which express CD40 ligand (CD40L).34 36 In the endothelium of the mucosa of patients with IBD, CD40 is activated by binding to CD40L, inducing the cells that express CD40L to produce inflammatory chemokines such as RANTES (regulated upon activation, normal T cell expressed, and secreted) and cell adhesion molecules, fostering and amplifying intestinal inflammation.37–40 Furthermore, leucocyte and platelet recruitment to the inflamed intestine is decreased in mice deficient in CD40 and CD40L. This indicates that the CD40–CD40L signalling pathway could be a relevant target for therapeutic intervention to inhibit the recruitment of leucocytes to the inflamed intestine.36–38 40 41
In addition to studying specific molecular pathways, mechanisms that lead to aberrant activation of endothelial cells are also now being characterised. In addition to the proinflammatory cytokines that enhance the expression of ICAM-1, VCAM-1 and MAdCAM-1, several other mediators have recently been described. For instance, nitric oxide (NO) is an inflammatory mediator for which a role in IBD has clearly been established.42 Generation of NO was therefore investigated in HIMECs, to determine whether this alternative pathway also influences the activation of VECs in the gut, including their capacity to bind circulating leucocytes. Generation of NO occurred in control HIMECs through both constitutive endothelial NO synthase (eNOS or NOS3) and inducible NO synthase (iNOS or NOS2); however, expression of the mRNA for iNOS was lost in HIMECs derived from patients with IBD, which corresponded to a decrease in NO generation and enhanced leucocyte binding.43 44 Finally, an increase in arginase activity is also observed in HIMECs exposed to an inflammatory milieu, which may contribute to the decrease in production of NO.45
HIMECs are also activated by homocysteine, a known player in microvascular inflammation in patients in IBD. Activation of VECs by homocysteine alone or in combination with tumour necrosis factor (TNF)α (which synergises with homocysteine), upregulates expression of VCAM-1, production of monocyte chemotactic protein-1 (MCP-1), and phosphorylation of p38 mitogen-activated protein kinase (MAPK).46
The MAPK signal transduction pathways are important players in a variety of cellular processes, including regulation of expression from genes for CAM and cytokine production. Scaldaferri et al have recently studied the functional role of the MAPK p38, p42/44 and JNK in the regulation of lymphocyte extravasation. The phosphorylation levels of MAPK are higher in both the fibroblasts and endothelium of the mucosa of patients with IBD.47 They also play an important role in regulation of expression of CAMs, chemokine production by HIMECs, and extravasation of lymphocytes into the tissue.47
Given the important role they play in processes central to the development and maintenance of chronic inflammation, MAPK inhibitors could be potentially important targets for anti-inflammatory therapeutics.48 Indeed, potent anti-inflammatory activity has been demonstrated for several MAPK inhibitors. For example, pyridinyl imidazole compounds, which can reduce synthesis of inflammatory cytokines,48 are selective inhibitors of p38 MAPK.48 In addition, a potent inhibitor of JNK/p38 MAPK, CNI-1493 (a synthetic guanylhydrazone), has shown clinical benefit and mucosal healing in clinical trials in patients with severe CD.49 However, several side effects related to liver and central nervous system toxicity have been observed in clinical trials with MAPK inhibitors, as a result of which the development of these agents has been suspended. Given that p38 MAPK plays such a pivotal role in processes other than inflammation, it is not surprising to find that their systemic use results in undesirable side effects. On the other hand, local inhibition of MAPK activation in mucosal endothelial cells of patients with IBD could specifically inhibit the aberrant homing of leucocytes to the gut. This promising possibility warrants the investigation of drug carriers that can specifically target MAPK inhibitors to inflamed tissues in such patients.
Studies of these pathways have already revealed many potential, as well as some actual, targets for therapeutic intervention, and it is likely that many more will be revealed.
Angiogenesis
Chronic inflammation is always accompanied by angiogenesis, the growth of new blood vessels, and indeed it is becoming clear that angiogenesis and inflammation are intertwined processes.50 Extensive angiogenesis and remodelling of the microvasculature are intrinsic to the tissue remodelling that occurs in inflamed intestine in patients with IBD.51 52 Although angiogenesis and remodelling are distinct phenomena that occur in response to different triggers, they often occur together in the tissues of patients with IBD, and both involve proliferation of endothelial cells.
The processes of inflammation and angiogenesis are linked on a number of levels. Hypoxia in inflammatory tissues is an important proangiogenic stimulus, as it upregulates angiogenic factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), TNFα and hypoxia-inducible factor 1.51 52 In addition, extravasated plasma fibrinogen is involved in stimulation of neovascularisation.51 52 Inflammatory cells, such as macrophages, lymphocytes, mast cells and fibroblasts, produce diverse angiogenic factors that stimulate vessel growth.51 Activated platelets have also recently been identified as potent inducers of intestinal angiogenesis,53 and shear stress on the endothelium due to increased blood flow may stimulate angiogenesis.54 Finally, recently, an active contribution of the bacterial flora to the induction of VEGF-A and angiogenesis has been demonstrated.55 56
Initially, the prevalent changes to the vascular endothelium are functional, including dilation, increased permeability, activation of the endothelium and diapedesis. The next step is structural changes, with capillary and venule remodelling and proliferation of endothelial cells.57 Indeed, in chronic inflammatory disorders, tissue damage and repair continue concurrently.58 With time, the endothelial cells in the inflamed capillaries respond to locally produced angiogenic factors and start to multiply, forming permanent remodelled vessels.58 This anatomical expansion of the microvascular bed combines with its increased activation state to foster further influx of inflammatory cells, a cyclical step that results in chronic co-dependence of angiogenesis and inflammation.
VECs that participate in the angiogenic process display an activated cell surface molecular pattern not found on resting vessels, with altered expression of endothelial adhesion molecules,57 58 and increased concentrations of soluble adhesion molecules24 observed in intestinal biopsy specimens from patients with IBD. Angiogenic vessels can be identified through the expression of specific integrins, in particular αvβ3 and αvβ5; several receptors for angiogenic factors are also measurably upregulated.6 55
Neoangiogenesis has been investigated in patients with CD and UC by quantifying the mucosal vascularisation state, assessing local expression of markers of angiogenesis, and determining the presence of functional proangiogenic activity in inflamed tissue from patients with IBD.59 The mucosal vascularisation state was quantified by counting the number of vessels that were positive for the vessel marker CD31. A significant increase in the number of vessels was found in tissues from affected areas of patients with IBD compared with controls, which suggests that active angiogenesis has taken place.59
Expression of the neoangiogenic marker αvβ3 by the mucosal microvasculature was most prominent in inflamed areas of the mucosa. Moreover, αvβ3 was upregulated in cultured HIMECs that were exposed to proinflammatory and proangiogenic factors that are overexpressed in inflamed tissue from patients with IBD—that is, TNFα, VEGF, interleukin (IL)-8, and basic FGF (bFGF), which probably act in a complementary fashion.59 Along similar lines, the novel marker of endothelial junction remodelling, CD146, is highly expressed on VECs in intestinal biopsy specimens from patients with IBD, which is associated with decreased concentrations of the soluble form of CD146.60 In addition, increased serum and/or tissue concentrations of several proangiogenic factors, such as bFGF, VEGF-A, angiogenin, angiopoietins, substance p and corticotrophin-releasing hormone, have been reported in patients with active IBD and in experimental colitis.61–69 Indeed, mucosal extracts from patients with IBD exhibited an augmented capacity to induce dose-dependent migration of HIMECs, suggesting that the angiogenic-enriched mucosa in patients with IBD is functionally active.
The extent and complexity of the network of signals in which endothelial cells and their angiogenesis is involved is exemplified by the critical role of the CD40–CD40L pathway in immune-driven angiogenesis.34 70 In addition to the direct role that soluble CD40L plays in mucosal angiogenesis,70 inflammation-activated CD40L-expressing T cells may activate intestinal fibroblasts, causing them to release angiogenic cytokines that activate HIMEC angiogenesis. Indeed, mice that are deficient in CD40 or CD40L have impaired, pathological angiogenesis in the gut.70 This would point to a dual mechanism underlying CD40-dependent angiogenesis in the inflamed gut.
The complexity of the interactions between inflammation and angiogenesis in experimental colitis is underlined by the overlap between some common mediators. For instance, the angiogenic factor, VEGF-A, can induce expression of CAMs on the intestinal endothelium and promote adhesion of leucocytes in a similar manner to more classical inflammatory agents such as TNFα.71 The importance of angiogenesis to the pathophysiology of experimental colitis has been demonstrated in both the dextran sodium sulfate (DSS)-induced and CD4+CD45RBhigh T cell transfer models of colitis.72 Upregulation of proangiogenic mediators such as matrix metalloproteinase 2 and 9, endothelial sphingolipid G-protein-coupled receptor 1, endoglin, prostaglandin-endoperoxide synthase 2, TNFα, chemokine (CXC) ligand 1, and hepatocyte growth factor, as well as downregulation of some antiangiogenic factors, including CD36 antigen and chromagranin A, was observed in both colitis models. Interestingly, the authors found differential regulation of numerous angiogenic, antiangiogenic and angiostatic genes between these two models, suggesting that angiogenesis occurs through different mechanisms in the two models: loss of angiogenic inhibition in the DSS model, and upregulation of proangiogenic mediators in the CD4+CD45RBhigh model. Both of these mechanisms could be potential sites for intervention for selective treatment of various forms of IBD.72
Indeed, angiogenesis has already proven to be an effective therapeutic target for the treatment of experimental colitis. In the IL-10 knockout model of colitis, blockade of αvβ3 effectively ameliorated colitis and decreased the production of inflammatory cytokines.73 Similarly, inhibition of VEGF-A was effective in both the DSS-induced74 and trinitrobenzenesulfonate (TNBS)-induced75 models of colitis, improving histological inflammation and inhibiting mucosal cytokine production in both.
Several of the factors involved in pathological angiogenesis in chronically inflamed tissues are regulated at specialised lipid rafts known as caveolae. The major structural protein of these caveolae, caveolin-1 (Cav-1), is involved in the regulation of angiogenesis,76 and its expression is increased after the induction of DSS colitis. In Cav-1(−/−) mice or mice administered a Cav-1 inhibitory peptide, the colitis histopathology scores, vascular densities and concentrations of inflammatory infiltrates were significantly decreased after induction of colitis compared with wild-type mice. In addition, lower concentrations of leucocyte and platelet rolling and adhesion colitis were observed in these mice compared with wild-type mice. Interestingly, Cav-1(−/−) mice that received transplants of wild-type bone marrow had a lower colitis score than wild-type mice, while disease was not attenuated in wild-type mice that received Cav 1(−/−) bone marrow. In addition, mice that overexpress Cav-1 only in the endothelium developed disease similar to that of wild-type mice. These data indicate that endothelial Cav-1 may play a critical role in the regulation of colitis. In addition to its effects on disease severity, specific deletion or blockade of endothelial Cav-1 decreased vascular densities and angiogenesis scores.76
Natural antiangiogenic molecules such as thrombospondin-1 (TSP-1) have also consistently been found to control colitis-associated angiogenesis. Indeed, TSP-1 knockout mice have increased susceptibility to DSS-induced colitis, whereas blockade of TSP-1 decreases disease severity, thus reinforcing the therapeutic potential of targeting angiogenesis for colitis,77 78 as well as specifically implicating TSP-1 as a novel target for the treatment of IBD.
However, the translational therapeutic implications of these findings are limited by the consideration that angiogenesis is an essential aspect of the tissue healing process, and therefore any therapy that targets angiogenesis may have potentially serious side effects. Indeed, the colon cancer drug, bavacizumab, which targets human VEGF, induces ulcers and gastrointestinal bleeding as a deleterious side effect. A case report that described administration of bavacizumab to a patient with UC also reported this side effect.79 In addition, administration of the antiangiogenic sorafenib (a receptor tyrosine kinase inhibitor that inhibits VEGFR1, 2, 3) to a patient with UC also exacerbated the disease.80
Despite the promising data on the potential for blockade of angiogenesis, profound alteration of wound healing through inhibition of angiogenesis could be deleterious for IBD depending on the stage of disease. This has been highlighted by a study in which knockout of the VEGF homologue placental growth factor significantly worsened disease severity and morbidity in mice with DSS- or TNBS-induced experimental colitis. Furthermore, angiogenesis was significantly decreased compared with wild-type mice in which colitis had been induced.81
Nonetheless, potentially promising effects of promotion of wound healing through stimulation of angiogenesis were suggested by a study of the probiotic Bacillus polyfermenticus. When HIMECs were exposed to B polyfermenticus conditioned medium, cell migration, permeability and tube formations were increased, as well as production of IL-8. In addition, inoculation of mice with B polyfermenticus facilitated their recovery from colitis, as well as increasing angiogenesis and production of IL-8 in the mucosal layer. B polyfermenticus therefore promotes angiogenesis in the mucosa during recovery of mice from colitis, suggesting clinical utility of this probiotic for intestinal wound healing.56
These data indicate that the potential utility of inhibitors of angiogenesis may be specific to the stage of disease, but that the risks of targeting angiogenesis should be carefully evaluated when considering whether preclinical findings can translate into clinical utility.
Angiogenesis is a constant feature in both human and experimental IBD
Many inflammatory mediators and cell types promote pathological angiogenesis
Blockade of angiogenesis can be therapeutically beneficial
Since angiogenesis is also relevant for tissue healing, its blockade could lead to mucosal ulcers depending on the disease stage
Coagulation
That IBD is associated with both a hypercoagulable state and a prothrombotic condition has been demonstrated in the clinic and by laboratory researchers. Indeed, coagulation abnormalities are intrinsic to IBD, while a significant proportion of the morbidity and mortality in patients with IBD is caused by thromboembolic disease.5 This haemostatic risk appears to be unique to IBD among the chronic inflammatory diseases, as no increased risk has been observed in patients with rheumatoid arthritis and other chronic bowel diseases, such as coeliac disease.
Besides thromboembolism, coagulation plays an essential role in inflammation, as they are closely linked and interdependent processes.5 82 Under physiological conditions, the tissue microcirculation rests in an anticoagulant and anti-inflammatory state. However, when inflammation occurs, coagulation is activated and participates in the spreading of inflammation. Recently, novel and unexpected roles of haemostasis in the humoral and cellular components of immunity have been described.5 82 For instance, platelets, which are typically considered as simply coagulative cells, play a role in the microcirculation of patients with IBD by fostering inflammation.83
The mucosa of patients with IBD also shows signs of both coagulation abnormalities and thromboembolic complications. This is exemplified by the fact that one of the earliest abnormalities that has been identified in the mucosa of patients with CD is the presence of platelet thrombi cross-linked with fibrin in the mucosal microvasculature.84 Other such crucial changes to the mucosal microvasculature have been identified in patients with IBD, including vascular injury, focal arteritis, fibrin deposition, micro-infarction and neoangiogenesis in patients with CD,85 as well as intracapillary clots in rectal biopsy specimens of patients with UC.86 Finally, injury and the resulting disruption of the endothelium could expose the subendothelial matrix, to which platelets are strongly attracted, further promoting the formation of micro thrombi.
Indeed, thrombi that adhere to the endothelium are easy to find in the microcirculation of patients with either UC or CD, using confocal microscopy. Both circulating and mucosal platelets express activation markers such as P-selectin and CD40L, and are able activate HIMECs in a CD40-dependent manner, resulting in increased surface expression of ICAM-1 and VCAM-1, enhanced production of IL-8, and increased adhesion of leucocyte endothelial cells. In addition, the activated platelets release many factors that probably play a role in colonic inflammation: RANTES binds to the endothelial cell surface to further promote leucocyte recruitment, while soluble CD40L, histamine, platelet activator factor (PAF) and cationic proteins may contribute to endothelial activation, barrier dysfunction and increased vascular permeability.36 87
A critical role for platelets has also been demonstrated in experimental animal models of colitis. DSS-induced colonic inflammation is marked by accumulation of platelets in colonic venules which coincides with enhanced adherence of leucocytes and worsening disease activity.87 Of these platelets, ∼20% are bound directly to endothelial cells, while ∼80% are attached to the surface of adherent leucocytes.88 After induction of colitis in mice with DSS, the expression of CD40 is increased on endothelial cells in the colonic microcirculation, while deficiency of either CD40 or CD40L attenuates leucocyte–endothelial cell adhesion.41 Platelet-associated CD40L (or its circulating soluble form) may also mediate interaction of platelets with both adherent leucocytes and the endothelium, as depletion of platelets significantly reduces the recruitment of leucocytes to the inflamed gut.
Another important receptor–ligand pair, P-selectin and P-selectin glycoprotein ligand-1 (PSGL-1), is also affected by induction of colitis with DSS. Expression of P-selectin is increased on the surface of platelets and endothelial cells, while PSGL-1 is increased on endothelial cells and leucocytes. The importance of the P-selectin interaction with PSGL-1 is reinforced by the demonstration that immunoblockade or genetic deletion of either significantly reduces the recruitment of both platelets and leucocytes after stimulation with DSS87 88 P-selectin on the surface of platelets also mediates binding to leucocytes, which constitutively express PSGL-1 in their surface; the overall increase in leucocyte adhesion therefore further serves to recruit platelets.89
Beside platelets, another major system that bridges inflammation and coagulation is the protein C (PC) system,90 which both participates in and controls mucosal microvascular inflammation in patients with IBD.91 92 The expression of the anticoagulant, thrombomodulin, and endothelial PC receptor is dramatically downregulated in microvessels of patients with IBD.92 93 Similarly, surface expression of thrombomodulin and endothelial PC receptor on HIMECs is downregulated by the inflammatory mediators, TNFα and IL-1β.92 In addition, the activation of PC that occurs constitutively under resting conditions in HIMECs is inhibited by stimulation with these proinflammatory cytokines.
On the other side of this equation, PC can also be potently anti-inflammatory, inhibiting the ability of TNF to upregulate CAMs and the secretion of chemokines from HIMECs. Activated PC was potently anti-inflammatory in cultured HIMECs, downregulating cytokine-dependent CAM expression and chemokine production, and inhibiting adhesion of leucocytes. In mice with DSS-induced colitis, activated PC ameliorated disease, with reduced weight loss, disease activity index, and histological colitis scores, as well as inhibition of leucocyte adhesion to inflamed intestinal vessels.92
In an experimentally induced model of extraintestinal thrombosis, the application of DSS enhanced thrombus formation. However, this could be attenuated by either the administration of activated PC, or overexpression of the endothelial PC receptor, while an antibody to activated PC enhanced thrombus formation. These data indicate a protective effect of activated PC against extraintestinal thrombosis, and further underline the complex link between inflammation and thrombosis.94
These data suggest that the PC pathway is functionally impaired in the mucosal microcirculation of patients with IBD, and demonstrate the overall importance of the coagulation cascade in intestinal inflammation. Restoring the PC pathway may represent a new therapeutic approach to suppressing intestinal inflammation in IBD, and is also under investigation in several forms of tissue inflammation including asthma, rheumatoid arthritis and atherosclerosis.90
A further example of overlap between inflammation and coagulation is represented by the procoagulant molecule tissue factor. Its expression is increased in the microvasculature of the inflamed mucosa, closely correlating with the degree of thrombosis in patients with CD.84 In mice with DSS-induced colitis, antibody blockade of tissue factor prevented several aspects of both coagulation and inflammation, including prevention of the increase in thrombin–antithrombin complexes, reduced leucocyte and platelet recruitment, and tissue injury, as well as blunted thrombus formation.95
Taken together, the above evidence suggests that targeting classically recognised coagulative molecules has a strong impact on inflammation and that coagulation should therefore be actively pursued as a potential target for translational therapy in IBD.
Coagulation and inflammation share multiple pathways and are both activated in patients with IBD
Classical coagulative cells, such as platelets, and coagulative pathways, such as the protein C pathway and tissue factor, promote intestinal inflammation
Modulation of coagulative cells and coagulative molecules could offer an entirely new way to treat IBD
Innate immunity
The innate immune response is triggered by the binding of pathogen-associated molecular patterns to ubiquitously expressed cell-surface pattern-recognition receptors (PRRs). The best studied of the PRRs are the toll-like receptors (TLRs), which can trigger the expression of proinflammatory genes, leucocyte chemotaxis, phagocytosis and cytotoxicity, as well as activating the adaptive immune responses.96 It has clearly been established that PRRs are expressed on the surface of cells of the immune system. However, more recently, evidence has started to accumulate that indicates that expression of PRRs on the surface of non-immune cells, including VECs, plays an important role in the innate immune response. For example, the accumulation of neutrophils may depend on TLR4 expressed on the surface of VECs, rather than leucocytes, as sequestration of neutrophils in the lung is deeply impaired in mice that lack expression of TLR4 in the endothelium.97 However, the latter observation contrasts with the significant decrease in leucocyte binding caused by lipopolysaccharide (LPS) in HIMECs. This may reflect tolerance of HIMECs to high concentrations of endotoxin, which they are constantly exposed to in the gut microenvironment.98
Expression of TLRs on the surface of VECs is upregulated by vascular inflammation, as well as more specifically by LPS.99 However, the expression patterns of different TLRs vary depending on the origin of the VECs. Expression of the bacterial flagellin receptor, TLR5, and the double-stranded RNA receptor, TLR3, has been specifically demonstrated on the surface of HIMECs.100 101 Indeed, TLR3 on the surface of HIMECs can modulate the expression of IL-12 and related molecules by these cells.101 Activation of TLR5 expressed on the surface of dermal VECs upregulated expression of ICAM-1, although this was not shown in HIMECs.100
Other PRRs involved in innate immunity include the nucleotide-binding oligomerisation domains (NODs) 1 and 2, cytosolic proteins that regulate inflammation in response to microbial peptides.102 Both NODs are expressed on the surface of VECs and are upregulated in response to LPS and proinflammatory cytokines.
The response of HIMECs to PRR ligands has been compared to that of monocytes and dendritic cells. Production of IL-6 and IL-8 by HIMECs was upregulated to a comparable extent to, or greater extent than, the immune cells. HIMECs were also shown to produce MCP-1, RANTES and VEGF, as well as to upregulate ICAM-1 and to enhance the adhesiveness of leucocytes. All of these responses were mediated through activation of NF-κB activation and phosphorylation of p38 and Erk1/2 MAPKs. Interestingly, while the monocytes and dendritic cells showed tolerance after repeated doses of the PRR ligands, with decreased production of IL-8, no evidence of tolerance by the HIMECs was observed, with IL-8 production remaining high. The production of IL-8 by HIMECs was further augmented after pretreatment with TNF, indicating sensitisation to bacterial products.103
In apparent contrast, Ogawa et al found that HIMECs developed tolerance in response to pretreatment with endotoxin.98 Indeed, leucocyte adhesion in response to LPS was significantly attenuated after pretreatment with LPS for 24–48 h. However, in accordance with the study by Scaldaferri et al,103 although LPS pretreatment inhibited expression of E-selectin, VCAM-1, IL-6 and CD86, the expression of ICAM-1, IL-8 and HLA-DR, on the other hand, were not altered.98
Taken together, the above data suggest that the intestinal endothelium is also actively involved in innate immunity, acting as a second checkpoint for bacterial entry. This task could be particularly important in the prevention of dissemination of pathogens and bacteria in patients with IBD, where epithelial permeability is increased.104
The intestinal endothelium is involved in innate immunity by expressing TLR and nod like receptors (NLR)
Because of high exposure to bacterial content, the intestinal endothelium is a key second checkpoint for bacterial entry
Stimulation of its ability to prevent bacterial entry could prevent dissemination of pathogens and bacteria in patients with IBD, where epithelial permeability is increased
Lymphatic endothelium
The two main tasks performed by the lymphatic vasculature are (1) the drainage of activated inflammatory cells into the draining lymph nodes (DLNs) and (2) complementing the vascular network by transporting extravasated fluid unidirectionally from tissues back to the blood circulation,15 105 including drainage of inflammatory elements from inflamed tissues. Mediation of leucocyte exit and clearance of chemokines by the lymphatic vasculature also prevents the development of oedema under physiological conditions.15 106 These functions underlie the fundamental role that the lymphatic vasculature plays in immunity,17 105 107 108 as acquired immune responses are triggered by migration of lymphocytes and antigen-presenting cells through peripheral lymphatic capillaries and into the DLNs.17 107 108
Given the essential role played by the lymphatics in the resolution of inflammation, it is possible to envisage a critical role for dysfunction of the lymphatic vasculature in the development and/or maintenance of diseases of chronic inflammation such as IBD. However, investigation of a role for the lymphatics at a molecular and cellular level has only recently gained attention, and such a role has yet to be clearly elucidated. It is therefore noteworthy that, since as early as the 1930s, pathologists have reported that the fundamental alteration in the mucosa of patients with CD is consistent with chronic lymphangitis.16 Then in the 1970s it was demonstrated that obstruction of the lymphatics of the small intestine in rats and pigs could generate fistulising intestinal disease, which shared many characteristics with CD16 109 110; indeed these models are felt to more closely resemble CD in humans than any generated subsequently.
Recently the lymphatic system has been identified as a critical controller of experimental colitis and inflammation-associated colon cancer through the decoy receptor D6. Expression of D6 is upregulated on the lymphatic endothelium and intestinal leucocytes of patients with IBD, with a further marked increase in patients with IBD-associated colon cancer.111 D6 is involved in the post-inflammatory clearance of β-chemokines, and deficiency of D6 results in greater inflammation and accumulation of CC chemokines in DSS colitic mice in which D6 had been knocked-out than in wild-type mice; this was also associated with increased leucocytic infiltration. The protective effect of D6 was found to be specifically a function of D6 expression, as disease severity was unchanged in mice deficient in D6 on the lymphatics but with wild-type leucocytes, while mice in which the lymphatics were wild-type for D6, but lacking expression on haematopoietic cells developed disease of the same severity as wild-type mice. These results therefore support a role for the lymphatic system in the control of intestinal inflammation through the decoy receptor D6.
Although researchers have only recently begun investigating the pathogenic role of the lymphatic vasculature in IBD, in other forms of tissue chronic inflammation, such as psoriasis and rheumatoid arthritis, the lymphatic system has received much more attention. For instance, in a recent study, Kataru et al demonstrated expansion of lymphatic vessels in a bacterial pathogen-induced model of acute skin inflammation, which was associated with significantly increased migration of macrophages and other inflammatory cells into the DLNs, as well as an overall increase in the lymph flow.106 There was concomitant upregulation of the lymphangiogenic factors, VEGF-A, -C and -D, blockade of which attenuated the lymphangiogenesis, migration of inflammatory cells, and antigen clearance. On the other hand, migration of inflammatory cells to the DLNs and resolution of inflammation were enhanced in VEGF-C transgenic mice.106 These findings clearly indicate the importance of the lymphatic vasculature in the resolution of skin inflammation, and it is likely that similar events could also occur in the gut.
In inflammation, the lymphatic system undergoes intense expansion through lymphangiogenesis.105 Lymphangiogenesis occurs in chronically inflamed tissues, such as in the inflamed synovium, psoriatic skin, kidney transplant undergoing rejection, the intima of atherosclerotic lesions, and the lung or corneal inflammation.15 105 112 113 This is true also in patients with IBD, although there are only limited data available regarding the involvement and regulation of lymphangiogenesis, with a few descriptions of aberrant increases in the lymphatic vasculature in patients with CD and UC.114 115 A significant increase in lymphatic vessels has been observed in patients with active UC compared with treated UC, in particular involving extension of lymphatic vessels into areas where they are not normally found under physiological conditions, such as the lamina propria.114 The increase in staining for lymphatic vessels was also found to be similar in patients with CD compared with UC.116
A comprehensive analysis of the lymphatic system and its functional role in the pathogenesis of IBD is therefore needed, as well as the therapeutic relevance of its blockade or stimulation. Indeed, a recent paper has reported functional changes in the lymphatics in the TNBS-induced model of ileitis in guinea pigs. The investigators found that the lymphatic function of isolated vessels was impaired in colitis compared with control guinea pigs.117 A correlation between the degree of inflammation and the functional impairment was demonstrated. This manifested in vivo, with a decrease in spontaneous constrictions in the lymphatics of animals in which ileitis had been induced.117 However, no data regarding lymphatic function in humans are available thus far.
The isolation and in vitro culture of human intestinal LECs has allowed characterisation of LECs and comparisons with VECs.118 Among other important findings, it has now been established that certain markers are unique to LECs compared with VECs.119 120 Examples of markers that are unique to LECs compared with VECs include the glomerular podocyte membrane mucoprotein podoplanin, Prox-1, a homeobox gene product that is involved in early lymphatic development, and LYVE-1, a receptor for hyaluronan.119 120 Definitive data on the characterisation and function of human intestinal LECs will help further our understanding of the role played by the lymphatic system in patients with IBD, keeping in mind the important role played by this system in immunity, bacterial clearance and absorption of oedema. Therefore, stimulating lymphatic function could be therapeutically relevant by promoting the exit of pathogenic leucocytes from the inflamed gut, clearing inflammatory chemokines, and resolving interstitial oedema.
Conclusion
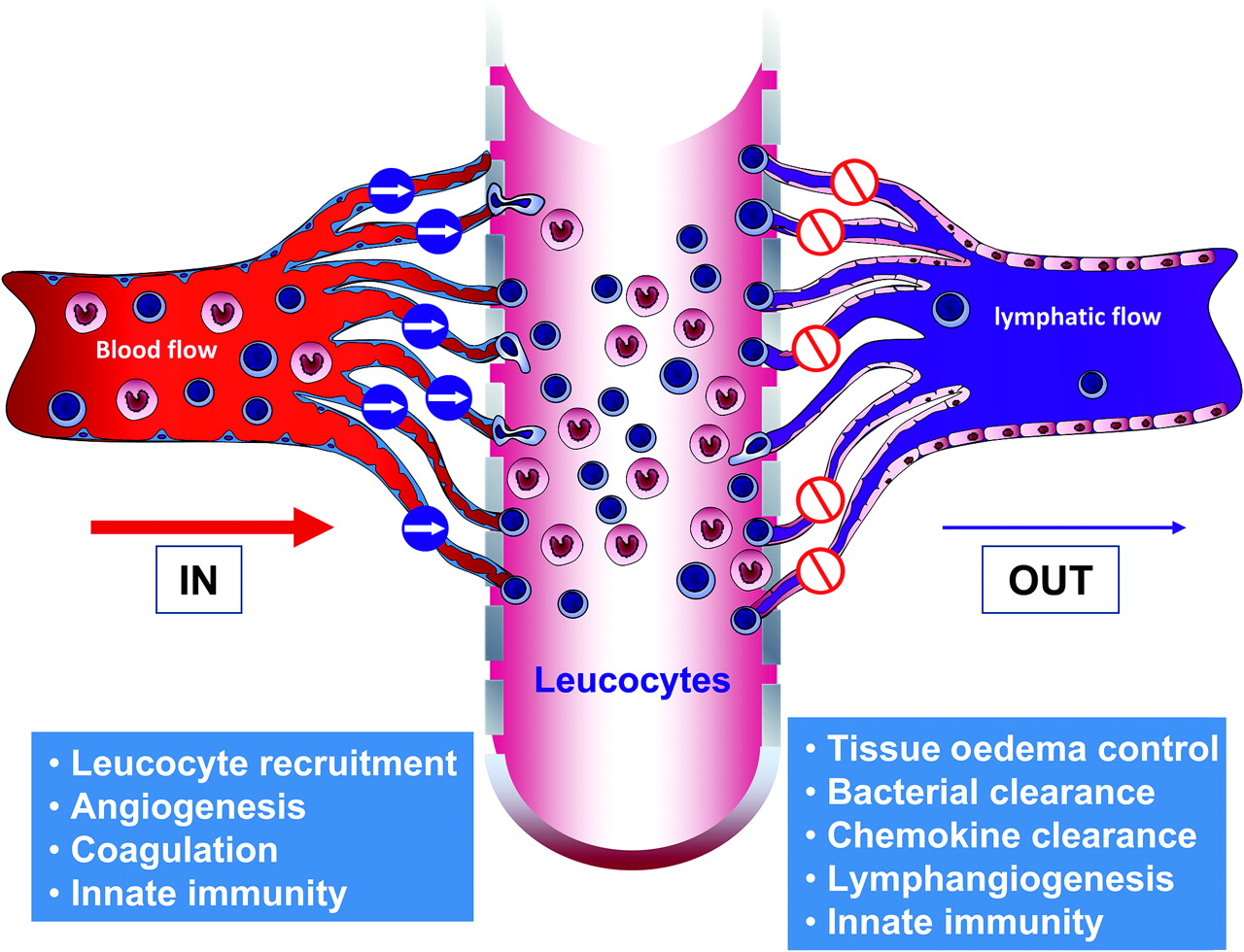
The majority of the research that has been carried out so far into the mechanisms that underlie the development and maintenance of chronic inflammation in patients with IBD has focused on the mechanisms of cell entry into the mucosa, bacterial and foreign antigen invasion, angiogenesis, and the control of gut inflammation through intestinal microvasculature. These mechanisms, the ‘IN’ of chronic inflammation in patients with IBD (figure 2), have provided several valid and effective targets, and promise many more—both on immune and non-immune cells—for the treatment of experimental inflammation, some of which have translated into patients. However, it is becoming increasingly clear that it is also important to focus on the ‘OUT’ of chronic inflammation, by studying the lymphatics and their role in controlling tissue oedema, leucocyte exit, bacterial clearance and oedema absorption, as this promises to be another rich source of non-immune cell targets for therapeutic intervention. The functions of the microvascular and lymphatic endothelium in the gut are therefore complementary, although opposite. In patients with IBD, a dysfunction in each would work together to amplify disease, with the microvascular endothelium overloading, and the lymphatic endothelium failing to relieve the intestinal mucosa. Just as one needs the other for proper physiological function, so dysfunction in one may support dysfunction in the other in disease pathogenesis, the proverbial ‘brother in arms’.
{kind=link}
{kind=link}
The ‘In’ and ‘Out’ of intestinal inflammation. The major tasks performed by the vascular and lymphatic endothelium are summarised.
Acknowledgments
I thank Sarah A De La Rue of Readable Science for her assistance with the manuscript.
References
Footnotes
Funding The studies reported in this review were supported by grants from the Broad Medical Research Program, the Italian Ministry of Health (Ricerca Finalizzata 2006, n.72 and Bando Giovani Ricercatori), Fondazione Cariplo, the Italian Association for Cancer Research (my first AIRC Grant and IG 10205) and AMICI Italia to SD. This work was conducted in the context, and with the support, of the Fondazione Humanitas per la Ricerca (Rozzano, Italy).
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.