Article Text

Abstract
Objective Advanced and recurrent diseases are the major causes of death in colon cancer. No standard preclinical model addresses advanced disease and spontaneous metastasis after orthotopic tumour growth. In this study, the authors report the establishment of such standardised orthotopic mouse models of colon cancer and their use in evaluating metronomic topotecan alone or in combination with standard chemotherapy.
Design Human colon cancer cell lines, transfected with human chorionic gonadotropin and luciferase, were injected orthotopically into the caecal wall of severe combined immunodeficient mice, intrasplenically or subcutaneously. For adjuvant therapy, caecal resections were performed 3–5 weeks after tumour cell injection. Chemotherapy drugs tested included uracil/tegafur, folinic acid, oxaliplatin, topotecan, pazopanib and various combinations.
Results Subcutaneous tumours showed exaggerated sensitivity to treatment by delayed tumour growth (p=0.002) and increased survival (p=0.0064), but no metastatic spread. Intrasplenic cell injection resulted in rapid and extensive but artefactual metastasis without treatment effect. Intracaecal cell injection with tumour take rates of 87.5–100% showed spontaneous metastases at clinically relevant rates. Metronomic topotecan significantly polonged survival and reduced metastasis. In the adjuvant setting, metronomic maintenance therapy (after FOLFOX-like induction) prolonged survival compared with vehicle controls (p=0.0003), control followed by topotecan (p=0.0161) or FOLFOX-like therapy (p=0.0003).
Conclusion The refined orthotopic implantation technique proved to be a clinically relevant model for metastasis and therapy studies. Furthermore, metronomic therapy with oral topotecan may be promising to consider for clinical trials of metastatic colon cancer and long-term adjuvant maintenance therapy of colon cancer.
- Metastasis
- orthotopic implantation
- adjuvant therapy
- cancer
- abdominal surgery
- colon carcinogenesis
- colorectal carcinoma
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
- Metastasis
- orthotopic implantation
- adjuvant therapy
- cancer
- abdominal surgery
- colon carcinogenesis
- colorectal carcinoma
Significance of this study
What is already known on this subject?
-
Local recurrence and metastatic disease remain the leading causes of death in colon cancer. This is likely to be a major factor in the disparity of therapy results that are so frequently obtained using localised, ectopically implanted tumour models with those observed in clinical investigations, clearly highlighting the need for improved preclinical models.
-
Metronomic cyclophosphamide plus uracil/tegafur was shown to be remarkably effective in prolonging the survival of mice with advanced metastatic human breast cancer (Munoz et al. Cancer Res 2006;66:3386–91). A similar regimen with bevacizumab is now undergoing randomised phase III clinical trial evaluation because of its efficacy and because of its minimal toxicity as reported in a prior phase II clinical trial (Dellapasua et al. J Clin Oncol 2008;26:4899–905 and http://www.clinicaltrials.gov, NCT01131195.
-
With respect to colon cancer, the concept of metronomic therapy appears to be highly promising and two randomised phase III trials (CAIRO3 und ACT2, http://www.clinicaltrials.gov; NCT 00442637 and NCT 01229813) are currently under way to evaluate metronomic maintenance therapy with the oral 5-fluorouracil prodrug capecitabine, combined with molecular targeted therapy (bevacizumab and erlotinib), following upfront standard maximum tolerated dose induction therapy.
-
Uncovering further potentially useful drugs or drug combinations for metronomic therapy of colorectal adenocarcinoma is therefore an important area of translational cancer research.
Significance of this study
What are the new findings?
-
This study presents an improved, reproducible, orthotopic and spontaneously metastatic preclinical xenograft model of colon cancer. Transfection of tumour cells with the genes for luciferase and β-human chorionic gonadotropin enables non-invasive in vivo monitoring of primary growth and metastasis at regular intervals. Caecal resections, combined with imaging, enable adjuvant therapy studies to be undertaken.
-
Comparing the new orthotopic model with existing preclinical colon cancer models, it is shown that subcutaneous tumours showed exaggerated sensitivity to treatment, but no metastatic spread. Intrasplenic cell injection resulted in rapid and extensive but artefactual metastasis without treatment effect. Intracaecal cell injection showed spontaneous metastases at clinically relevant rates.
-
In the orthotopic model, metronomic topotecan significantly prolonged survival and reduced metastatic spread in primary and adjuvant treatment protocols.
-
It is shown, for the first time, that metronomic therapy with oral topotecan may be promising to consider for clinical trials of advanced metastatic colon cancer, and for long-term adjuvant maintenance therapy after FOLFOX induction.
How might it impact on clinical practice in the foreseeable future?
-
Using modified orthotopic intracaecal cell implantation models can improve and facilitate translating preclinical therapy results into appropriate clinical trial designs and, hence, more successful outcomes.
-
Several possible phase II clinical trials testing metronomic oral topotecan in colon cancer are suggested.
Introduction
Colorectal adenocarcinoma (CRC) is the second most common cancer worldwide.1 Although the majority (85%) of CRC patients in developed countries can undergo initially curative local resection,2 leading causes of death are local recurrence and metastatic disease. Therefore, adjuvant therapy and treatment of advanced metastatic disease are two critically important fields of research in CRC. Sixty per cent of metastases in CRC patients occur in the liver, and up to 35% have metastases exclusively in this organ.3 While surgical management of colorectal liver metastases has undergone enormous improvements during the past years,4 non-surgical approaches are still being developed. For example, standard neoadjuvant, or conversion therapy regimen before hepatic resection, needs to be established, as do adjuvant therapy after hepatic resection and optimal liver-specific chemotherapy.
‘Metronomic’ chemotherapy usually refers to low(er)-dose administration of conventional chemotherapy at close (often daily) intervals over prolonged periods of time without drug-free intervals. Mechanisms of action of metronomic therapy include suppression of tumour-angiogenesis, stimulation of anticancer cytotoxic T-cell immune responses, inhibition of the hypoxia-inducible factor-1 α, direct tumour cell targeting effects, perhaps including cancer stem cells, and the induction of tumour dormancy.5–8 With respect to CRC, two randomised phase III trials (CAIRO3, ACT2, http://www.clinicaltrials.gov; NCT 00442637, NCT 01229813) are currently under way to evaluate metronomic maintenance therapy with the oral 5-fluorouracil (5-FU) prodrug capecitabine, combined with molecular targeted therapy (using bevacizumab or erlotinib), following standard maximum tolerated dose (MTD) induction therapy. Uncovering additional potentially promising drugs or drug combinations for metronomic therapy of CRC is an important area of translational cancer research.
The pyrimidine-analogue 5-FU and the topoisomerase-inhibitor irinotecan are two approved drugs used in conventional MTD therapy in CRC. While two oral prodrugs of 5-FU (uracil/tegafur (UFT) and capecitabine) are approved for MTD treatment of CRC, irinotecan has to be administered intravenously and is therefore not ideal for highly frequent metronomic administration. However, the availability of the oral topoisomerase-inhibitor topotecan,9 along with two extremely encouraging studies combining metronomic topotecan with pazopanib in preclinical models of advanced ovarian cancer,10 ,11 suggest that oral topotecan may be an ideal candidate for metronomic therapy in CRC.
Preclinical CRC therapy studies in mice are mostly performed using localised, ectopically implanted (subcutaneous) primary tumours.12–14 To study metastatic disease, investigators often use so-called experimental metastasis models by injecting CRC cells intraveneously, intrasplenically or directly into secondary sites (eg, liver, peritoneum).15–17 Major discrepancies are frequently observed between encouraging preclinical therapy results and subsequent, much less impressive or negative clinical trial results.18 Only very few investigators use orthotopic injection of tumour cells into the caecal wall.19–24 It is likely that one reason for the failure of most other investigators to routinely use intracaecal tumour cell injections is that many technical obstacles can compromise the successful establishment of an orthotopic model. There is an urgent need to develop an improved, reliable method to increase the routine use of these models in preclinical research.18 ,25
In this study, we present a detailed description of the technical establishment of orthotopic intracaecal injection of CRC cells along with, for the first time, a thorough cross-comparison with three existing human xenograft models of CRC. We then use the orthotopic model to evaluate the efficacy of metronomic UFT, topotecan, and, since targeted therapies (eg, using the monoclonal antibodies bevacizumab or cetuximab) are part of the standard therapy for metastatic CRC,26 an oral anti-angiogenic targeting agent, pazopanib. Pazopanib targets vascular endothelial growth factor (VEGF) receptors, platelet-derived growth factor receptors (PDGFR) and c-kit.27 ,28 The combination of UFT, oral topotecan and pazopanib enabled us to evaluate a metronomic multidrug regimen—composed entirely of orally bio-available drugs. To our knowledge, this is the first time that such a study has been conducted preclinically in orthotopic and adjuvant settings. We show that metronomic topotecan significantly prolongs survival and reduces metastasis. Furthermore, this regimen could be continued daily uninterrupted for 28 weeks without overt signs of toxicity.
Methods
Cell lines
The human colon cancer cell line HT29 was kindly provided originally by Dr Laferte (University of Saskatchewan), HCT116 was kindly provided by Dr Vogelstein (Johns Hopkins University). Cells were maintained in Dulbecco's modified Eagle medium (DMEM) and 10% fetal bovine serum (Hyclone). Cell lines were transfected with a firefly luciferase vector and a hCG.pIRES vector as described.29 ,30 Six clones, highly expressing both markers, were pooled for each cell line and used for experiments.
Animals
Female CB17 severe combined immunodeficient (SCID) mice (Charles River, Quebec, Canada), aged 6 weeks, were used. Procedures were carried out in accordance with the guidelines by the Sunnybrook Research Institute Animal Care Committee (animal protocol 10-268), accredited by the Canadian Council of Animal Care.
In vivo monitoring
β-Human chorionic gonadotropin (β-hCG) levels in the mouse urine were assessed weekly as previously described.30 ,31 For bioluminescence imaging, mice were intraperitoneally injected with 150 mg/kg of luciferin (Caliper Life Sciences, MA), and imaged following the manufacturer's recommendations with the tumour facing the camera using an IVIS200 (Caliper Life Sciences).
Tumour implantation
Cells were dissociated with trypsin, washed once in serum-containing DMEM and twice in serum-free DMEM, and re-suspended to the desired concentration in DMEM. All animals receiving tumour cell injections were injected with the same quantity of tumour cells (0.5 million; subcutaneous, intrasplenic or intracaecal).
Subcutaneous (ectopic) implantation of tumour cells
Tumour cells numbering 5×105 (100 μl) were injected subcutaneously into SCID mice. Tumour size was measured with calipers and tumour volume calculated as width2×length/2. Endpoint was 1700 mm3.
Orthotopic (intracaecal) implantation of tumour fragments
A ‘donor mouse’ received a subcutaneous tumour cell implantation. When the tumour size reached 100 mm3, it was aseptically removed, transferred into ice-cold DMEM, and cut into 1 mm3-sized fragments. Abdominal access in recipient mice was through a 1 cm hypogastric midline skin and peritoneal wall incison. The caecum was gently exteriorised. A tumour fragment was sutured onto the caecal wall. The caecum was returned to the peritoneal cavity, and peritoneum and skin closed by running sutures and wound clips.
Orthotopic (intra-caecal) injection of tumour cells
Injection technique was first established by injections of trypan blue into the caecal wall (figure 1A). Access to the caecum was as described above. The caecum was placed on a scalpel holder, flattened and stabilised with a forceps (figure 1B). This manoeuvre is crucial to prevent leakage of tumour cells into the caecal lumen or peritoneal cavity. A volume of 5 μl (5×105 cells) was injected into the caecal wall under 4× magnification and using a 10 μl Hamilton syringe and 30G needle. The caecum was returned to the peritoneal cavity; peritoneum and skin were closed as described above.

(A) An injection technique was established by injections of trypan blue into the caecal wall (excised pieces are shown on a petri dish). (B) Exposure of the caecum for orthotopic injections; for successful intra-caecal cell injection, the caecum was placed on a scalpel holder, flattened and stabilised with a forceps. (C) In vivo monitoring: quantification of bioluminescence, (D) ß-human chorionic gonadotropin (ß-hCG) secretion in the urine. (E) subcutaneous tumour sizes. (F) Survival curves. cc, caecal cell implantation; cc-t, caecal cell implantation plus treatment; ct, caecal tissue implantation; ct-t, caecal tissue implantation plus treatment; is, intrasplenic cell implantation; is-t, intrasplenic cell implantation plus treatment.
Ectopic (intrasplenic) injection of tumour cells and splenectomy
Abdominal access was obtained via a left subcostal skin and peritoneal wall incision. The spleen was gently exteriorised. Tumour cells numbering 5×105 (5 μl) were injected with a 10 μl Hamilton syringe and 30G needle. The needle was slowly retracted and the injection site pressed with a moist cotton swab to prevent leakage. The spleen was returned to the peritoneal cavity; peritoneum and skin were closed as described above.
In some animals, splenectomy was performed 1 min after intrasplenic injection. The hilus vessels were ligated with 4/0 synthetic absorbable suture and the spleen was removed using a cauterizer (Fine Science Tools, Foster City, CA).
Tumour resections
For the adjuvant model, primary tumour resections were performed 3–7 weeks after tumour cell injection. The caecum was ligated with 4/0 synthetic absorbable suture and excised (see figure 2). The dissection site was swabbed with iodine.

Adjuvant therapy model. (A) Bioluminescence monitoring served as non-invasive proof of successful complete tumour respectability. (B) Caecal resection: The caecum was ligated with 4/0 synthetic absorbable suture and excised. Complete tumour resection was shown by (C) H&E staining of the resected caecum including (*) tumour. (D) Quantification of bioluminescence. (E) ß-human chorionic gonadotropin levels. (F) Necropsy: nine weeks after tumour cell implantation, a local recurrence causing bowel obstruction (*) and multiple liver metastases were found.
Drugs and treatment schedules
Topotecan and pazopanib were obtained from GlaxoSmithKline. UFT were obtained from Taiho Pharmaceutical Co, Ltd (Tokyo, Japan). Pazopanib, topotecan and UFT were gavaged daily at 150 mg/kg, 1 mg/kg and 15 mg/kg, respectively. Folinic acid and oxaliplatin were purchased from the institutional pharmacy. The combination of UFT, folinic acid and oxaliplatin will henceforth be refered to as ‘FOLFOX-like’. Animals treated with this therapy received daily gavage of 15 mg/kg UFT and 4mg/kg folinic acid and intraperitoneal injections of 10 mg/kg oxaliplatin every other week. Oxaliplatin was stopped after three injections to prevent host toxicity. Control animals received gavage of 0.1% hydroxypropylmethyl cellulose and injections of normal saline. Endpoint criteria were 20% weight loss, bowel obstruction or the development of ascites. Primary tumour and organs were dissected, weighed, analysed by bioluminescence and formalin fixed.
Immunohistochemistry
Immunostaining for human Ki67 using standard protocols for formalin-fixed tissues was performed to identify hepatic and pulmonary micrometastases in sections serially cut at 100 μm intervals. For organs with micrometastases, four or fewer sections were necessary to detect metastasis. Organs with no metastases in eight sections were defined as being tumour-free. Caecal sections were H&E stained to verify complete tumour resection in the adjuvant model. Sections were analysed at 100× magnification with a Zeiss Axioplan 2 microscope under bright-field conditions. Pictures were taken with a Zeiss Axiocam camera connected to the microscope using AxioVision V.3.0 software. Extent and size of pulmonary and hepatic metastases were assessed as percentage of metastasis covering tissue per high-power field (100× magnification) using ImageJ64 software.
Statistical analysis
Results are reported as mean and SE of the mean. Statistical significance was assessed by analysis of variance (ANOVA). Kaplan–Meier survival curves were generated and the significance assessed by log rank tests. Statistics were generated using GraphPad Prism 4.00 (GraphPad Software, San Diego, CA). The level of significance was set at p<0.05.
Experimental design
(1) Five mice were injected with trypan blue solution into the caecal wall to determine optimal technique and injection volume. (2) Thirty-two mice were intracaecally implanted with HT29.hCG.Luc, HCT116.hCG.Luc, SW620.hCG.Luc or CaCo2.hCG.Luc (n=8) to determine tumour take rate and metastatic potential. (3) Forty mice received HT29.hCG.Luc cells/tumour fragments (n=10): (i) subcutaneous injection, (ii) intrasplenic injection, (iii) intracaecal injection and (iv) orthotopic tumour fragment implantation. Seven days after tumour injection, drug-treatment with UFT, folinic acid and oxaliplatin, or vehicle treatment was initiated (n=5/group respectively). Abbreviations are as follows: sc = subcutaneous cell implantation, sc-t = subcutaneous cell implantation plus treatment, cc = caecal cell implantation, cc-t = caecal cell implantation plus treatment, ct = caecal tissue implantation, ct-t = caecal tissue implantation plus treatment, is = intrasplenic cell implantation, is-t = intrasplenic cell implantation plus treatment. (4) To determine whether selecting hCG/luciferase positive clones had an influence on tumour growth and metastasis, and to evaluate the impact of splenectomy after intrasplenic injection of tumour cells, 20 mice were injected with parental HT29 colon cancer cells as follows (n=5): (i) orthotopic intracaecal injection, (ii) orthotopic implantation of a tumour fragment, (iii) intrasplenic injection and (iv) intrasplenic injection followed by splenectomy. (5) Eighteen mice were injected intracaecally with HT29.hCG.Luc cells. The primary tumour was resected at different time-points. Animals were monitored for completeness of resection and scored for primary tumour regrowth and metastasis. (6) Twelve mice were intracaecally implanted with HT29.hCG.Luc cells. After 1 week, treatment with control vehicle, topotecan or topotecan + pazopanib was begun (n=4). Animals were sacrificed after 10 weeks and analysed for primary tumour weight and metastatic spread. (7) Forty-two mice were intracaecally implanted with HT29.hCG.Luc cells. After 1 week, treatment with (i) control vehicle, (ii) UFT, (iii) topotecan, (iv) pazopanib, (v) UFT plus topotecan, (vi) UFT plus pazopanib, (vii) topotecan plus pazopanib or (viii) UFT plus topotecan plus pazopanib (n=5–6) was started. Animals were sacrificed when they reached endpoint criteria. (8) Thirty-one mice were intracaecally implanted with HCT116.hCG.Luc cells. After 2 weeks, treatment with (i) control vehicle, (ii) FOLFOX-like, (iii) topotecan, (iv) topotecan plus pazopanib, (v) FOLFOX-like plus topotecan, (vi) FOLFOX-like plus pazopanib and (vii) FOLFOX-like plus topotecan plus pazopanib (n=4-5) was started. Animals were sacrificed when they reached endpoint criteria. (9) Twenty-eight mice were intracaecally implanted with HT29.hCG.Luc cells. Primary tumours were removed when bioluminescence levels reached 0.5×106 photons/s. After 1 week, mice received control vehicle or the FOLFOX-like therapy for 3 weeks (n=14). Then, the mice were randomised within their respective groups to either continue the previous treatment or to switch over to metronomic oral topotecan maintenance therapy. Thus, the groups were as follows: (i) control–control, (ii) control–topotecan, (iii) FOLFOX-like–FOLFOX-like and (iv) FOLFOX-like–topotecan (n=7).
Results
Establishment of an intracaecal orthotopic model of CRC
Initially, the injection technique was established using trypan blue injections (figure 1A). When injected too deeply, the dye was diluted by the bowel content. A too superficial injection resulted in peritoneal leakage. Injection volumes exceeding 5 μl increased the risk of leakage. When carefully inserting a microsyringe into the flattened and stabilised caecal wall using a microscope, blebbing within the caecal wall can be observed as a quality control for the injection technique. (figure 1B). Tumour take rate was reproducible at 87.5–100% for both cell lines. Spontaneous metastases to lymph nodes, liver, lungs and peritoneum were found within 16 weeks after injection.
Comparison of tumour growth, response to treatment and survival
Bioluminescence and β-hCG quantification were used to non-invasively monitor tumour burden. The relative levels of these surrogate tumour markers correlate with tumour burden.32 ,33 Bioluminescence imaging was performed weekly. The online supplementary figure 1 shows two representative animals of each group for every other week. Quantification of bioluminescence and β-hCG are shown in figure 1C,D, caliper measurements of subcutaneous tumours in figure 1E. Treatment was well tolerated in all models and did not result in overt signs of toxicity, nor weight loss.
Following subcutaneous tumour cell injection, luciferin signal remained localised to the injection site. By weeks 4 and 9 we observed significant growth delays in drug-treated compared with vehicle-treated mice (p=0.002 and 0.018, figure 1E).
After intracaecal cell injection, animals showed a markedly slower increase in bioluminescence compared with all other groups (p<0.05, figure 1C). Anti-tumour treatment efficacy was very pronounced as evident by significantly less (p=0.0021) bioluminescence than vehicle-treated animals. Analysis of β-hCG levels confirmed these findings (figure 1D). However, due to high SEs in β-hCG results, the observed treatment effect was not statistically significant (p=0.2659).
After orthotopic implantation of a tumour fragment, no treatment effect could be shown by bioluminescence or β-hCG analysis. By contrast, one day after intrasplenic injection, the bioluminescence signal was evident in the left upper quadrant (location of the spleen), and in the right upper quadrant (location of the liver). Such a rapid localisation to the liver can only be explained by embolisation, not by a process of authentic spontaneous metastasis. No significant treatment effect was observed.
Survival curves are shown in figure 1F. Therapy-treated animals with subcutaneous tumours (sc) showed a longer survival than vehicle-treated controls (median sc 54 days, median subcutaneous tumours plus treatment (sc-t) 74 days, p=0.0064). Other groups showed no significant treatment benefits with respect to survival (cc vs cc-t p=0.1945, ct vs cc-t p=0.489, is vs is-t p=0.8188). Mice with caecal cell implantation lived significantly longer (median 86.5 days) than those with subcutaneous cell implantation (median 54 days, p=0.0049), intrasplenic cell implantation (median 43 days, p=0.0045), or orthotopic tumour fragment implantation (median 57 days, p=0.0047).
Assessment of metastatic capacity
The frequency of metastasis in each model was evaluated by three methods: necropsy, bioluminescence imaging of individual organs and immunohistochemistry (figure 3). Following subcutaneous tumour cell injection, primary growths were observed in all animals but no macroscopic or microscopic metastases were evident (figure 3A). There was no significant difference in endpoint tumour weight (p=0.1457) between treated and control groups (online supplementary figure 2A).
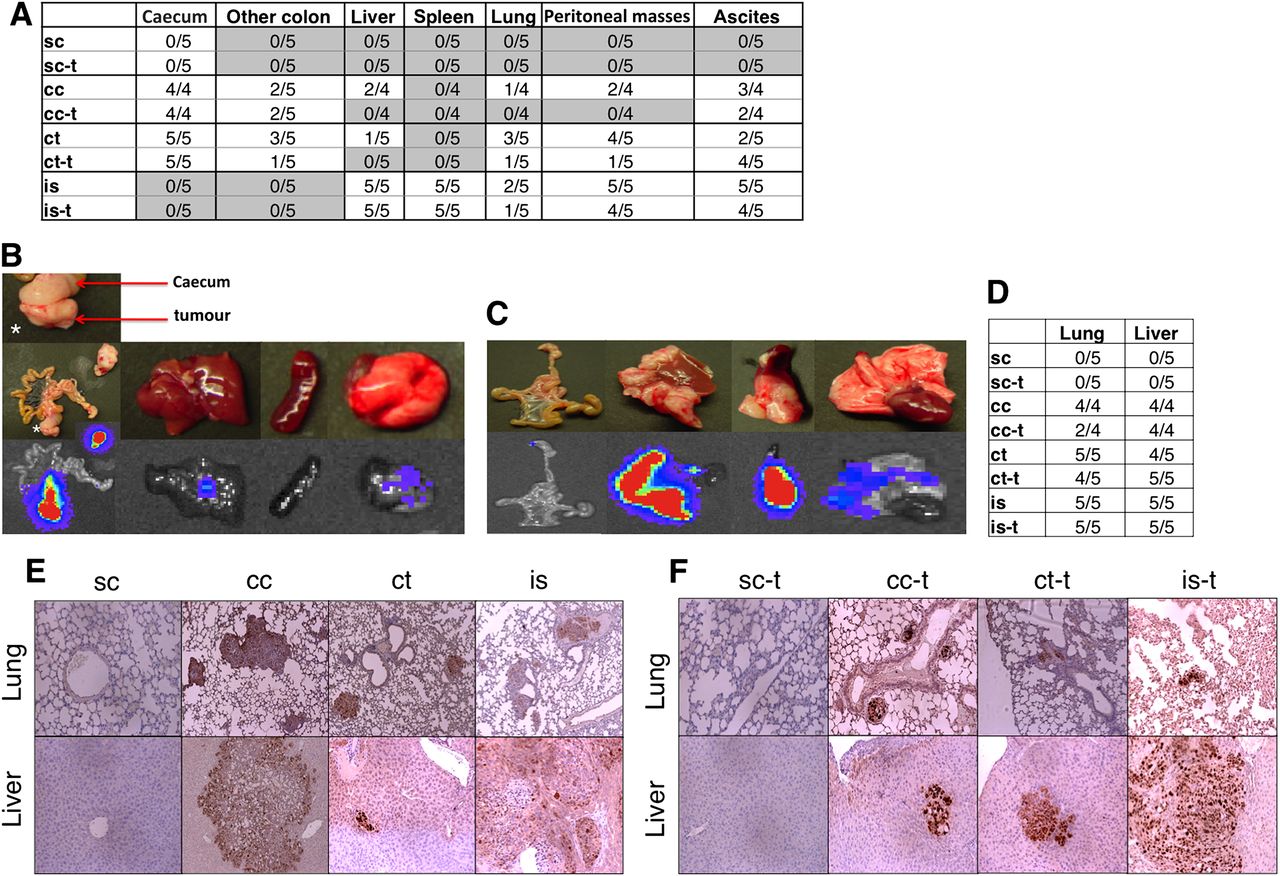
Postmortem evaluation. (A) Bioluminescence evaluation of metastasis. Treatment reduced metastatic spread after intracaecal tumour cell injection. (B) Representative photographs after caecal tumour fragment implantation and after (C) intrasplenic cell injection. (D) Microscopic quantification of metastasis after Ki67/haematoxylin staining, 100× magnification. Lungs and livers were screened for metastasis. No metastases were found in mice bearing subcutaneous tumours. After caecal cell implantation, 100% incidence of hepatic and pulmonary metastasis was found in vehicle-treated animals whereas only 50% of FOLFOX-like treated animals showed lung metastasis. After caecal tumour fragment implantation, hepatic and pulmonary metastasis was detected with no treatment effects. After intrasplenic injection, massive and treatment-resistant ‘metastasis’ was observed. Characteristic pictures are shown in (E) (untreated animals). (F) (treated animals). Refer to figure 1 and text for details of abbreviations.
After intracaecal tumour cell injection, necropsy and bioluminescence analysis showed that fewer drug-treated animals had metastases compared with vehicle-treated animals (figure 3A). Microscopic analysis showed fewer pulmonary micrometastases in treated animals compared with vehicle-treated animals (figure 3D–F). Hepatic micrometastases were observed in all treated and vehicle-treated mice. However, pulmonary and hepatic metastases were fewer in number (p=0.028 and 0.038), and liver metastases were smaller in size (p=0.017) in drug-treated compared with vehicle-treated animals (online supplementary figure 2A,B,C).
After tumour fragment implantation, necropsy and bioluminescence analysis showed that fewer drug-treated mice had metastases compared with vehicle-treated mice (figure 3A). Representative autopsy and bioluminescence images are shown in figure 3B. No impact of treatment was evident in terms of prevalence or size of pulmonary or hepatic micrometastases (figure 3 and online supplementary figure 2A,B,D).
For the intrasplenic model, extensive hepatic and pulmonary metastasis and ascites were observed, and no treatment effect on number or size of metastasis was observed (figure 3 and online supplementary figure 2A,B,E). The number of hepatic metastasis could not be assessed in this model since metastasis was often widespread throughout the entire organ. Splenic tumours weighed significantly more than spleens of mice in other models (p<0.01, data not shown). Due to the extensive hepatic metastasis, liver weights were higher than in all other models (p<0.05, data not shown). Representative images and bioluminescence are shown in figure 3C.
Impact of selection and splenectomy
As observed with HT29.hCG.Luc-injected animals, mice receiving intracaecal cell implantations of parental HT29 cells lived significantly longer (median 80 days) than animals receiving caecal tumour fragment implantation, intrasplenic tumour cell injection or intrasplenic injection followed by splenectomy (median 60, 45, and 58 days, p<0.003 for all, see online supplementary figure 3A,B). Relative survival of all groups was similar to that observed with HT29.hCG.Luc cells. Importantly, survival of animals receiving splenectomy was not significantly different from intrasplenic injection of tagged (p=0.1268) or untagged (p=0.2764) tumour cells without splenectomy.
Macroscopic and microscopic analysis showed that hepatic and pulmonary metastasis occurred with the same frequency as observed with the HT29.hCG.Luc cells. Animals that underwent splenectomy showed similar patterns of distribution and tumour burden of metastases compared with mice that had not undergone splenectomy (data not shown).
Establishing a resectable CRC model for adjuvant therapy studies
Caecal resections were performed 3–7 weeks after orthotopic intracaecal injection of HT29.hCG.Luc cells. Surgery between weeks 3 and 5 resulted in complete resections of the primary tumour (ie, tumour-free resection margin and loss of bioluminescence and β-hCG signals). By contrast, resections at later time points were not complete. The feasibility of carrying out a complete resection correlated with the relative bioluminescence data. Tumours with a bioluminescence reading of 5×106 photons/s or less could be completely resected. The relative β-hCG levels at this early stage of tumour growth were too low to reliably serve as a biomarker for optimal timing of surgery. A representative example is shown in figure 2. Nine weeks after tumour cell implantation, this animal showed a local recurrence causing bowel obstruction and extensive liver metastases (figure 2F). Based on these findings, nine mice were injected orthotopically with HT29.hCG.Luc cells, and the primary tumours were resected when the bioluminescence signal reached 4.5×106 photons/s. Three mice (33%) developed metachronous liver and lung metastases, five mice (56%) developed peritoneal- and lymph node metastases and six mice (67%) developed local regrowths. Three mice (33%) were cured by removing the primary tumour.
Short-term evaluation of topotecan and topotecan plus pazopanib
Twelve mice were intracaecally implanted with HT29.hCG.Luc and treated with control vehicle, topotecan or combined topotecan plus pazopanib (n=4). Animals were sacrificed after 10 weeks. Both topotecan and the topotecan plus pazopanib combination produced significantly smaller primary tumours than vehicle-treated controls (p=0.0276 and 0.0022, figure 4A). The combination treatment showed reduced tumour growth compared with topotecan monotherapy, but this did not reach statistical significance (p=0.1467). Necropsy and bioluminescence analysis showed that three of four control mice had widespread liver metastases (figure 4B), whereas no liver metastases were found in any of the treated animals. In addition, lymph node and peritoneal metastasis were reduced in treated animals.

Evaluation of oral metronomic topotecan (topo). Twelve mice were orthotopically injected with HT29 human colon cancer cell and treated with control vehicle, topotecan or topotecan plus pazopanib (pazo) (n=4). Mice were sacrificed after 10 weeks. Primary tumour weights (A) and metastatic spread (B) were significantly reduced by topotecan mono- or combination therapy. Green arrows indicate hepatic metastases
Long-term treatment of HT29.hCG.Luc orthotopic tumours: in vivo monitoring and survival
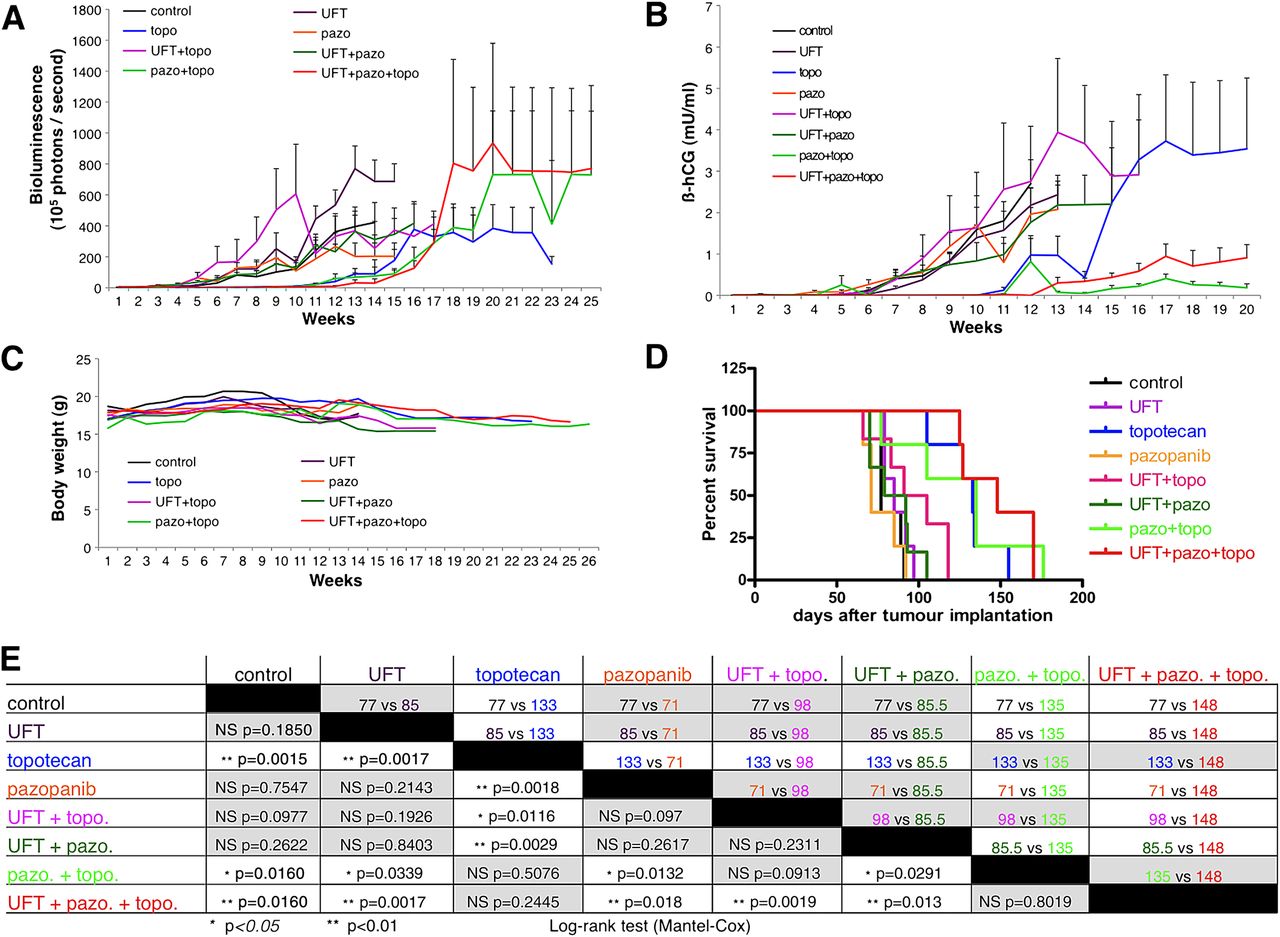
Forty-two mice were intracaecally implanted with HT29.hCG.Luc cells. After one week, tumour burden was analysed by bioluminescence and mice were randomised into the following groups of daily oral treatment (1) control vehicle, (2) UFT, (3) topotecan, (4) pazopanib, (5) UFT plus topotecan, (6) UFT plus pazopanib, (7) topotecan plus pazopanib, or (8) UFT plus topotecan plus pazopanib. All groups had five mice, except groups UFT plus topotecan and UFT plus pazopanib which both had six mice. Metronomic UFT, or pazopanib alone and the combination of these two drugs did not delay the increase in bioluminescence signal when compared with controls (figure 5). By contrast, all groups treated with a regimen containing topotecan showed a delayed increase in bioluminescence. Animals treated with topotecan alone, topotecan plus pazopanib and UFT plus topotecan plus pazopanib showed a significantly reduced signal during the first 12 weeks of treatment, as compared with all other groups (p<0.05, see figure 6A). Among these three topotecan-based regimens, no significant difference in bioluminescence was observed. These results were confirmed by quantification of secreted β-hCG in the mouse urine, which was lower in animals treated with topotecan alone, topotecan plus pazopanib and UFT plus topotecan plus pazopanib during the first 10 weeks of treatment, as compared with all other groups (p<0.05, figure 6B). No regimen produced any signs of overt toxicity or weight loss (figure 6C).

In vivo bioluminescence monitoring of orthotopic HT29.hCG.Luc tumours. All groups treated with topotecan mono- or combination therapy showed a slower increase in bioluminescence signal. pazo, pazopanib; topo, topotecan; UFT, uracil/tegafur.

In vivo monitoring of orthotopic HT29.hCG.Luc tumours. Analysis of (A) bioluminescence, (B) ß-human chorionic gonadotropin (ß-hCG) secretion in the urine, (C) body weight. Survival curves and their statistical analyses are shown in panels D and E including p values (lower-left side) and median survival in days (upper-right side). Grey boxes contain results that are not considered significant. pazo, pazopanib; topo, topotecan; UFT, uracil/tegafur.
Control animals showed a median survival of 77 days after tumour implantation. Survival was significantly prolonged in animals treated with topotecan alone (133 days, p=0.0015), topotecan plus pazopanib (135 days, p=0.0160) and UFT plus topotecan plus pazopanib (148 days, p=0.0015, figure 6D,E). No significant difference in survival was observed between these treatment regimens. Animals treated with UFT alone, pazopanib alone, UFT plus topotecan, and UFT plus pazopanib, showed no difference in survival compared with controls.
Long-term treatment of HT29.hCG.Luc orthotopic tumours: postmortem analysis
Results of necropsy and hepatic single organ bioluminescence are shown in figure 7A,B. All control animals had lymph node metastases, and four out of five mice had extensive liver, diaphragmatic and peritoneal metastases. Two control mice also showed lung metastases and malignant ascites. All treatment groups that received topotecan (as mono- or combination therapy) showed reduced metastasis. This result was most pronounced for liver metastasis: of 21 mice treated with control vehicle or regimens lacking topotecan, 15 mice (=71%) had liver metastases. By contrast, of the 21 mice treated with topotecan (mono- or combination therapies), only four mice (=19%) showed hepatic metastases. All livers were paraffin embedded and stained for human Ki67. Quantification of the number of hepatic micrometastases per section and the average diameter of metastases are shown in figure 7C,D. Control mice had an average of 6.1±1.1 metastases per section. This was significantly reduced in animals treated with topotecan (0.33±0.31, p=0.0002), UFT (1.9±0.8, p=0.007), UFT plus topotecan (1.75±0.57, p=0.0016), UFT plus pazopanib (1.64±0.73, p=0.0033), pazopanib plus topotecan (2±0.87, p=0.01), and UFT plus pazopanib plus topotecan (1.75±0.88, p=0.01). Mice treated with pazopanib alone did not show a significant reduction in the incidence of liver metastases compared with controls (5.09±1.35, p=0.59).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Postmortem necropsy and single-organ bioluminescence analysis of metastatic spread. Topotecan significantly reduced metastatic spread (A), most pronounced in the liver (B). Microscopic quantification of the number of hepatic metastases per section and the average diameter of metastases are shown in panels C and D. pazo, pazopanib; topo, topotecan; UFT, uracil/tegafur.
For all liver metastases confirmed by histology, the average size was analysed. Only mice treated with topotecan-containing therapies showed a reduction compared with controls. Liver metastases in control mice had an average diameter of 2338±959 μm. Confirming our necropsy and bioluminescence data, only one of the five mice treated with topotecan had microscopic metastases. These metastases had an average diameter of 50±5 μm. The size of hepatic liver metastases was also reduced in animals treated with UFT plus topotecan (314±146 μm), pazopanib plus topotecan (67±12 μm), and UFT plus pazopanib plus topotecan (50±5 μm). In some groups, only one animal showed hepatic metastases; therefore, statistical analysis could not be determined for the size of hepatic metastases.
To confirm our treatment results in a second colon cancer cell line, mice were intracaecally implanted with HCT116.hCG.Luc cells. Here, UFT monotherapy and UFT combination therapy was replaced by a UFT-based FOLFOX-like regimen (see online supplementary results and supplementary figures 4 and 5).
Postoperative adjuvant treatment of HT29.hCG.Luc orthotopic tumours
Twenty-eight mice were intracaecally implanted with HT29.hCG.Luc cells. Primary tumours were removed when the bioluminescence level reached 0.5×106 photons/s (ie, 3–5 weeks after tumour implantation). When wound healing was stable at 1 week after resection, 12 animals had developed recurring bioluminescence signals and were randomised to receive control vehicle or FOLFOX-like therapy for 3 weeks (n=14). Mice were then randomised once again for bioluminescence signal within their respective groups to either continue the previous treatment or to switch over to oral metronomic topotecan maintenance therapy (online supplementary figure 6A). Control animals showed a median survival of 46 days after tumour resection. Survival was significantly prolonged in animals treated with the FOLFOX-like therapy followed by topotecan (74 days, p=0.0003), which was also significantly longer than animals treated with metronomic topotecan after prior control vehicle treatment (56 days, p=0.0073) or continued FOLFOX-like therapy (46 days, p=0.0003, online supplementary figure 6B). By contrast, treatment with the continued FOLFOX-like therapy and control vehicle treatment followed by metronomic topotecan failed to reach significance compared with control animals (p=0.6381 and p=0.2812).
Once again, treatment groups receiving metronomic topotecan showed reduced metastatic spread and again, this result was most pronounced with respect to liver metastasis: Of a total of 14 mice treated with control vehicle or regimens without topotecan, 10 mice (=71%) had liver metastases. By contrast, only four of the 14 mice (=29%) treated with metronomic topotecan-containing therapy showed hepatic metastases (online supplementary figure 6C).
Discussion
In the present study, we established a reproducible, standardised and improved orthotopic human xenograft model of colon cancer. Transfection of tumour cells with the genes for luciferase and β-hCG enabled in vivo monitoring at regular intervals after orthotopic intracaecal tumour cell injection and proved to be an excellent marker for completeness of tumour resection/resectability. In the first part of our study, we rigorously cross-compared this refined orthotopic model with the more frequently used preclinical models of CRC. Subcutaneous (ectopic) injection of human colon cancer cells into nude and SCID mice is convenient to set up, tumour growth can be monitored by caliper measurements and endpoints, defined by tumour size, are reached within a reasonable time frame. We showed a significant delay of tumour growth, and a significant survival benefit in drug-treated versus vehicle-treated animals bearing subcutaneous tumours. By contrast, necropsy did not show significances in differences of tumour size or weight. As subcutaneous tumour-bearing mice did not show symptoms of deterioration, endpoint was here defined according to our animal care guidelines by maximum tumour size of 1700 mm3. Control animals reached endpoint earlier, but tumours of treated mice eventually reached the same tumour size. Subcutaneous cell injections of HT29 cells did not lead to metastasis, nor cause clinical impairment. Similar to our study, many other preclinical drug-testing studies have successfully been performed using such subcutaneous models.12–14 However, many of these highly encouraging results seldom produce a benefit in the clinic, and when they do, they are often with far less impressive outcomes.
Intrasplenic tumour cell injection results in rapid colonisation of tumour cells in liver and lungs. Bioluminescence imaging showed that even as soon as one day after cell injection, a clear hepatic signal could be detected, which clearly cannot be due to the development of authentic spontaneous metastases. Mice succumb to extensive tumour burden within a few weeks. Perhaps not surprisingly, no treatment efficacy on metastasis was observed under such extremely aggressive growth conditions.
Orthotopic injection of tumour cells into the caecal wall is a more complex procedure requiring a degree of skill and appropriate technical equipment. We show here for the first time that intracaecal tumour cell injection can be performed with a reproducibly high tumour take rate of 87.5–100%. Drug treatment of animals with intracaecal tumour cell implantation showed a significant reduction of bioluminescence and reduced metastasis compared with vehicle-treated animals. We therefore considered this model as clinically relevant for analysing treatment protocols in the following experiments. One critical aspect might be that survival times of treated animals were not significantly different from vehicle-treated mice since survival was limited by bowel obstruction. Thus, survival analysis in this model is a marker for the effect of treatment on the primary tumour, whereas endpoint analyses are necessary as marker for the effect of treatment on metastatic spread.
We also applied a second orthotopic CRC model where a subcutaneously grown tumour fragment is sutured to the caecal wall. No significant treatment benefits were observed in treated mice compared with vehicle-treated mice. As shown in figure 3B, the tumour fragment grows mainly on the outside of the caecal wall and into the peritoneal cavity. This, together with the shorter survival, gives the tumour less time to invade host tissue and initiate metastasis. However, this model proved to be locally invasive into the caecal wall and to produce metastases to distant organs as described previously.19 ,34 Further analysis of this model is needed to determine its clinical relevance.
Presently, thousands of patients are enrolled in clinical phase III trials testing new therapies including treatments using molecularly targeted drugs in the adjuvant setting. The recently announced results of the first two clinical phase III trials testing the benefit of adjuvant bevacizumab were disappointing (NSABP-C-08 and AVANT).35–37 These results raise numerous questions. What are the reasons for these failures? Can biomarkers identify subgroups who, nevertheless, benefit from adjuvant targeted therapies? In this regard, we present here a potentially promising preclinical model for adjuvant therapy studies of CRC. In our model, complete resection of the primary tumour can be performed within a defined time after tumour cell injection and within a defined range of bioluminescence signal. Tumour recurrence and metastatic spread can be monitored in vivo. The rate of metachronous hepatic metastasis proved to be in the same range as the published rates of metachronous hepatic metastases in CRC patients.38 These are clinically relevant findings which in the follow-up experiments enabled us to examine the effect of various metronomic primary tumour and microscopic metastasis adjuvant therapies.
The concept of metronomic therapy may be highly promising for adjuvant treatment of CRC patients and treatment of metastatic CRC following the failure of standard therapy. Many metronomic chemotherapy regimens produce minimal host toxicity and can be given over extended periods.39 ,40 In addition, metronomic therapy has sometimes been shown to be active against chemoresistant tumours.41 As one of the first examples of translating metronomic chemotherapy from bench to bedside, we recently reported that metronomic cyclophosphamide plus UFT was remarkably effective in prolonging the survival of mice with advanced metastatic human breast cancer.42 A similar regimen with capecitabine plus cyclophosphamide in combination with bevacizumab is now undergoing randomised phase III clinical trial evaluation because of the efficacy of this regimen as reported in a prior phase II clinical trial,43 and because of its minimal associated toxicity,43 and http://www.clinicaltrials.gov, NCT01131195.
A preclinical study of colon cancer treated with metronomic irinotecan has shown reduced proliferation of colon cancer cells and endothelial cells in vitro and reduced growth of HT29 subcutaneous xenografts.44 In a first pharmacological clinical phase II trial, heavily pretreated and chemoresistant CRC patients received metronomic irinotecan, which showed promising results, including a marked increase in plasma concentrations of the angiogenesis inhibitor thrombospondin-I.41 Interestingly, Merritt et al and we have both concurrently and independently reported excellent antitumour activity of metronomic oral topotecan in preclinical models of ovarian cancer.10 ,11 Furthermore, oral metronomic topotecan has shown promising activity in a clinical phase II study for recurrent childhood brain cancer.45 These results suggest that oral metronomic topotecan may be a potentially promising treatment regime in CRC. In this study, we evaluate for the first time, preclinical therapy outcomes of oral metronomic topotecan in CRC, using the improved preclinical models we developed.
Our initial experiment using metronomic topotecan showed a striking reduction in size of primary tumours and also of metastatic spread, after oral metronomic topotecan administration. This led us to undertake the two subsequent treatment experiments using the human HT29 and HCT116 models. After orthotopic implantation of HT29 cells, topotecan monotherapy and the combinations topotecan plus pazopanib and UFT plus topotecan plus pazopanib significantly delayed tumour growth, prolonged survival and reduced metastasis (especially hepatic metastasis). Surprisingly, topotecan combined with metronomic UFT did not show an equivalent therapeutic benefit. These results may be consistent with those of a study by Bocci et al, where metronomic irinotecan alone, but not metronomic 5-FU, or the combination of both drugs and metronomic oxaliplatin, reduced proliferation of HT29 and endothelial cells in vitro.44 Our results show that oral metronomic topotecan has potentially potent antitumour activity in CRC, and that it can lose this activity when combined with certain other drugs. As combination therapies are standard in MTD treatment of CRC, this highlights the need for thorough preclinical and pharmacological analyses prior to clinical testing of metronomic therapy.
Given at standard MTD (1.5 mg/m2/d for 5 days every 21 days), topotecan has shown only minor anti-tumour activity and considerable toxicity in phase II clinical trials of CRC.46–48 However, our results suggest the possibility that topotecan, given metronomically, that is, more frequently at lower doses, can cause potent anti-tumour and anti-metastatic activity along with reduced host toxicity. For clinical application of metronomic topotecan, prior determination of an appropriate metronomic dose is necessary. A first phase I trial addressing this issue has been completed in relapsed ovarian cancer patients (http://www.clinicaltrials.gov NCT00382733).
The microscopic evaluation of hepatic metastases revealed that all groups treated with topotecan (mono- or combination therapy), but also metronomic UFT alone and UFT plus pazopanib, reduced the number of hepatic micrometastases compared with control animals. Together with the size of the hepatic micrometastases (which were found reduced in all topotecan-containing groups, but not in the UFT, the pazopanib or the UFT plus pazopanib groups), the necropsy and bioluminescence results, this suggests that metronomic topotecan might not prevent the initiation of the metastatic process, but may reduce the subsequent growth of microscopic metastases. These results are highly translational, as localised CRC liver metastases can be successfully resected in up to 30% of cases.4 Prior phase III clinical trial results of metronomic-like regimens of UFT administered daily for 2 years in early stage non-small cell lung cancer or breast cancer patients have shown clinical benefits on disease-free survival.39 ,40 Our results suggest similar benefits might not be attained in early-stage CRC patients.
We tested the same therapies in the HCT116 model, that is, an independently derived CRC cell line. All groups treated with a regimen containing oral topotecan, once again, showed a significantly delayed increase in bioluminescence, prolonged survival, and reduced metastasis.
In contrast to our results, and results of others obtained in ovarian cancer,10 ,11 co-treatment with pazopanib did not enhance the antitumour activity of metronomic topotecan. Pazopanib has been approved for the treatment of advanced renal cell carcinoma and has shown promising preliminary results in phase II clinical trials of nasopharyngeal carcinoma, breast cancer, cervical cancer, ovarian cancer, non-small cell lung cancer, glioblastoma and soft tissue sarcoma.27 ,49–54 To date, there is no data on the efficacy of pazopanib in CRC, although two clinical phase I trials are under way (http://www.clinicaltrials.gov NCT00387387 and NCT00540943). Approved targeted biological agents in CRC include the anti-VEGF antibody bevacizumab, and the anti-epidermal growth factor receptor (anti-EGFR) antibodies cetuximab and panitumumab.26 While no biomarkers are known to predict successful bevacizumab treatment, K-Ras and B-Raf mutations serve as biomarkers for non-response to EGFR inhibition. The cell lines used in our study have mutations in K-Ras (HCT116) or B-Raf (HT29).55 However, a recent publication has shown that pazopanib acts as a ‘pan-Raf-inhibitor’, inhibiting wild type and K-Ras or B-Raf mutated breast cancer cells.56
Our adjuvant study showed that metronomic topotecan maintenance following FOLFOX-like induction therapy increased survival compared with control vehicle treatment, topotecan maintenance after control vehicle treatment or continued FOLFOX-like therapy. Topotecan maintenance following control vehicle treatment did not show any benefit in survival or reducing metastatic spread, which may not be surprising in light of 3 weeks without drug treatment and a total survival of 56 days after resection, Surprisingly, Folfox-like therapy alone also did not improve survival. However, as discussed in the online supplementary material, mimicking FOLFOX preclinically in mice is complicated, for example, oxaliplatin treatment had to be discontinued after only three doses to avoid excessive toxicity. Suppression of metastatic spread by metronomic topotecan in our adjuvant model was not as pronounced as in the orthotopic primary tumour model, which may be consistent with our hypothesis, that metronomic topotecan does not prevent the initiation of micrometastasis (which in this model occurs before treatment is started), but instead may reduce the subsequent growth of metastases. If so, this would translate into prolonged overall survival times in the adjuvant setting.
In summary, we present a reproducible, standardised orthotopic human xenograft model of colon cancer. Our treatment results show, for the first time, that metronomic therapy with oral topotecan may be promising to consider for clinical trials of advanced metastatic colon cancer, and for long-term adjuvant maintenance therapy after FOLFOX induction. We suggest three possible phase II clinical trials testing metronomic oral topotecan (1) in metastatic colon cancer after failure of multiple-line standard therapy and in comparison to best supportive care, (2) in metastatic colon cancer as maintenance therapy versus observation after successful (ie, stable disease, partial response, or complete response) standard first-line therapy until disease progression and introduction of second-line therapy and (3) as adjuvant maintenance therapy versus observation in stage III CRC patients after prior standard FOLFOX therapy.
Acknowledgments
We thank Cassandra Cheng for her excellent secretarial assistance.
References
Supplementary materials
HTML Page - index.htslp
Supplementary Data
Files in this Data Supplement:
- Data Supplement 1 - SUPPLEMENTAL RESULTS
- Data Supplement 2 - SUPPLEMENTAL DISCUSSION
- Data Supplement 3 - Online figure 3
- Data Supplement 2 - Online figure 2
- Data Supplement 4 - Online figure 4
- Data Supplement 1 - Online figure 1
Footnotes
-
Funding This study was supported by grants to R S Kerbel from the National Institutes of Health, USA (CA-41233), the Ontario Institute for Cancer Research, and a sponsored research agreement with GlaxoSmithKline, Philadelphia, USA. RSK holds a Tier I Canada Research Chair in Tumour Biology, Angiogenesis and Antiangiogenic Therapy. CH was supported by a Research Fellowship from the Deutsche Forschungsgemeinschaft (German Research Foundation). CM is supported by the Ontario Ministry of Research and Innovation Post-Doctoral Fellowship, the Research Excellence Fellowship for Women in Science (L'Oreal Canada and Canadian Commision for UNESCO) and the 2010 Brenda M Williams Young Investigator Award.
-
Competing interests RSK is a consultant to Taiho Pharmaceuticals, Japan, and GSK, USA. He receives funding from GSK to undertake studies using oral topotecan and pazopanib. CH, SM, GF, CM and PX have no competing interests.
-
Provenance and peer review Not commissioned; externally peer reviewed.