Article Text

Abstract
Objective Etrolizumab (rhuMAb β7, anti-β7, PRO145223) is a humanised monoclonal antibody targeting the β7 subunit of the heterodimeric integrins α4β7 and αEβ7, which are implicated in leucocyte migration and retention in ulcerative colitis (UC). This randomised phase I study evaluated the safety and pharmacology of etrolizumab in patients with moderate to severe UC.
Design In the single ascending dose (SAD) stage, etrolizumab (0.3, 1.0, 3.0, 10 mg/kg intravenous, 3.0 mg/kg subcutaneous (SC) or placebo) was administered 4:1 (n=25) in each cohort. In the multiple dose (MD) stage, new patients received monthly etrolizumab (0.5 mg/kg SC (n=4), 1.5 mg/kg SC (n=5), 3.0 mg/kg SC (n=4), 4.0 mg/kg intravenous (n=5)) or placebo (n=5). The pharmacokinetics was studied and Mayo Clinic Score evaluated at baseline, day 29 (SAD), and days 43 and 71 (MD).
Results In the SAD stage, there were no dose limiting toxicities, infusion or injection site reactions. Two impaired wound healing serious adverse events occurred in two patients receiving etrolizumab. In the MD stage, there were no dose limiting toxicities, and no infusion or injection site reactions. Headache was the most common adverse event, occurring more often in etrolizumab patients. Antietrolizumab antibodies were detected in two subjects. The duration of β7 receptor full occupancy was dose related. A clinical response was observed in 12/18 patients, and clinical remission in 3/18 patients treated with etrolizumab in the MD stage, compared with 4/5 and 1/5 placebo patients, respectively.
Conclusion Etrolizumab is well tolerated in moderate to severe UC. Further investigation is warranted.
- Etrolizumab
- rhuMAb β7
- ulcerative colitis
- drug development
- safety
- pharmacokinetics
- IBD clinical
- IBD basic research
- IBD
- IBD models
- apoptosis
- cell cycle
- immunology
- inflammatory bowel disorders
- dendritic cells
- Crohn's disease
- cytokines
- genetics
- signal transduction
- IBD–genetics
- antibody targeted therapy
- arthritis
- autoimmune disease
- integrins
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
- Etrolizumab
- rhuMAb β7
- ulcerative colitis
- drug development
- safety
- pharmacokinetics
- IBD clinical
- IBD basic research
- IBD
- IBD models
- apoptosis
- cell cycle
- immunology
- inflammatory bowel disorders
- dendritic cells
- Crohn's disease
- cytokines
- genetics
- signal transduction
- IBD–genetics
- antibody targeted therapy
- arthritis
- autoimmune disease
- integrins
Significance of this study
What is already known on this subject?
-
Migration of lymphocytes to the gut is a key feature of inflammation in inflammatory bowel disease.
-
Selective antimigration therapies are currently developed and specific inhibition of homing of lymphocytes to the gut is currently aimed at for therapy.
-
These new therapies target the α4β7–MadCam-1 interaction.
What are the new findings?
-
Etrolizumab (antibodies to β7) does inhibit the migration of lymphocytes to the gut and antagonises the retention of lymphocytes in the mucosa by blocking the αEβ7–E-cadherin interaction.
-
This phase I study in severe ulcerative colitis shows suitable pharmacokinetics and the early safety of this treatment.
-
Clinical improvement of the disease observed in treated patients warrants further phase II studies.
How might it impact on clinical practice in the foreseeable future?
-
We hypothesise that a therapy that blocks migration of lymphocytes to the gut and recruiting of effector lymphocytes from the gut would be more effective than therapies blocking migration alone.
Introduction
Ulcerative colitis (UC) is a chronic inflammatory condition of the colon characterised by mucosal ulceration, rectal bleeding, diarrhoea, abdominal pain and sometimes extraintestinal manifestations.1 UC may be complicated by severe bloody diarrhoea and toxic megacolon that requires colectomy.2 Pharmacological management of UC currently relies on 5-aminosalicylates, corticosteroids, purine antimetabolites, calcineurin inhibitors such as tacrolimus and cyclosporine, and antitumour necrosis factor (anti-TNF) therapy. Corticosteroid dependence and probability of colectomy remain clinically significant challenges, the latter most commonly due to failed medical therapy.
Sustained remission in inflammatory bowel disease (IBD) with currently available therapies is achieved in no more than 20%–30% of patients with chronic disease.3 ,4 Patients treated with anti-TNF therapy eventually lose response and are at risk of serious side effects.5 ,6 Therefore, there is a need to develop more targeted therapies optimised for chronic use that can modify the disease while decreasing dependence on corticosteroids and the probability of progression to surgery, without compromising immune competence. New therapies should provide improved safety profiles with sustained remission and mucosal healing, thereby preventing long-term complications in a greater proportion of patients, including patients who do not respond to standard of care.
Integrins are cell surface glycoproteins that play a role in leucocyte adhesion, signalling, proliferation and migration,7 aiding in leucocyte recruitment from blood into both lymphoid and non-lymphoid tissues in a highly regulated manner.8 Migration into the gastrointestinal tract is highly dependent on interactions between the α4β7 integrin expressed on the surface of the leucocyte and its ligand, mucosal addressin cell adhesion molecule (MAdCAM-1) expressed on endothelial cells.9 α4β7 Binds preferentially to MAdCAM-1 expressed on the high endothelial venules of mucosal-associated lymphoid tissues, and has limited binding to vascular cell adhesion molecule 1 (VCAM-1) and the fibronectin fragment CS-1.10–12 α4β7+ Cells and MAdCAM-1 expression are elevated at sites of chronic inflammation such as in the intestinal tract of patients with UC.13 ,14
The αEβ7 integrin, another member of the β7 integrin family, is expressed exclusively on mucosal intraepithelial T lymphocytes. αEβ7 binds selectively to E-cadherin on epithelial cells and has been proposed to play a role in the retention of T cells in mucosal tissue,15–17 which exhibit cytotoxicity against stressed or infected epithelial cells.18–20 The expression of αEβ7 is elevated in UC and Crohn's Disease (CD) in the active phase of the disease,21 ,22 and anti-αEβ7 treatment attenuates experimental colitis in mice, implicating a role for αEβ7+ lymphocytes in experimental models of IBD.23 Interestingly, genetic polymorphisms at the CDH1 locus, which encode E-cadherin, have been associated with susceptibility to UC.24 ,25 In addition, CDH1 polymorphisms associated with CD may result in the generation of a truncated E-cadherin protein with abnormal intracellular accumulation and impaired plasma membrane localisation.26 Studies of gene targeted animals with E-cadherin defects in the gut epithelium have demonstrated that this protein is critical for maintenance of intestinal epithelial integrity and defense.27 ,28 Thus, while alterations in the E-cadherin homotypic interactions likely underlie the increased disease susceptibility associated with CDH1 polymorphisms, dysregulated E-cadherin expression or function may also result in altered αEβ7+ cell responses.
Etrolizumab is a humanised IgG1 monoclonal antibody which targets the β7 subunit of the α4β729 ,30 and αEβ715 integrins with high affinity, blocking binding to their ligands, MAdCAM-1 and E-cadherin, respectively. Murine and primate studies indicate that etrolizumab provides selective targeting of lymphocyte homing to mucosal sites, with limited to no effects on migration to non-mucosal tissues.31 ,32 Amelioration of IBD in nonclinical models is associated with downmodulation and/or disappearance of α4β7 and αEβ7 cells from the inflamed mucosa.33–38
Etrolizumab provides a dual approach mechanism of action for the treatment of IBD that involves inhibition of leucocyte trafficking to the gut (through blocking of α4β7–MAdCAM-1 interactions) as well as inhibition of retention of leucocytes in the intraepithelial lining of the gut (through blocking αEβ7–E-cadherin interactions), which may provide improved efficacy over currently available therapies.
Materials and methods
A randomised, placebo-controlled, double-blind within-cohort study was designed to evaluate the safety and tolerability of etrolizumab in patients with moderate to severe UC. Secondary objectives included the characterisation of the pharmacokinetic (PK) profile, and to conduct an initial assessment of immunogenicity. Exploratory objectives were to evaluate pharmacodynamic (PD) properties and evaluate potential clinical activity. Study participants were recruited from 15 centres in Canada, the USA and Europe (the UK, Belgium, The Netherlands, Germany). Institutional review boards at each study site approved the protocol, and all subjects provided written informed consent. The trial was registered under US National Institutes of Health ClinicalTrials.gov identifier NCT00694980.
Patients
Eligible patients included male and female subjects, 18–70 years of age, with a diagnosis of UC and a Mayo Clinic Score (MCS)39 of ≥5 points at screening. Patients had a disease duration of ≥12 weeks at screening, and were on stable doses of oral 5-aminosalicylic acid (2.4–4.8 g/day) or oral steroids (≤20 mg/day of prednisone or prednisone equivalent), or azathioprine, 6-mercaptopurine or methotrexate. Key exclusion criteria included hospitalisation due to severity of UC; evidence of moderate to severe anaemia (haemoglobin <10 g/dl); risk of infection including presence of JCV DNA in serum, such as ongoing or history of severe infections; recent live vaccinations; concomitant medications including anti-TNF, cyclosporine or tacrolimus within 3 months; and any manifestation of UC or other conditions likely to require, in the investigator's judgement, treatment with >20 mg/day of prednisone or prednisone equivalent.
Study design
We initiated a randomised within cohort single ascending dose (SAD) stage followed by a randomised within cohort, multiple dose (MD), parallel-treatment stage. The interactive voice response system conducted the randomisation/treatment assignment through the use of a process designed by the biostatistician. In the SAD stage of the study, patients (n=5/cohort; 4 etrolizumab to 1 placebo; total n=25) were enrolled sequentially, in ascending doses, into five cohorts (Cohorts A–E) and treated with single intravenous or subcutaneous (SC) doses at one of four dose levels (0.3, 1.0, 3.0 or 10.0 mg/kg) (figure 1). Enrolment in one cohort was completed before the next cohort opened. In the first dose cohort (Cohort A, 0.3 mg/kg intravenous) no more than one patient was treated per 24 h, and each patient was monitored for 24 h for acute adverse reactions before the next patient was treated. Within subsequent cohorts (Cohorts B–E), dosing of the first two patients in each cohort was staggered by 24 h.

Study design and patient flow. IV, intravenous; SC, subcutaneous; SAO, single ascending dose; DE, dose escalation.
Safety was evaluated after a cohort had completed enrolment and all patients in the cohort had completed follow-up through day 15. If the safety data were acceptable as determined by the number of dose limiting toxicities (DLTs), then enrolment of the subsequent cohort was initiated. Patients were followed for 18 weeks after the single dose of study drug was administered, with follow-up visits at days 1, 2, 4, 8, 15, 22, 29, 43, 57, 71, 99 and 127.
The MD parallel stage was commenced ∼10 weeks after dosing was completed for the last patient treated in Cohort E (3.0 mg/kg SC) of the SAD stage, and was conditional on adequate PK properties being observed in the SAD stage and establishment of a maximum tolerated dose (MTD) for etrolizumab of >10 mg/kg intravenous. In the MD stage, patients were enrolled concurrently and randomised within cohort to receive placebo (n=5) or etrolizumab at the following doses: 0.5 mg/kg SC (Cohort F; n=4), 1.5 mg/kg SC (Cohort G; n=5), 3.0 mg/kg SC (Cohort H; n=4) or 4.0 mg/kg intravenous (Cohort I; n=5). In the MD stage, no patients were enrolled who participated in the SAD stage.
Patients were treated with single intravenous or SC doses every 4 weeks for three cycles, at days 1, 29 and 57. Patients were followed for safety for 20 weeks (through day 197) after the final dose of study drug was administered during the MD stage of the study.
Assessments
Safety
Safety data were assessed at each visit and graded according to the National Cancer Institute Common Toxicity Criteria Adverse Event (NCI CTCAE), V.3.0. A DLT was defined as any NCI CTCAE Grade ≥3 adverse event (AE) occurring within 14 days of study drug administration during the SAD stage of the study. Grade 3 infusion related toxicities were considered DLTs. Exacerbation of UC was not considered a DLT. The MTD was defined as the dose level below the one at which ≥2 patients treated with etrolizumab experienced a DLT. It was determined in the protocol that if no more than one patient experienced a DLT in each of the five SAD cohorts, the MTD will not have been defined and the highest tested dose will be 10 mg/kg intravenous.
All patients were required to have undetectable JCV DNA in the serum in order to take part in the study. Each patient underwent regular cognitive assessment and completed a progressive multifocal leucoencephalopathy (PML) symptom assessment interview during the treatment and safety follow-up stages of the study. Any patient who developed detectable JCV DNA in the serum during the study had neurological assessment by a neurologist and a brain MRI. There was also a PML expert panel available to evaluate and adjudicate in real time any suspected cases of PML. After completion of the safety follow-up stage of the study, as per the study risk-monitoring plan, patients were monitored for signs and symptoms of PML for a total of 2 years following initial dosing with the study drug using the PML Symptom Assessment Interview. Total and differential white cell count was also measured during the 2-year follow-up.
Pharmacokinetics
In the SAD stage of the study, PK serum samples were obtained predose, postdose on study day 1 (1 h-intravenous only and 4 h) and then on study days 2, 4, 8, 15, 22, 29, 43, 57, 71, 99 and 127 (or at early treatment termination). In the MD stage, PK serum samples were obtained on days 1 (predose, 1 h postdose-intravenous only, 4 h postdose), days 2, 15 and 29 (predose, 1 h postdose-intravenous only, 4 h postdose), days 30, 43 and 57 (predose, 1 h postdose-intravenous only, 4 h postdose) and days 58, 60, 64, 67, 71, 85, 99, 113, 141, 169 and 197 (or at early treatment termination).
Etrolizumab concentration was determined using a validated ELISA assay with a lower limit of quantification of 0.0125 μg/ml. The following PK parameters were assessed: maximum serum concentration (Cmax), area under concentration–time curve from time 0 to infinity, SAD (AUC), area under concentration–time curve during a dosing interval, MD (AUCτ), total body clearance at steady state after intravenous doses (CLss) or apparent total body clearance at steady state after SC doses (CLss/F) and elimination half-life (t1/2z). PK parameters were calculated using a standard non-compartmental method with the computer program WinNonlin V.5.2.1 (Pharsight Corporation, Mountain View, California, USA).
For antitherapeutic antibody (ATA) measurement, serum samples were collected at study days 1 (predose) and days 15, 29, 57, 99 and 127 in the SAD stage. For the MD stage, samples were collected at days 1 (predose), day 29 (predose), day 57 (predose) and days 64, 85, 113, 141, 169 and 197.
Serum was analysed for the development of ATAs using a validated antibody-bridging electrochemiluminesence assay. Serum samples were first screened for the presence of ATAs. Samples that screened positive for ATA were confirmed by immunodepletion. Samples with confirmed ATA responses were further diluted to obtain titre values. ATAs in serum were detected with a minimum reportable titre of 1.30 titre units (corresponding to a minimum 1/20 sample dilution).
PD evaluations
Drug occupancy on target CD4+ lymphocytes (‘available’ β7 receptors) following single or multiple intravenous or SC administration of etrolizumab was evaluated on subsets of CD4+ T cells in peripheral blood of patients. CD4+ T cells were assessed by flow cytometry according to expression of CD45RA and β7, and phenotypically subdivided according to homing properties into subsets of CD4+CD45RA-β7high T cells (mucosal-homing), CD4+CD45RA-β7low T cells (peripheral homing) and CD4+ CD45RA+ T cells (cells that traffic well to both intestinal and peripheral lymph nodes and tissues).40
In the SAD stage of the study, peripheral blood samples were obtained at screen, day 1 (predose) and days 2, 8, 15, 22, 29, 43, 57, 71, 99 and 127. In the MD stage, peripheral blood samples were obtained at screen, day 1 (predose), days 2, 15 and 29 (predose), days 30, 43 and 57 (predose) and days 58, 64, 72, 85, 99, 113, 141, 169 and 197.
Occupancy of etrolizumab was assessed using fluorescently-labelled etrolizumab. The absolute number (ABS) of T lymphocyte subsets was assessed at each time point and expressed as a percentage of predose baseline (% BL) T lymphocytes. Baseline was defined as the average of the predose (screen and day 1 predose) values. For each cohort, the mean absolute count (% BL) for patients treated with etrolizumab was calculated. Placebo patients from the single and MD cohorts were combined and used as the controls for the single and MD analyses, respectively.
Correlative analysis of pharmacokinetics and pharmacodynamics
A total of 38 etrolizumab treated patients were evaluated to determine the minimal serum etrolizumab concentration that is required to maintain full occupancy of the β7 integrin. Five of the 38 treated patients were excluded from the PK/PD analysis due to missing PK and PD values as they terminated early from the study.
Clinical activity
Exploratory analyses were conducted for the SAD and MD stages to evaluate the effect of etrolizumab on the MCS compared with baseline on days 29 (SAD only) and days 43 and 71 (MD stage only). A clinical response was defined as ≥3 points and 30% reduction from baseline in MCS, and ≥1 point decrease in rectal bleeding subscore or absolute rectal bleeding score of 0 or 1. Clinical remission was defined as MCS ≤2 with no individual subscore exceeding 1 point.
Statistical analysis
The cohort sample size was determined by estimating the minimum number or patients required to safely escalate at different DLT rates to the next dose level in the SAD. All patients who received at least one dose of study drug or placebo were included in the safety, PK and PD analyses. AEs, vital signs, laboratory tests, PK, PD and disease activity measurements were descriptively compared across the various treatment groups with no formal statistical testing.
Results
Study population
Patient characteristics were similar between active drug and placebo recipients (table 1) in both the SAD and MD stages of the study. Information on previous therapy with anti-TNF was not available for patients who took part in the SAD stage of the study. In the MD stage, concomitant treatment for UC was imbalanced between placebo and the active treatment arms with a greater proportion of placebo patients receiving concomitant corticosteroid therapy (80% placebo vs 44% for etrolizumab). In addition, among patients who received active treatment with etrolizumab, there were a greater proportion of patients who had been treated with anti-TNF therapy (76% etrolizumab vs 40% on placebo). The mean MCS for patients participating in the phase I study was 9.0±1.7 (SD) indicating they had severe UC.
Patient demographics and baseline characteristics
The demographics and baseline characteristics for patients enrolled in the SAD and MD stages, per cohort, are presented in Supplementary table 1. Although each cohort has data for only four or five patients, these covariates appear balanced across cohorts. The only covariate with a high variability is disease duration, higher in the 1 mg/kg intravenous and 3 mg/kg intravenous than the other SAD cohorts.
Patient disposition
Overall, 26 patients in the etrolizumab group and seven patients in the placebo group completed the study. Of the 25 subjects enrolled in the SAD stage, seven subjects discontinued early from the study, five in the etrolizumab treated groups and two in the placebo group. Of the 23 subjects enrolled in the MD stage, eight subjects discontinued early from the study: seven in the etrolizumab treated groups and one in the placebo group. Subject disposition and the reasons for premature discontinuation for subjects treated during the SAD and MD stages of the study are presented in table 2.
Study completion status
Safety
Etrolizumab was well tolerated during the study. The overall incidence of treatment-related AEs was marginally higher in the active treatment group (table 3), and occurred in the first 24 h of treatment. There was no dose-dependent increase in the incidence of AEs. Headache was the most common AE occurring more often in actively treated patients. No DLTs or clinically significant infusion reactions or injection site reactions were reported.
Safety outcomes
Eight patients had serious AEs: seven in the SAD stage (6:1; etrolizumab:placebo) and one in the MD stage (1:0; etrolizumab:placebo). Exacerbation of UC was the most common serious AE. There were two patients with impaired wound healing posturgent colectomy for exacerbation of UC. Both patients had severe UC with baseline MCS of 9 and 11. Each patient received a single dose of etrolizumab at dose levels of 1 mg/kg intravenous and 3 mg/kg SC, respectively. There were significant confounding factors for impaired wound healing such as the use of high dose steroids, hypoalbuminemia, type 2 diabetes mellitus and obesity in one patient, and hypoaluminemia and obesity in the second patient. Two other patients also underwent colectomy after exposure to single doses of etrolizumab in the phase I study without any postoperative complications.
Two patients, one in the SAD stage and one in the MD stage, developed transient low level JCV shedding into the serum at levels of 20–30 viral copies/ml. Neither patient developed any symptoms suggestive of PML and both had no significant abnormalities on brain MRI.
Pharmacokinetics
The PK behaviour of etrolizumab was generally consistent with that of a typical humanised IgG1 monoclonal antibody (figure 2). The PK profile of etrolizumab is generally linear and dose proportional with regard to Cmax and AUC (SAD) or AUCτ (MD) within the dose range tested. A slight trend of nonlinearity was observed at doses ≤1.0 mg/kg, possibly due to target-mediated clearance at lower etrolizumab serum concentrations. The slight non-linearity of PK at doses ≤1.0 mg/kg resulted in a shorter mean t1/2z at lower doses (4.8–7.4 days at 0.3–1 mg/kg) compared with higher doses (10.6–13.1 days at 3–10 mg/kg). These slight differences in half-life and PK linearity had no impact on the exposures (Cmax, AUC) and the overall PK dose proportionality across the dose ranges tested. The apparent bioavailability following a single SC administration, based on cross-cohort comparison with intravenous single dose, was estimated to be approximately 67% at 3 mg/kg. Mean CLss/F ranged from 5.1 to 10.1 ml/day/kg following multiple SC doses and showed no time-dependent changes as compared with a single SC administration. The repeated monthly dosing for three doses resulted in moderate drug accumulation over time (accumulation ratio for AUCτ of approximately 1.2-fold for intravenous and 2.0-fold for SC).
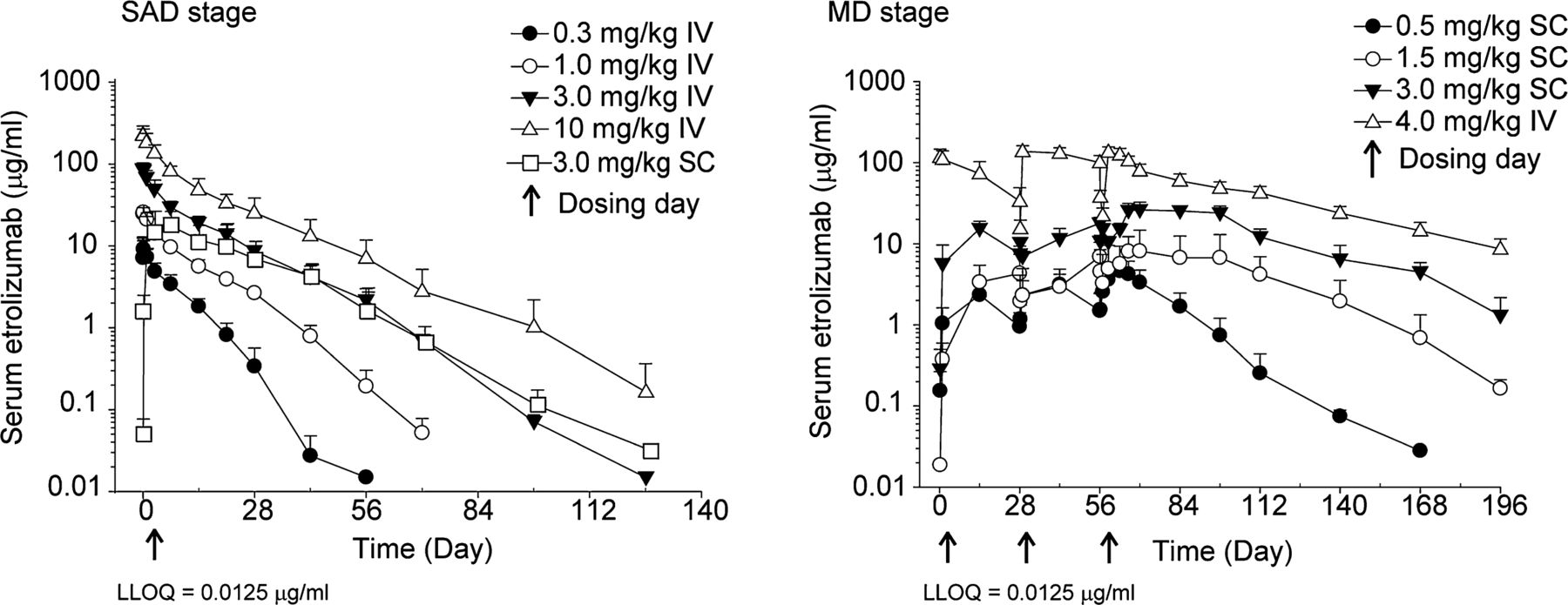
Concentration–time profiles following single or multiple doses given by IV or SC administration. The lower limit of quantification is 0.0125 μg/ml. IV, intravenous; MD, multiple dose; SAD, single ascending dose; SC, subcutaneous; LLOQ, lower limit of quantitation.
Antietrolizumab antibodies were detected in two out of a total of 38 drug-treated patients. One patient was from the lowest dose cohort in the SAD stage (0.3 mg/kg intravenous) with positive ATA detected in a range of 1.6–2.02 titre units on study day 29 and all subsequent ATA sample collection days up to day 197. Another patient was from the lowest dose cohort in the MD stage of the study (0.5 mg/kg SC) with positive ATA confirmed by immunodepletion but with < 1.3 titre units on study day 169 and 1.44 titre units on study day 197. In these two ATA positive patients, the ATA titre levels were low and close to the minimum reportable level (1.3 titre unit). There were no effects on PK profile for either of these patients nor were there any AEs suggestive of an allergic reaction.
Pharmacodynamics
Etrolizumab occupied β7 integrin on CD45RA-β7high CD4+ T cells in all cohorts (ABS, % BL, figure 3). There was an observed dose-dependent trend in the duration of occupancy in the actively treated patients. For the cohorts receiving single intravenous doses of 0.3, 1.0 and 10 mg/kg, cohort mean occupancy was maintained for approximately 2 weeks, 6 weeks and 10 weeks, respectively. The extent and duration of β7 occupancy was similar in the 3 mg/kg SC and 3 mg/kg intravenous cohorts, with cohort mean occupancy maintained for approximately 10 weeks.

Occupancy (mean ± SD) of β7 integrin on CD4+CD45RA-β7high T lymphocytes following administration of etrolizumab or placebo. BL, baseline; IV, intravenous; MD, multiple dose; SAD, single ascending dose; SC, subcutaneous.
For the cohorts administered multiple SC doses of 0.5 mg/kg and 1.5 mg/kg etrolizumab, cohort mean occupancy was maintained for ∼2–4 weeks postfinal dose (day 57), whereas in the cohorts dosed with 3 mg/kg SC and 4 mg/kg intravenous, there was an extended duration of occupancy which was maintained for ∼8–12 weeks postfinal dose. The duration of occupancy was also longer when comparing the cohort receiving a single SC dose of 3 mg/kg etrolizumab (∼10 weeks) compared with those receiving multiple SC doses of 3 mg/kg (∼12 weeks).
Recovery of unoccupied β7 receptors on gut-homing T lymphocytes was associated with decreasing serum drug concentrations. A consistent PK–PD relationship was observed in both the single dose and MD cohorts in which a minimum serum concentration range of 0.05–2.7 μg/ml of etrolizumab (mean ± SD =0.5±0.6 μg/ml, median =0.3 μg/ml) maintained occupancy of CD4+ CD45RA-β7high T lymphocytes (see supplemental figure 1).
There was no change from baseline in the total lymphocyte count in patients who received etrolizumab (data not shown).
Clinical activity
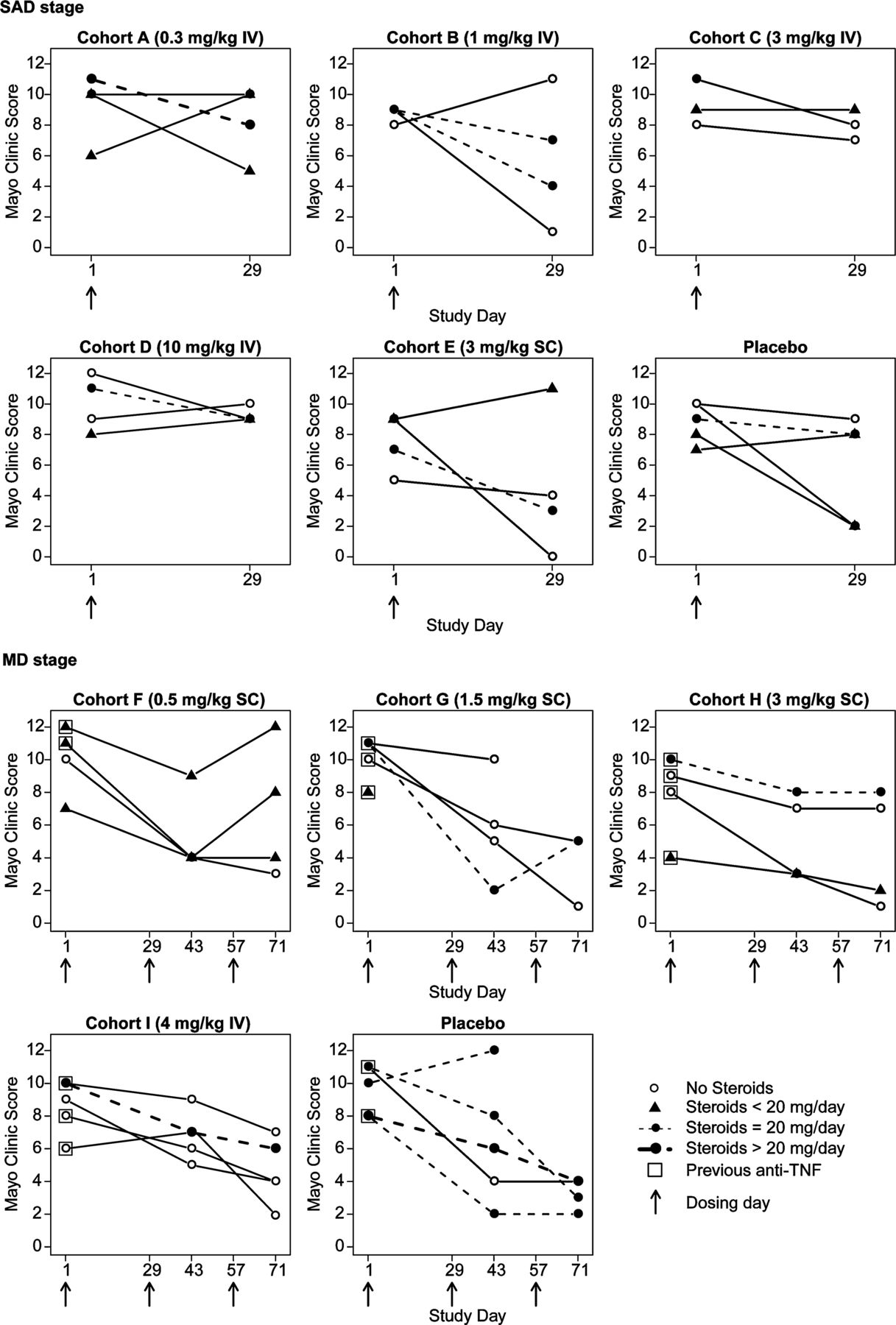
In the SAD stage, individual changes in MCS show a trend for a decrease in the MCS in the low dose etrolizumab treated groups, especially in those where etrolizumab was administered SC (figure 4). In the MD stage, there was a similar trend for decrease in MCS in the low dose SC groups treated with etrolizumab (figure 4). The trend for decrease in the MCS in the etrolizumab treated groups was not different from that observed in the placebo treated patients and is confounded by the imbalanced use of oral corticosteroids in the placebo treated patients compared with the etrolizumab treated groups (table 1). Endoscopy scores were evaluated as an exploratory end point and tracked well with the overall MCS (supplementary figure 2).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Individual patient changes in Mayo Clinic score. IV, intravenous; MD, multiple dose; SAD, single ascending dose; SC, subcutaneous; TNF, tumour necrosis factor.
Discussion
The humanised monoclonal antibody etrolizumab targets the β7 subunit of the α4β7 and αEβ7 integrin heterodimer receptors. In this phase I study, etrolizumab was safe and well tolerated in patients with moderate to severely active UC. PK data indicate that etrolizumab has acceptable PK parameters to enable monthly SC administration. There were no early indications that etrolizumab is immunogenic on repeated intravenous or SC administration. Etrolizumab achieves immediate occupancy of β7 integrin expressed on peripheral blood T cells, the duration of which is dose-dependent. Although the numbers are small, and there is an imbalance in UC concomitant medication between placebo and the active treatment arms in the MD stage of the study, there is an early indication of clinical activity of etrolizumab in patients with severe UC.
Clinical studies have demonstrated efficacy with an anti-α4 integrin antibody (natalizumab) for the treatment of CD.4 Unlike natalizumab, which blocks the function of both α4β1 and α4β7 heterodimers, etrolizumab does not interfere with the function of α4β1, an integrin heterodimer critical for the homing of leucocytes to the central nervous system through interactions with VCAM-1.41 Notably, treatment with natalizumab has been associated with confirmed cases of PML.42 The selectivity of etrolizumab was confirmed by its inability to inhibit adhesion of α4β1+α4β7− Ramos cells to VCAM-1 at concentrations as high as 100 nM (data not shown). Consistent with these in vitro data, preclinical rodent studies have demonstrated that blockade ofβ7 integrin can prevent the recruitment of T cells to the inflamed colon in a mouse model of IBD while having no effect on lymphocyte homing to the brain in a mouse model of multiple sclerosis.31 ,34 ,43 In addition, healthy cynomolgus monkeys treated with etrolizumab had an increase in peripheral blood mucosal-homing CD45RA-β7high CD4+ T cells with no substantial effect on CD4+ T cell subsets that home preferentially into non-mucosal sites.31 Following 26 weeks of treatment, etrolizumab induced a dose-dependent reversible decrease in gut-associated lymphoid tissue in cynomolgus monkeys and lymphocytes in murine intestinal lamina propria.32 These findings point to the ability of etrolizumab to specifically inhibit trafficking of mucosal-associated leucocytes implicated in disease pathogenesis of IBD while sparing the function of T cell subsets expressing other integrins subunits, such as α4β1, which are critical for normal host protective immune responses.
Clinical efficacy has also been demonstrated with the anti-α4β7 antibody vedolizumab (MLN-0002, LDP-02) in UC and CD.35 ,36 As with vedolizumab, etrolizumab binds α4β7 but is unique in that it also blocks binding of αEβ7 to its ligand E-cadherin. Thus, etrolizumab could affect leucocyte composition within the gut mucosa through multiple mechanisms. First, as discussed above, it could block α4β7-expressing leucocytes from entering the gut by inhibiting their extravasation through MAdCAM-1-expressing mucosal endothelial venules. Notably, MAdCAM-1 expression is increased in patients with UC and CD,13 and antibodies against β7 or α4β7 are effective in blocking migration of lymphocytes into the inflamed gut mucosa in animal models.31 ,33 ,34 ,43 Second, by inhibiting αEβ7 interactions with E-cadherin, it could directly affect retention of leucocytes within the intestinal mucosa.17 αE integrin expression is very low in peripheral blood and is found primarily on gut resident cells, including intraepithelial lymphocytes44 and dendritic cells.45 More than 90% of intraepithelial lymphocytes and 50% of T cells in the human intestinal lamina propria express αEβ7 integrin, suggesting a distinctive role in mucosal immunology.46 ,47 In addition, αEβ7 is expressed on intestinal dendritic cells, which have been implicated in the generation of gut-tropic effector T cells.48 Importantly, intraepithelial lymphocytes may exhibit cytotoxic activity against epithelial cells19 ,20 and cells expressing αEβ7 integrin are pathogenic in mouse models of colitis and acute graft versus host disease.23 ,49 Thus, etrolizumab represents a novel therapeutic approach for UC by virtue of its unique ability to inhibit both recruitment and retention of potentially pathogenic immune cells in the inflamed gut mucosa.
α4β7 And αEβ7 can regulate trafficking and retention of immune cells in both the intestine and other mucosal tissues such as the lung. Blockade of these integrins could theoretically impact immune function at these sites, potentially leading to opportunistic infection. Intraepithelial lymphocytes expressing αEβ7 play an important role in intestinal immunity to Toxoplasma gondii.50 However, αEβ7 has recently been reported to be dispensable for immunity to the intestinal helminth pathogen Trichuris muris.51 Thus, αEβ7 and intraepithelial lymphocytes participate in mucosal immune responses but their relative importance is unclear and would likely need to be evaluated on a pathogen-by-pathogen basis. We have not directly evaluated the effect of β7 integrin blockade in animal models of opportunistic or enteric infection, but in our 26-week toxicology studies in mice and cynomolgus monkeys, there were no instances of enteric infection despite an etrolizumab-induced decrease in gut-associated lymphoid tissue.32 Similarly, there were no pulmonary safety signals in our study. In addition, our observation of no change from baseline in the total blood lymphocyte count in patients who received etrolizumab reflects the relatively small percentage of leucocytes affected by binding of etrolizumab.
The favourable safety profile observed with etrolizumab treatment in this trial warrants larger and longer-term studies to confirm our findings with etrolizumab in moderate to severe UC. A definitive phase II study is currently underway.
Acknowledgments
We wish to thank the patients and investigators who participated in this study. We thank Dr Banmeet Anand for his help in PK data analysis in the early stages of this study. Medical writing assistance was provided by Genentech, Inc.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement 1 - Online figure 1
- Data Supplement 2 - Online figure 2
- Data Supplement 3 - Online figures and tables
Footnotes
-
Funding This study was sponsored by Genentech, Inc.
-
Competing interests PR: honoraria for participating in scientific advisory boards by Genentech. RF: research grant funding from Genentech. DH: advisory board funding from Genentech. AS: payment for talks or consultancy or scientific research projects from Abbott, ArdeyPharm, Bayer-Schering, Centocor, Essex, Dr. Falk Pharma, Ferring, Fresenius, Shire, Merckle-Recordati, MSD, Travacare, UCB, Vifor and Genentech. DCB: honoraria for participating in scientific advisory boards organised by Genentech. All of his activities and contracts are in conformity with the ‘FSA-Kodex Fachkreise’ (voluntary self-monitoring code for expert consultants to the pharmaceutical industry), have been checked by the Medicolegal Department of Charité and have been approved by the directorate of the Faculty of Medicine. BB: Abbott, Merck, GSK, BMS, Amgen, Millenium Pharmaceuticals and Genentech. SS: Ad board services for Genentech. JM: Ad board services for Genentech. MW, MT, JV, XW, MK, DL, DD, JE and SO are employees of Genentech.
-
Patient consent Obtained.
-
Ethics approval Ethics approval was provided by Institutional review boards at each study site approved the protocol.
-
Provenance and peer review Not commissioned; externally peer reviewed.