Article Text

Abstract
P-21 activated kinases (PAKs) are effectors of Rac1/Cdc42 which coordinate signals from the cell membrane to the nucleus. Activation of PAKs drive important signalling pathways including mitogen activated protein kinase, phospoinositide 3-kinase (PI3K/AKT), NF-κB and Wnt/β-catenin. Intestinal PAK1 expression increases with inflammation and malignant transformation, although the biological relevance of PAKs in the development and progression of GI disease is only incompletely understood. This review highlights the importance of altered PAK activation within GI inflammation, emphasises its effect on oncogenic signalling and discusses PAKs as therapeutic targets of chemoprevention.
- INFLAMMATORY BOWEL DISEASE
- COLON CARCINOGENESIS
- GASTROINTESTINAL CANCER
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Most GI disorders are triggered by three main denominators: the environment (Western lifestyle), gut microbiota and genetic susceptibility. Such genes control various gut functions such as control of GI inflammation and tumourigenesis. Alterations in GI homeostasis via disruption of cell polarisation, proliferation, differentiation and apoptosis are associated with disease. One, albeit not fully appreciated, group of key regulators of diverse gut functions includes the p-21 activated kinases (PAKs). PAKs are involved in the innate immune response, barrier function, maintenance of crypt proliferation, epithelial cell migration and cell survival.1 ,2 As PAKs are nodal kinases orchestrating multiple signalling cascades, they are attractive targets for therapeutic intervention.3
PAKs are highly conserved serine/threonine kinases from Saccharomyces cerevisiae to Homo sapiens.4 ,5 Mammals have six different PAKs which are classified based on their homology into group I (PAK1–PAK3) and group II (PAK4–PAK6)1 (table 1). Interestingly, Drosophila only has three PAKs, in which two are homologous to group I, and one homologous to group II.6 Overall, PAK1 and PAK4 are the most investigated of all PAKs, and have been brought into focus due to their association with various malignancies.1 PAK overexpression contributes to tumour invasiveness in uveal melanoma,7 ,8 neurofibromatosis,9 breast,10 ,11 cervical,12 colon,13 ,14 oesophageal,15 gastric,16 ,17 hepatic,18 lung,19 ovarian,20 prostate21 ,22 and thyroid cancer.23 PAKs were also correlated to inflammatory diseases such as rheumatoid arthritis24 and asthma.25 Here we highlight the importance of PAK activation and their role in the pathogenesis of GI inflammation and malignant transformation. Our focus is built around PAK1, although the significance of other PAKs in GI disease has also been brought into context.
Overview of groups I and II P-21 activated kinase (PAK) chromosomal location
Structure and activation of PAKs
The PAK1 gene in H sapiens is located on chromosome 11q13.5 and includes 20 different splice variants.26 The largest isoform consists of 14 exons and encodes 545 amino acids.26 ,27 It is under transcriptional control of Forkhead Homeobox type O (FOXO) transcription factors which directly interact with the PAK1 promoter.28 PAK4 is located on 19q13.2, includes 13 splice variants, the largest of which consists of eight exons, and encodes 591 amino acids.26
Crystal structure revealed three conserved domains in all group I PAKs. The N-terminus contains a regulatory domain comprising a protein binding domain (PBD) that overlaps with an auto-inhibitory domain (AID) and a kinase domain at the C-terminus (figure 1). The AID provides an auto-inhibited homodimer where the AID of one PAK molecule overlaps with the kinase domain of the other.29 Recently, it was shown that group II PAKs also contain a sequence-related AID, although they do not form auto-inhibited homodimers.30 Additionally, proline-rich sequences within the regulatory domain facilitate interactions with SH3 domain containing adapter molecules (figure 1A).

Structure and activation of P-21 activated kinase (PAK)1. (A) Based on structural and biochemical studies, the mechanism of PAK activation is conserved in group I. The N-terminal auto-inhibitory domain (AID) keeps PAK1 as a dimer in an auto-inhibited state, blocking substrate binding through stabilisation of an inactive conformation. The AID partially overlaps with the GTPase binding domain. (B) GTP-bound Rho GTPases (Cdc42/Rac1) releases PAK1 from its auto-inhibitory conformation, allowing its auto-phosphorylation (Thr-423). Phosphorylation at Thr-423 is critical for PAK1 activity. Subsequently, additional residues are phosphorylated at N-terminus, blocking auto-inhibition.
PAK activation is complex and is initiated via GTPases, membranous phoshoinositides, adaptor proteins, growth factors and effectors of intestinal bacteria such as the enterohaemorrhagic Escherichia coli O157:H7 type III effector EspG.1 ,31 ,32 Rho GTPases Cdc42/Rac1, as well as Wrch-1,33 directly bind to group I PAKs within the N-terminal PBD, also referred as the GTPase binding domain or Cdc42/Rac1-interactive binding (CRIB).5 Subsequent auto-phosphorylation at multiple N-terminal residues releases the dimer and initiates C-terminal kinase activation (figure 1B). Phosphorylation at residue Thr-423 is critical for the maintenance and stability of PAK1 activation.5 Group II PAKs have a higher specificity to Cdc42 in comparison with Rac1. Interestingly, PAK4 is constitutively phosphorylated at Ser474, but only becomes activated upon Cdc42 binding.30 Cdc42 also regulates the cellular localisation of PAK4.34 ,35
Selyunin and Alto recently identified an alternative mechanism of group I PAK activation by EspG, a virulence effector protein from enterohaemorrhagic E coli.32 EspG directly binds the AID in a region independent from the CRIB domain to disrupt the homodimer and initiate PAK activation.31
Membranous phosphoinositides such as phosphatidylinositol 4,5-bisphosphate bind to the N-terminal basic region of PAK1 and assist in Rac1-mediated activation.36 Sphingosine disrupts PAK1's auto-inhibited state and the enzyme 3-phosphoinositide-dependent kinase-1 (PDK1) directly phosphorylates PAK1 at Thr-423.37
Adaptor proteins such as growth factor receptor-bound protein 2 (GRB2),38 PAK-interactive exchange factor (PIX)39 and Nck40 interact with proline-rich motifs of PAK1, thereby recruiting PAK1 dimers to the membrane and increasing their interaction with GTPases and lipids. Adaptor proteins also influence PAK1's localisation within the cytoplasm or within the nucleus.41 Growth factors such as epidermal growth factor (EGF) or hepatocyte growth factor (HGF) also contribute to PAK activation. EGF activates the protein kinase CK2 which phosphorylates PAK1 at S-223 and contributes to its kinase activation42 whereas HGF initiates PAK4 kinase activation through a phospoinositide 3-kinase (PI3K)-AKT2 mechanism which lacks clarity.43 ,44 The extracellular matrix glycoprotein vitronectin also activates PAK4, and stimulates its migration from the cytoplasm to the membrane.45 ,46
PAK physiology: kinase versus scaffolding functions
PAKs are predominantly located in the cytoplasm, although they have roles at the membrane and within the nucleus. PAK1 has both kinase dependent and independent functions in controlling cytoskeletal dynamics, cell migration, influencing mitosis and survival.1
Cytoskeletal dynamics
PAK1's role in cytoskeletal remodelling includes the phosphorylation of multiple targets as well as protein–protein interactions independent of its kinase activity. PAK1 facilitates actin stabilisation through phosphorylation of myosin light chain (MLC), LIM kinase, filamin A and dynein light chain 1 (DLC1).47 ,48 PAK1 contributes to microtubule disassembly via phosphorylating stathmin (STMN) and tubulin B.48 Lamellipodia formation and membrane ruffling still occur in PAK1 kinase mutants; thus, PAK1's PBD regulates cytoskeletal remodelling in absence of an activated kinase.49 The role of PAK4 in cytoskeletal reorganisation has also been demonstrated and is mechanistically different from group I PAKs. PAK4 does not phosphorylate MLCK; instead, Cdc42 recruits PAK4 to the Golgi and induces polymerisation of actin and the formation of filopodia.50 PAK4 kinase dead mutants were shown to block filopodia formation; however, stimulation of PAK4 kinase activity was not sufficient to alter cytoskeletal dynamics in the absence of Cdc42.35
Switching from adherence to migration
Cell migration involves cytoskeletal rearrangements in addition to turnover of focal adhesion contacts to the stroma at the leading edge of the cell.51 PAK1 translocation to the membrane via adaptor proteins influences such turnover of focal adhesion contacts via phosphorylation of focal adhesion kinase (FAK).52 Wound healing assays have demonstrated that a multiprotein complex including PAK1, PIX and G-protein receptor kinase interactor 1 (GIT1) translocates from focal adhesion contacts to cell-to-cell contacts in order to initiate wound closure and contact inhibition.53 Intestinal epithelial cells must also balance signalling between focal adhesion contacts at the stroma and cell-to-cell contacts such as adherens junctions (AJ). Cadherins such as E-cadherin are a critical component of AJ, and control the localisation of the PAK1–PIX–GIT1 complex at focal adhesion contacts. This complex may contribute to a crosstalk between AJ and focal adhesion contacts.54 PAK1 regulates E-cadherin transcription, albeit through a separate pathway. PAK1 phosphorylates and activates Snail, a transcriptional repressor of E-cadherin.55 PAK4 overexpression leads to the disassembly of stress fibres and focal adhesion contacts thereby promoting anchorage independent growth.50 HGF induces an interaction between GRB2-associated binding protein 1 (Gab1) and PAK4 at the cell periphery, and disruption of the Gab1–PAK4 complex impeded cell migration and invasion.56 PAK4 activation by vitronectin increases cell motility. Specifically, PAK4 increases integrin αvβ5 turnover, disrupts actin–integrin complex and weakens cell adhesion.45
PAK nuclear translocation and its regulatory effects on transcription
Growth factors such as EGF induce the translocation of PAK1 to the nucleus. PAK1 has three nuclear localisation signals (NLS1–NLS3).57 Its nuclear translocation requires binding of DLC1 to activated PAK1.58 Activation of PAK1 facilitates an interaction between NLS2 and importin to induce its nuclear translocation.57 PAK4 includes both nuclear localisation and nuclear export signals. Nuclear import of PAK4 requires an interaction with α5 importin, and its export is through the chromosome region maintenance-1 pathway.59 PAK1 has differential effects on gene transcription. Chromatin immunoprecipitation assays revealed that PAK1 directly binds to and represses transcription of the nuclear factor of activated T cell (NFAT-1) gene, as well as upregulating transcription of phosphofructokinase, although indirectly as a component of a transcriptional complex.57 PAK1 can inhibit and activate transcriptional corepressors and transcription factors. The corepressor C-terminal binding protein 1 is inhibited by PAK1, while SMRT/HDAC1-associated repressor protein is activated.48 PAK1 also inhibits FOXO by phosphorylation, which acts as a negative feedback loop for its own transcription.60 Phosphorylation of histone H3 by PAK1 may provide an unspecific mechanism by which PAK1 influences gene transcription.1 ,48 Both PAK1 and PAK4 are involved in post-transcriptional regulation; for example, PAK1 phosphorylates Poly-c-RNA binding protein (PCBP1) which increases its nuclear localisation and activity.61 PAK4 includes a ribonucleoprotein (RNP) interacting region which associates with RNPs and RNA binding proteins to regulate translation as well as nuclear/cytoplasmic transport.62 Within the nucleus, PAKs have diverse actions in modulating transcription of multiple targets. However, in the context of GI disease, the positive and negative cellular consequences of nuclear PAK have yet to be defined.
Downstream of RAS
Receptor tyrosine kinase activation of RAS leads to the activation of multiple signalling cascades, including the activation of PAK1.1 Inversely, PAK1 knockout (KO) mice (which have no distinct phenotype) display reduced expression of RAS key effectors including ERK and AKT within the colon.63 ,64 PAK4 KO mice are embryonically lethal.65 Cellular knockdown of PAK4 also impeded ERK signalling in vitro.66 PAK1 activates mitogen activated protein kinase (MAPK)/ERK signalling via phosphorylation of RAF, MEK and ERK, as well as forming protein–protein interactions in the absence of its kinase activity.67 ,68 PAK4 is located downstream of AKT;43 however, PAK1 is centralised between RAS and PI3K/AKT. Within the PI3K/AKT cascade, PAK1 plays another key role via scaffolding PDK1, thereby facilitating phosphorylation of AKT.69 In a positive feedback loop, both PDK1 and AKT phosphorylate and activate PAK137 ,70 (figure 2).

P-21 activated kinase (PAK)1 signalling downstream of receptor tyrosine kinases (RTK). RAS activation initiates PAK1, RAF/mitogen activated protein kinase (MAPK) and phospoinositide 3-kinase (PI3K)/AKT signalling. Activation of PAK1 by epidermal growth factor induces its nuclear translocation. PAK1 activation by RAS may provide a positive feedback loop to further activate MAPK and PI3K. Importantly, PAK1 allocates crosstalk between the PI3K and MAPK pathways. PAK1 may induce MEK1/2 or ERK1/2 independent of RAF, as well as increase PI3K/AKT signalling by scaffolding PDK1 to increase phosphorylation of AKT. In turn, PDK1 may also directly phosphorylate and activate PAK1. PAK1 signalling may activate pro-inflammatory or pro-survival pathways by facilitating nuclear activation of NF-κB.
Activation of β-catenin
The Wnt/β-catenin pathway is a key process in GI development and homeostasis.71 Intestinal LGR5+ stem cells use Wnt signalling for continual regeneration.71 ,72 β-Catenin is the key effector of the Wnt signal, and regulates transcription of Wnt target genes when bound to the T cell factor (TCF) or lymphoid enhancer factor (LEF) family of transcription factors. Importantly, β-catenin drives canonical Wnt signalling through Rac1.73 The Rac1 effector, PAK1, directly interacts with β-catenin and regulates its transcriptional activity through phosphorylation at Ser-663 and Ser-67513 ,74 (figure 3). PAK1 KO mice in an adenomatous polyposis coli (APC) wildtype background displayed a marked reduction in nuclear β-catenin expression.63 PAK4 phosphorylates β-catenin at ser-675, and colocalises with β-catenin in the nucleus thereby increasing TCF/LEF transcriptional activation.59 β-Catenin is an effector of Wnt, and plays a role in cell-to-cell contacts as a complex with E-cadherin at AJ.75 Inhibition of PAK1 increased the membranous localisation of both β-catenin and E-cadherin,76 thereby increasing the formation of AJ and reducing intestinal permeability. This further underlines the role of PAK1 in intestinal integrity and mucosal barrier function.

P-21 activated kinase (PAK)1 facilitates Wnt/β-catenin signalling. (A) In the absence of the Wnt ligand, β-catenin signalling is regulated and β-catenin is degraded through a multiprotein destruction complex. (B) Upon binding of the Wnt ligand to the membrane-bound Frizzled receptor, β-catenin accumulates in the cytoplasm and translocates to the nucleus. Phosphorylation of β-catenin by PAK1 within the cytoplasm occurs at two different residues, Ser-663 (red) and Ser-675 (blue), although only Ser-663 is specific to PAK1. As a consequence, β-catenin stability, nuclear translocation and transcriptional activity are increased. (C) In the absence of PAK1 activity, β-catenin is restored at adherens junction (AJ) where it anchors E-cadherin to the cytoskeleton through an interaction with α-catenin.
Upstream of NF-κB
Rac1 is also a key activator of the NF-κB, which orchestrates a cellular inflammatory response and promotes tumourigenesis.77 ,78 Tumour necrosis factor (TNF)α or interleukin (IL)-1β stimulate canonical NF-κB signalling, whereas, lipopolysaccharide (LPS) or lymphotoxin B activates the non-canonical pathway (figure 4).79 LPS has been shown to activate both canonical and non-canonical NF-κB, albeit through different cascades.80 Both pathways regulate transcription of multiple genes which activate or suppress proliferation, apoptosis or cytokine production. Activation of NF-κB has also been correlated to mucosal barrier dysfunction81 ,82 further implicating the importance of NF-κB in GI homeostasis.

P-21 activated kinase (PAK)1 in canonical and non-canonical NF-κB signalling. NF-κB signalling regulates transcription of target genes via two separate cascades known as the canonical and non-canonical pathways. In the canonical pathway, TNFα elicits PAK1 and c-Jun N-terminal kinase (JNK) phosphorylation. Subsequent phosphorylation of IKKα/β by JNK initiates phosphorylation of IκB. Consequently, RelA is released from the IκB complex and translocates into the nucleus. IκB is tagged by ubiquitin for proteasomal degradation. LPS is a known activator of non-canonical NF-κB signalling. LPS may elicit PAK1 activation through an unknown mechanism. PAK1 phosphorylates and activates NF-κB interacting kinase (NIK), which activates IKKα. In turn, IKKα phosphorylates p100 and the p100–RelB complex is released. P100 is processed into p52 which dimerises with RelB and translocates to the nucleus to activate target genes.
Several studies have investigated the interaction of PAK1 and NF-κB activation. The AKT-PAK1 cascade is required for NF-κB signalling in epithelial cells stimulated with HGF, which itself stimulates PAK1.83 ,84 Another study implicated that PAK1 regulates multiple NF-κB signalling cascades in fibroblasts.85 PAK1 was also found to regulate non-canonical signalling via activation of NF-κB interacting kinase (NIK).86 PAK1 activates caspase-1 cleavage to promote maturation of IL-1β in LPS stimulated macrophages.87 PAK4 is involved in canonical TNFα/NF-κB pro-survival signalling in epithelial cells. PAK4 assists TNFR-associated death domain binding to the TNFR1 complex via both kinase and scaffold functions.66 Preliminary data indicate that PAK1 plays a critical role in canonical NF-κB signalling in normal diploid human colon epithelial cells.88 TNFα treatment was sufficient to activate PAK1 through phosphorylation at Thr-423. PAK1 activation initiated RelA translocation to the nucleus, and its inhibition via a specific kinase inhibitor, IPA-3, or via overexpression of a kinase dead PAK1 mutant (KD-PAK1), inhibited nuclear translocation of RelA.88
PAKs in GI diseases
PAK activation affects NF-κB,16 ,87 MAPK,89–91 PI3K92 and Wnt/β-catenin signalling13 linking inflammation and malignant transformation in the gut. Further, PAK signalling promotes epithelial–mesenchymal transition and invasiveness,1 ,3 ,93 both of which are common features of colitis-associated cancer (CAC). PAK activation has been associated with point mutations and subsequent gene amplification,43 while mutations in activators upstream of PAK, such as Rac1/Cdc42, may constitutively activate PAKs.2 Multiple alterations in PAK signalling were identified in inflammatory bowel diseases and various GI cancers (table 2).
Effect of PAKs on cell signalling cascades in GI disease
Infectious diarrhoea
Bacterial enterotoxins use PAK1 as a cellular effector to break intestinal tight junctions (TJ) and impede epithelial barrier.94 Enteropathogenic E coli (EPEC), a human intestinal attaching and effacing pathogen, is a major cause of diarrhoea in the developing world.94–96 EPEC uses a type-three secretion system to penetrate the epithelial barrier. EPEC injects EPEC-secreted proteins (EspG or EspF) within enterocytes and causes diarrhoea by interfering with intestinal transport and disrupting TJ. EspG inhibits the anion exchanger downregulated-in-adenoma, while EspF downregulates the Na+/H+ exchanger (NHE3) and Na+/glucose transporter (SGLT1) within the gut.94 TJ proteins such as claudin-1, ZO-1 and occludin are continually maintained at cell-to-cell contacts through mechanisms which require endocytosis and constant remodelling of the cytoskeleton. Both microtubule disruption and actin depolymerisation impair TJ thereby disrupting epithelial barrier.94 EspG1 and EspG2 disrupt microtubules as well as impairing protein secretion and trafficking from the Golgi apparatus. Both groups I and II PAKs facilitate cytoskeletal rearrangements through modification of actin. Interestingly, EspG1, directly binds PAK2, disrupts the PAK auto-inhibited dimer and activates its kinase activity almost eightfold.31 This suggests that EPEC uses host's PAK signalling to induce barrier dysfunction within the gut.97
IBD and CAC
UC is a chronic inflammatory disease of the large bowel.98 Even though the pathogenesis of UC is multifactorial and complex, it is widely accepted that chronic inflammation is a precursor to neoplastic transformation in CAC,99 which is multifocal, accelerated and with a mutational sequence that differs from sporadic colorectal carcinoma (CRC).100 P53 and RAS mutations have been identified as rather early steps in this process,101 and may initiate Rac1, NF-κB, PI3K and MAPK signalling. Alternatively, loss of APC and subsequent activation of nuclear β-catenin signalling are late events.102
PAK1 has been correlated to a variety of inflammatory diseases such as arthritis, asthma as well as intestinal inflammation (figure 5).24 ,103–105 One likely driver of PAK1 activation in IBD is TNFα.88 PAK1 activation triggers signalling pathways which drive proliferation, initiate cell migration or block apoptosis, and may contribute to a loss of intestinal barrier function. PAK1 expression was higher upon disease progression to CAC. A similar pattern was observed in IL-10 KO mice, a common model for studying CAC.105 It is tempting to speculate that PAK1 might be responsible for fuelling various signalling cascades involved in CAC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
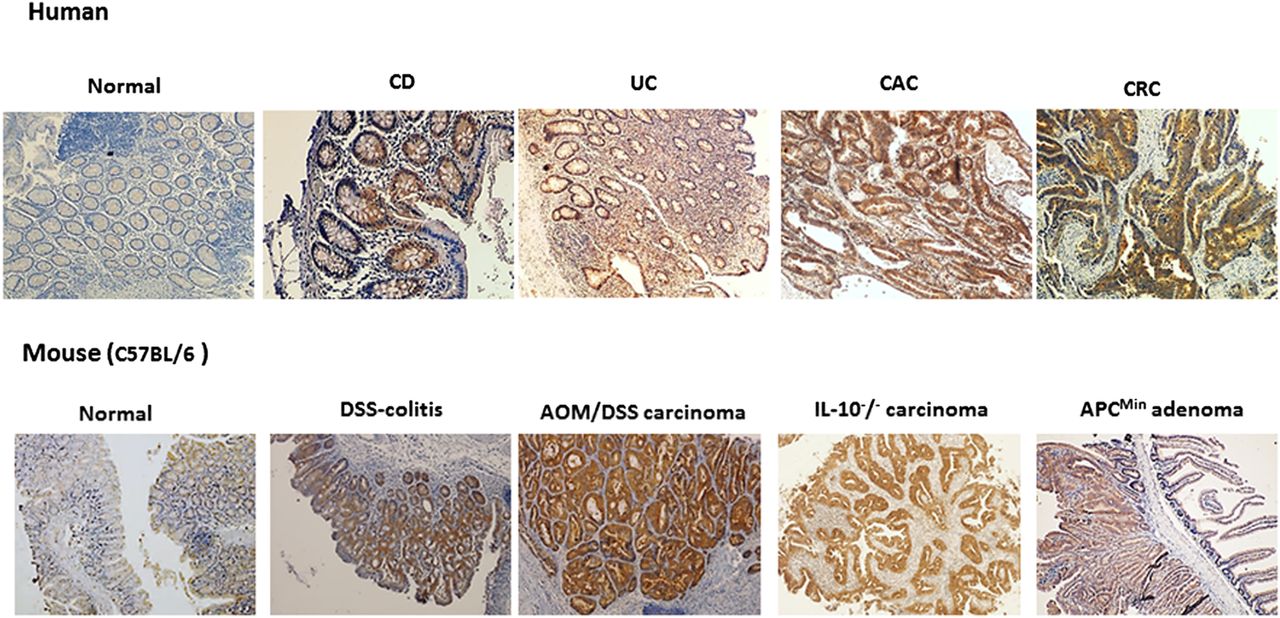
Immunohistochemical expression of P-21 activated kinase (PAK)1 in intestinal inflammation and colorectal cancer. In humans, PAK1 expression is low within normal colon mucosa. Chronic inflammation of the gut as observed in Crohn's disease (CD) and UC increases PAK1 expression. PAK1 is highly expressed in both colitis-associated cancer (CAC) and colorectal cancer (CRC). PAK1 expression was also found to be elevated in an animal model of experimental colitis (DSS-colitis) and AOM/DSS-induced colorectal cancer. IL-10 knockout mice which show spontaneous enterocolitis progressing to CAC also express higher levels of PAK1. APCmin mice also display high PAK1 levels in adenomas.
The CDH1 locus encoding E-cadherin is one of the barrier function genes implicated in the pathogenesis of UC.106 As described above, E-cadherin is important for assembly of AJ. Inhibition of PAK1 expression by mesalamine increased cell-to-cell adhesion through membranous restoration of E-cadherin at AJ.76 Therefore, the effect of mesalamine on PAK1 inhibition and restoration of intestinal barrier function may be a critical mechanism of this drug to achieve mucosal healing in UC.
Colorectal cancer
In comparison with other malignant GI diseases, CRC has the highest incidence and the third highest mortality rate due to cancer worldwide.107 Molecular mechanisms of CRC include chromosomal instability, microsatellite instability or the CpG-island methylator phenotype, all of which are also found in CAC.102 ,108 CAC involves DNA damage (double strand breaks and point mutations) likely from reactive oxygen species, and an altered DNA damage response.108 Mutations of APC, KRAS, BRAF and p53 drive the adenoma-to-carcinoma sequence in sporadic CRC.109 Carter and colleagues were the first to report that PAK1 is overexpressed in CRC studying biopsies from patients with various stages of disease.110 Immunohistochemical analysis revealed PAK1 expression was cytoplasmic within the colonic epithelium. Normal epithelial cells had significantly lower PAK1 staining than neoplastic cells, and the expression increased with disease progression. PAK1 staining was highest in lymph node metastases and was associated with decreased patient survival.110 ,111 Other investigations confirmed that both PAK1 and phospho-PAK1 were highly expressed in human and mouse CRC tissue samples105 ,111 (figure 5). Advanced CRC showed nuclear PAK1 expression indicative of poor patient prognosis.91 PAK4 mutations have also been identified in CRC; however, evidence is lacking to validate its role as driver of tumourigenesis.2 ,43 A separate study reported that PAK5 expression increased with CRC progression and implicated its role in metastasis.112 Considering that PAK1 expression increases throughout the adenoma-to-carcinoma sequence, it is plausible that PAK1 may be used as a biomarker for tumour grading. As multiple PAKs are overexpressed in CRC, future studies should compare the expression patterns between groups I and II PAKs to identify which PAK will serve as a better molecular target.
One important step of early colorectal tumourigenesis comprises the loss of APC function. Constitutive Wnt signalling provides high levels of nuclear β-catenin to fuel proliferation within stem cells of the crypts.71 Loss of APC was shown to induce crypt proliferation and activate Rac1, while epithelial-specific deletion of both APC and Rac1 reduced crypt proliferation specifically through repression of the LGR5+ population.77 Inhibition of Rac1 also impedes RAS-driven proliferation.77 PAK1 as a key effector of Rac1 is expected to be activated upon loss of APC as well, which may cause amplification of β-catenin's transcriptional activity. A functional relation between PAK1 and β-catenin has been established by inhibiting PAK1 in tumour cells: small interfering RNA (siPAK1) and KD-PAK1 significantly reduced β-catenin and cyclin D1 at the protein level.13 Moreover, PAK1 and β-catenin were found to be associated together in the cytoplasm.13 ,113 The functional significance of this protein interaction was shown by direct phosphorylation of β-catenin by PAK1.13 Activated PAK1 phosphorylates the C-terminus of β-catenin at Ser-675.13 It is important to note that β-catenin's Ser-675 domain is not exclusive to PAK1 since PAK4114 and PKA115 also phosphorylate the same residue. Instead, Ser-663 is an exclusive target of PAK1.74 The consequence of multiple phosphorylation sites increases β-catenin stability, as well as transcriptional activation of Wnt target genes. Both sites were phosphorylated in CRC tumours and cell lines. Inhibition of PAK1 with IPA-3, a PAK1 kinase inhibitor, specifically reduced Ser-663 whereas a PI3K inhibitor (LY294002) and a PKA inhibitor (H-89) had no such effects.74 These findings established that PAK1 plays a direct role in the activation of Wnt/β-catenin signalling in colonic epithelial cells.
RAS and RAF mutations lead to MAPK and PI3K pathway activation, and contribute to the adenoma-to-carcinoma sequence in CRC.90 ,116–118 Inhibition of both PAK1 and PAK4 blocks RAS-mediated cell proliferation and survival.14 RAS mutations hyperactivate PAK1 and initiate PAK1/ERK or PAK1/PI3K cascades.92 PAK1 knockdown reduced proliferation and survival, although pharmacological inhibition of ERK or PI3K alone could not. Interestingly, inhibition of PAK1 and ERK or PI3K lacked synergism,92 and this line of evidence suggested the crosstalk of ERK/PI3K observed upon mutated RAS or PI3K pathways can be effectively inhibited by blocking PAK1 in CRC.
Various studies provide insight concerning PAK1's role within CRC establishing that PAK1 activation promotes an invasive phenotype.92 PAK1 activation was shown to activate signalling cascades which contribute to invasiveness such as the MAPK and PI3K cascades.92 Interestingly, it was shown that PAK1 knockdown inhibited both PI3K and MAPK signalling thereby blocking the secretion of vascular endothelial growth factor (VEGF), an important mediator of tumour angiogenesis and cell survival.92 Another potential mechanism of VEGF activation includes a PAK1-HIF1α cascade.119 Specific inhibition of PAK1 kinase activity reduced colony formation in HCT116 and SW480 cells, and KD-PAK1 initiated G1 arrest in SW480 cells.13 LoVo cells transfected with shPAK1 also induced a G1 arrest via downregulation of CDK4/6 and cyclin D1. PAK1 knockdown increased apoptosis and was correlated to reduction in phospho-BAD, Bcl-2, Bcl-xL and XIAP proteins.91
To better understand how PAK1 contributes to ERK-mediated cell migration and tumour invasion, the regulation of focal adhesion contacts via ERK phosphorylation of FAK at Ser-910 was studied.111 PAK1 overexpression increased FAK Ser-910 phosphorylation, whereas both KD-PAK1 and siPAK1 had inhibitory effects. Activation of PAK1 via EGF stimulated phosphorylation of PAK1, ERK and FAK. Downregulation of MEK1/2 with a pharmacological inhibitor, U0126, reduced the migration and invasion of HCT116 and SW116 cells; however, a more potent reduction in migration was observed using siPAK1. These observations imply that targeting PAK1 is a viable option to decrease the migration and invasion of tumour cells.111
In conclusion, PAK overexpression is associated with increased cell proliferation and migration thereby promoting invasion and inhibition of apoptosis in CRC. The effect of PAK1 overexpression in normal colon epithelium remains to be investigated in order to explain if activation of oncogenic signalling promotes PAK1 activity or PAK1 itself contributes to neoplastic progression.
Peutz–Jeghers syndrome
Hereditary intestinal polyposis or Peutz–Jeghers syndrome (PJS) is characterised by the development of hamartomatous GI polyps and predisposes to cancer susceptibility. Germline mutations in the tumour suppressor gene LKB1 have been linked to this syndrome.120 LKB1 is downstream of p53 and may regulate PAK1 activity. Suppression of PAK1 by LKB1 is mediated by its phosphorylation at Thr-109,121 suggesting the role of LKB1 in suppression of cell migration through inhibition of PAK1. LKB1 regulates cellular responses by AMPK/mTOR signalling, which is reflected in PJS lesions that are associated with mTOR hyperactivation.122 With recent evidence indicating the role of PAK1 in mTOR signalling,123 it is imperative to investigate PAK1 expression in PJS polyps as well as PAK1's role in the activation of mTOR in this disease.
PAK1 and gastrin
GI peptide hormones, such as gastrin, play fundamental roles in gastric acid secretion,124 as well as increasing cell transformation,125 proliferation126 and evasion of apoptosis127 within GI cancer.128 ,129 Gastrin is post-translationally modified into amidated gastrin (Gamide) and glycine extended gastrin (Gly-gastrin). Gastrin KO mice display marked reduction in expression of total and phosphorylated PAK1 within the colorectal mucosa,63 and both Gamide and Gly-gastrin increased Rho GTPase activity and increased the phosphorylation status of PAK1 within a transgenic mouse gastric mucosal cell line.128 ,130 ,131
He et al128 investigated PAK1's relationship to the Wnt/β-catenin pathway and established that PAK1 regulates β-catenin activity downstream of gastrins. PAK1 and β-catenin's interaction was revealed by immunoprecipitation, while immunostaining confirmed their colocalisation. Treatment with gastrins stimulated migration of the complex to the nucleus; however, β-catenin remained membranous upon treatment with both the KD-PAK1 mutant and transient silencing of PAK1 (siPAK1).128 Treatment with gastrins reduced the association of β-catenin with E-cadherin; however, KD-PAK1 abrogated this effect.128 These observations indicated the role of PAK1 in the nuclear translocation of β-catenin. Gastrin treatment stabilised nuclear β-catenin, increased the association with the transcription factor TCF4, and increased the transcription of c-myc and cyclin D1; however, transfection with KD-PAK1 inhibited this effect.128 Gastrins stimulated migration within a wound-healing assay, and KD-PAK1 abolished this effect. This study established a novel role of PAK1 in the activation of Wnt/β-catenin signalling downstream of gastrins.
Helicobacter pylori and GC
Helicobacter pylori infection is implicated in chronic gastritis, duodenal ulcer, gastric metaplasia, gastric MALT lymphoma and gastric cancer (GC).132 Activation of Rho GTPases Rac1/Cdc42 by H pylori lead to PAK1-mediated nuclear responses, representing an early event in epithelial colonisation by this strain.133 Moreover, activation of the NF-κB pro-inflammatory pathway by H pylori involved direct interaction of PAK1 and NIK.86
Worldwide GC has the fourth highest cancer-associated mortality rate.134 Early GC is asymptomatic, and difficult to identify by endoscopy. Thus, patients are often diagnosed at late tumour stage and with a poor survival.134 PAK mRNA and protein expression were found to be upregulated in GC cancer cells.135 ,136 PAK4 and PAK1 were correlated with increased tumour growth, invasion, metastasis and poor prognosis in GC patients.16 ,136–138 Dysregulation of transcription growth factor-β (TGF-β) signalling plays a key role in GI inflammation and cancer.139 TGF-β transmits signals to the nucleus via a network of multiprotein complexes using the SMA and mothers against decapentaplegic (MAD; SMAD) family of proteins. Loss of SMAD was correlated to GC susceptibility,140 and HGF activation of PAK4 induced SMAD2 phosphorylation and proteasomal degradation.17 One mechanism in which PAK4 may promote GC metastasis is through the modification of microtubule homeostasis proteins such as superior cervical ganglia 10 (SCG10). PAK4 phosphorylates SCG10 and initiates cell migration and invasion in vivo.141
PAK1 knockdown in a GC xenograft model reduced tumour size.16 Stable and transient knockdown of PAK1 within GC cell lines (BGC823 and ACS) induced G2 cell cycle arrest, reduced proliferation and inhibited anchorage independent cell growth.16 Overexpression of the catalytically active (CA-PAK1) mutant enhanced the expression of cyclin B1, a critical component of the G2/M transition of the cell cycle, whereas PAK1 inhibition decreased the transcription of cyclin B1. PAK1 deficient clones had reduced activity of NF-κB, and pharmacological inhibition of NF-κB reduced the activity of cyclin B1. Interference with NF-κB activity, through a mutant IκBα vector, interfered with CA-PAK1-induced upregulation of cyclin B1. Liu et al16 concluded that overexpression of PAK1 in GC may regulate proliferation through an increase in cyclin B1 via NF-κB. A recent report suggested that PAK1 overexpression in GC activates the ERK and c-Jun N-terminal kinase (JNK) pathways, thereby playing a role in the metastatic phenotype.137 These studies implicate both PAK1 and PAK4 in GC. PAK4 modification of SMAD in the TGFβ pathway and PAK1's role within the NF-κB, ERK and JNK pathways contribute to proliferation, invasion and metastasis of GC.
Hepatocellular carcinoma
Similar to CRC, overexpression of PAK1 in hepatocellular carcinoma (HCC) is associated with advanced tumour stages. A recent report by Wong and colleagues demonstrated that PAK1 activates NF-κB in HCC cells and IPA-3 abrogated this effect. Within a HCC xenograft model, IPA-3 treatment significantly reduced tumour volume, and also inhibited phosphorylation of PAK1 and JNK.142 Increased cell motility and migration was observed in HCC cell lines stably expressing PAK1. Cell lines with higher expression of PAK1 showed JNK activation and inhibition of JNK reduced migration in HCC cells.18 Activation of VEGFR2/PAK1 signalling was found to promote resistance to anoikis in hepatoma patients with high expression of Klotho, an ageing suppressor gene.143 Inhibition of PAK1 activity by shRNA or IPA-3 reversed this effect in hepatoma cells. PAK1 is also upregulated by HBV protein HBx that renders hepatoma cells resistant to anoikis.144
Oesophageal cancer
Oesophageal squamous cell carcinoma (OSCC) is marked by poor prognosis, and has increasing prevalence in eastern society.107 ,145 In order to identify genetic changes within oesophageal tumour cells, one study used an ESCC xenograft model.15 ,146 Lymph fluid collected from patients with ESCC was injected into nude mice, and analysed via array based comparative genomic hybridisation. Interestingly, the authors identified PAK1, which increased fivefold during tumour growth.15
The MAPK pathway was investigated in ESCC cancer cell lines derived from bone marrow metastasis in ESCC patients. Overexpression of the ERK pathway was found to be independent of MEK1/2145 as pharmacological inhibitors of MEK1 (PD98059), MEK1/2 (U0126) and dominant negative DN-MEK1/2 failed to abolish ERK activity.145 However, pharmacological inhibition of the PI3K pathway using LY294002 or overexpression of KD-PAK1 DN-PAK1 mutants clearly reduced ERK activity.145 Although the PAK1 mutants did not affect total protein levels of ERK1/2 or alter the JNK pathway, this report demonstrated that interplay of PI3K and PAK1 stimulated ERK pathway independently of MEK. Continual crosstalk among multiple cell signalling cascades in ESCC may present a challenge in therapeutic inhibition of MAPK signalling. However, targeting PAK1 in ESCC may present an opportunity to inhibit cell signalling interplay thereby increasing the specificity of pharmacological ERK inhibition.
Pharmaceutical modulation of PAK1
Through its participation in multiple signalling pathways, PAK1 overexpression in GI cancer promotes cell proliferation, survival and metastatic progression evading apoptosis (table 3).
P-21 activated kinase (PAK) overexpression in GI tumourigenesis
Considering the contribution of PAK1 to GI disease, future strategies aim to target PAK1 expression or inhibition of its kinase activity for prevention or treatment.
Inhibition of PAK expression
In a microarray analysis performed to examine molecular targets of mesalamine, PAK1 was identified as a common mediator of pathways modulated in colorectal epithelial cells.76 In vivo, PAK1 was overexpressed in intestinal polyps of APCmin mice, and mesalamine blocked both PAK1 expression and tumour progression.76 In vitro, mesalamine blocked PAK1 at the protein level.76 As outlined above, PAK1 expression is elevated in gut inflammation105 and plays a role in the activation of the NF-κB pro-inflammatory pathway.88 Inhibition of PAK1 and subsequent NF-κB downregulation by mesalamine may be one mechanism in which mesalamine blocks inflammation and CAC altered innate immunity such as activated neutrophils-induced cell stress.147 PAK1 is required for degranulation148 of mast cells and neutrophil activation.149 Downregulation of PAK1 by mesalamine within innate immune cells may provide an effective therapeutic approach in reducing inflammation in active UC.
Another approach to inhibit expression of PAK1 involves overexpression of miRNA-29, which negatively regulates the cdc42 and PI3K pathways.150 MiRNA-29 inhibited PAK1 at the protein level, reduced proliferation, migration and cell invasion in GC cells.151 MiRNA-145 was shown to target PAK4 and impedes cell growth in CRC cells.152 Although, future studies are required to understand if miRNA-29 or 145 can be used to pharmacologically modulate PAK expression in vivo.
Bradykinin, which is known to be overexpressed in cancer, is implicated in angiogenesis.153 Furthermore, PAK1 has been correlated to angiogenesis154 and is a known effector of the bradykinin pathway.155 It was shown that overexpression of the IRX1 gene significantly downregulated both bradykinin and PAK1 protein levels in GC cells.155 It is noteworthy that the IRX1 gene was identified as a tumour suppressor gene in GC156 ,157 and restoration of IRX1 in vitro and in vivo reduced the metastasis of GC cells.155 Prospective studies should investigate if pharmacological modulation of IRX1 is effective in blocking angiogenesis and PAK1 within other GI diseases.
The complexity of targeting PAK1 is highlighted by the fact that PAK1 has a plethora of kinase dependent and independent cellular functions. Therefore, inhibition of total PAK1 expression may be an effective approach in PAK1 intervention. Future studies should identify whether downregulation of PAK1 expression by mesalamine, miRNA-29 or modulation of IRX1 is more effective in blocking inflammation than inhibition of PAK1 kinase activity alone.
PAK1 kinase inhibition
The Rho GTPase Rac1 plays a key role in PAK1 kinase activation,29 as well as T cell proliferation and differentiation.158 Tiede et al reported that azathioprine and 6-mercaptopurine (6-MP) induced apoptosis in activated CD4 T cells through inhibition of Rac1. Rac1 inhibition blocked downstream pro-survival signalling via downregulation of NF-κB, MAPK and bcl-xL.159 Considering that PAK1 mediates all three of these pathways, it is important to investigate the effect of azathioprine and 6-MP on PAK1 kinase activation and downstream signalling within both inflammatory and epithelial cells.
Synergistic effects with 5-FU
In CRC, 5-flurouracil (5-FU) is a mainstay chemotherapeutic drug; however, its effectiveness is limited due to drug resistance.160 Therefore, it is of great interest to investigate a possible synergism in PAK1 inhibition and 5-FU. Interestingly, in vitro, knockdown of PAK1 increased apoptosis in 5-FU treated LoVo cells.91 Additionally, it was investigated if PAK1 knockdown and 5-FU synergistically inhibits tumour growth in a xenograft model. LoVo cells harbouring shPAK1 constructs were injected into nude mice and PAK1 knockdown significantly reduced tumour growth and proliferation.91 A further reduction in tumour growth was observed in combination with 5-FU.
Small molecule PAK1 inhibitors
Analysis of PAK1's crystal structure has allowed for the identification and design of several PAK1 inhibitors.29 Initial studies analysed complexes of non-specific ATP-competitive inhibitors, such as staurosporine derivatives, with PAK1 to conceptualise kinase inhibition.161 However, identifying a selective inhibitor for PAK1's ATP binding pocket has proved to be difficult. Initially, CEP-1347 was found to inhibit PAK1 but further studies revealed its higher affinity for mixed lineage kinase-3 than PAK1 itself.162 The celecoxib derivative OSU-03012 was originally identified as a PDK1 inhibitor. Interestingly, it was found to inhibit group I PAKs although not completely specific to PAK1.163 At present, the most potent and selective ATP-competitive inhibitor of PAK1 is the organouthenium compound FL172, which has been shown to be effective within the nanomolar range, respectively.162 ,164 Recently identified PAK1 ATP inhibitors include 2-arylamino-4-aryl-pyrimidines.165 These selective compounds blocked proliferation of multiple CRC cell lines at nanomolar concentrations, and their non-cytotoxicity provides a rational to investigate their effectiveness in vivo.165
Although ATP inhibitors are thought to be highly potent, they are often non-specific.162 Alternatively, allosteric inhibition of PAK1 would be less potent, but more specific. IPA-3 is an allosteric small molecule inhibitor of PAK1. It blocks PAK1 kinase activation by covalently binding to the PBD and inhibiting activation by Rac1/Cdc42. However, IPA-3 cannot inhibit PAK1 which is already activated.162 IPA-3 is widely used in cell culture, although its efficiency in animal models may be limited due to a disulfide bond within the molecular structure.3 Nevertheless, IPA-3 has served as a platform to design allosteric inhibitors which have increased selectivity and therapeutic effectiveness within in vivo systems.
Future directions
With accumulating evidence establishing the role of PAK1 in GI inflammation and CRC, it is reasonable to examine the effect of PAK1 inhibitors in disease prevention and therapy. However, considering that PAK1 orchestrates multiple signalling cascades, it is important to understand the underlying cause of PAK1 overexpression in order to target this molecule. Future studies are requisite to validate PAK1 as a biomarker and pharmaceutical target.
Acknowledgments
The financial support by the Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development is gratefully acknowledged.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online methods
Footnotes
-
KD and VK contributed equally.
-
Funding The study was supported by Austrian Science Fund (P24121 to Christoph Gasche).
-
Competing interests CG has a research collaboration with Shire Pharmaceuticals and received research support, lecturing or consulting honoraria from Ferring and Dr Falk Pharma.
-
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Corrections