Article Text

Abstract
Objective STK33 has been reported to play an important role in cancer cell proliferation. We investigated the role of STK33 in hepatocellular carcinoma (HCC) and its underlying mechanisms.
Design 251 patients with HCC were analysed for association between STK33 expression and clinical stage and survival rate. Tamoxifen (TAM)-inducible, hepatocyte-specific STK33 transgenic and knockout mice models were used to study the role of STK33 in liver tumorigenesis. HCC cell lines were used to study the role of STK33 in cell proliferation in vitro and in vivo.
Results STK33 expression was found to be frequently upregulated in patients with HCC. Significant associations were found between increased expression of STK33 and advanced HCC staging and shorter disease-free survival of patients. Overexpression of STK33 increased HCC cell proliferation both in vitro and in vivo, whereas suppression of STK33 inhibited this effect. Using a TAM-inducible, hepatocyte-specific STK33 transgenic mouse model, we found that overexpression of STK33 resulted in increased hepatocyte proliferation, leading to tumour cell burst. Using a TAM-inducible, hepatocyte-specific STK33 knockout mouse model, we found that, when subjected to the diethylnitrosamine (DEN) liver cancer bioassay, STK33KOflox/flox, Alb-ERT2-Cre mice exhibited a markedly lower incidence of tumour formation compared with control mice. The underlying mechanism may be that STK33 binds directly to c-Myc and increases its transcriptional activity. In particular, the C-terminus of STK33 blocks STK33/c-Myc association, downregulates HCC cell proliferation, and reduces DEN-induced liver tumour cell number and tumour size.
Conclusions STK33 plays an essential role in hepatocellular proliferation and liver tumorigenesis. The C-terminus of STK33 could be a potential therapeutic target in the treatment of patients with STK33-overexpressed HCC.
- HEPATOCELLULAR CARCINOMA
- CANCER
- CELL BIOLOGY
- HEPATOCYTE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
STK33 is a new HSP90 client and is essential for mutant KRAS-dependent acute myeloid leukaemia cancer cell proliferation; therefore STK33 is a context-dependent therapeutic target.
What are the new findings?
Overexpression of STK33 in primary hepatocellular carcinoma (HCC) tumours correlates with advanced pathological features and shorter survival of patients. Overexpression of STK33 induced mouse liver tumour burst, while knockout of STK33 suppressed diethylnitrosamine (DEN)-induced mouse liver tumorigenesis. In addition, we found that STK33 bound directly to c-Myc and increased its transcriptional activity. Moreover, we found that the C-terminus of STK33 blocked STK33/c-Myc association, downregulated HCC cell proliferation, and reduced DEN-induced liver tumour number and tumour size.
How might it impact on clinical practice in the foreseeable future?
The C-terminus of STK33 might be used as a therapeutic target to treat patients with STK33-overexpressed HCC. The STK33/c-Myc complex may work as a target for early screening tests.
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common solid tumour worldwide and the third leading cause of cancer-induced death.1 In spite of significant technical improvements in curative treatments such as surgical resection and transplantation, the prognosis of patients with HCC remains very poor. Like molecularly heterogeneous tumours, the development of HCC is regulated by various oncogenes and signalling pathways.2–4 It is important to understand the molecular characteristics and related biological mechanisms in the proliferation, migration and metastasis of HCC for the development of earlier screening markers and therapeutic targets.
STK33 is a serine/threonine kinase that belongs to the calcium/calmodulin-dependent family of kinases,5 ,6 discovered by sequencing the human chromosome 11 region 11p15 and mouse chromosome 7. STK33 is expressed in a variety of tissues, including testis, fetal lung and heart, and retina, and is also weakly expressed in the liver.7 Recently, STK33 was found to be critical for the survival of KRAS-dependent haematopoietic cancer cell lines (acute myeloid leukaemia, multiple myeloma and T cell acute lymphoblastic leukaemia) and epithelial cancer cell lines (colon, breast, pancreatic and lung cancer). Using mutations in the ATP-binding loop, the kinase activity of STK33 was inferred to be required for the survival of KRAS-dependent cancer cell lines.8–11 However, the function and mechanism of STK33 in HCC are poorly studied and remain unclear.
In this study, we showed that frequent overexpression of STK33 in primary HCC tumours correlates with advance pathological features and shorter survival of patients. Hepatocyte-specific overexpression of STK33 in adult mice resulted in increased hepatocyte proliferation and liver tumour burst. When subjected to the diethylnitrosamine (DEN) liver cancer bioassay, hepatocyte-specific knockout of STK33 mice produced a markedly lower incidence of tumour. We found that the C-terminus of STK33 inhibited HCC cell proliferation and liver tumour growth.
Materials and methods
Ethics statement
Informed consent was obtained from all patients involved in the study, and the study protocol was approved by the Clinical Research Ethics Committee of the Second Military Medical University. The study protocol was approved by the Clinical Research Ethics Committee of Second Military Medical University for all animal experiments. Our policy is consistent with the US Public Health Service Policy on humane care and use of laboratory animals, available from the Office of Laboratory Animal Welfare, National Institutes of Health, and Department of Health and Human Services.
Patients and immunohistochemical staining
Tumorous tissue and adjacent non-tumorous liver tissues were collected from 251 patients who underwent curative surgery for HCC at the Eastern Hepatobiliary Surgery Hospital. Informed consent was obtained from each patient, and the study protocol was approved by the Clinical Research Ethics Committee of Second Military Medical University. A diagnosis of HCC was confirmed on histological examination. Tumour stage was classified according to American Joint Committee on Cancer criteria.
A 5 μm thick tissue section was obtained from each patient and incubated with primary antibodies, followed by incubation with a horseradish peroxidase anti-rabbit IgG antibody. Staining was developed by incubation with a DAB Substrate kit (Pierce). After being washed, tissue sections were counterstained with haematoxylin. The specificity of STK33 antibody (Abcam, ab110090) was tested using L02/STK33 overexpression cells and L02/V control cells.
To determine the immunoreactivity of STK33, cytosolic or nuclear staining of the yellowish or brownish granules were graded using the following scale: 0 for background staining, 1 for faint staining, 2 for moderate staining, and 3 for strong staining. The intensity was judged by comparison with L02/STK33 cells, which served as positive-staining controls (level 3 staining). In addition, the ratios of positive cell number and total cell number (as a percentage) among the tissue sections were graded according to the following scale: 0 for <5%, 1 for 5–25%, 2 for 26–50%, 3 for 50–75%, and 4 for 75–100%. These two parameters were multiplied to produce a weighted score for each tumour specimen; 0–2 and ≥3 were considered negative and positive staining, respectively.
DEN-induced hepatocarcinogenesis
At postnatal day 20, STK33KOflox/flox, Alb-ERT2-Cre mice, the littermate STK33KOflox/flox control mice and Alb-ERT2-Cre mice were given a single intraperitoneal injection of DEN (50 μg/g body weigh; Sigma-Aldrich, St Louis, Missouri, USA). Ten months later, the mice were killed and necropsied. The livers were then separated into individual lobes and analysed for number and size of hepatic tumours.
At postnatal day 20, C57BL/6 mice were given a single intraperitoneal injection of DEN (50 μg/g body weight) for Ad-STK33-C tumour inhibition assay. One week later, the DEN-challenged mice were given intraperitoneal injections of Ad-GFP or Ad-STK33-C (1×108 plaque-forming units (pfu)) once every week until being killed after 10 months. The livers were then separated into individual lobes and analysed for number and size of hepatic tumours.
Nude mice tumour growth
Nude mice (NU/J) were purchased from SLAC Laboratory Animal (Shanghai, China). L02/V, HepG2/V, L02/STK33, HepG2/STK33, HKCL/control shRNA, SMMC7721/control shRNA, HKCL/STK33 shRNA and SMMC7721 shRNA stable cell lines were resuspended at 1×107 cells/mL and a 0.1 mL aliquot of cell suspension was injected subcutaneously into athymic nude mice (n=10/group). Tumour volume was measured at different time points. Tumour volumes were determined by external measurements and calculated according to the following equation: V=[L×W2]×0.52 (V=volume, L=length, and W=width). Data were analysed by Student's t test, and p<0.05 was considered significant.
Immunofluorescent staining
Sections were blocked with 1% bovine serum albumin (Sigma) and incubated with appropriate primary antibody at 37°C for 1 h. After being washed extensively, they were incubated with Alexa Fluor-488 goat anti-mouse IgG (Invitrogen) and Fluor-Cy3 goat anti-rabbit IgG (Jackson Immunoresearch) at 37°C for 1 h. Sections were then washed and mounted for observation under a scanning confocal microscope (TCS SP2 Leica).
Pull-down assay
GST-STK33 or GST control protein (each 3 μg) was incubated with 3 μg His-c-Myc, His-c-Myc-N or His-c-Myc-C in 1 mL phosphate-buffered saline (PBS) with 0.1% bovine serum albumin at 4°C for 2 h. Then 20 μL glutathione beads (GE Healthcare) was added, and the samples were incubated for another 1 h at 4°C. The samples were washed three times with PBS and then analysed by immunoblotting assay.
Adenovirus injection for intrahepatic expression of STK33-N or STK33-C
The forward primer 5′-AAGAATTCATGGCTGATAGTGGCTTAGAT-3′ and the reverse primer 5′-AACTCGAGTTAGTTTAAGTTTATTTCATTG-3′ were used to amplify the N-terminus of STK33 (amino acids (aa) 1–260), while the forward primer 5′-AAGAATTCATGATAAAGGTGACTGATTTTGG-3′ and the reverse primer 5′-AACTCGAGTTAGAGTTTCTTTTTGGTTCTGG-3′ were used to amplify the C-terminus of STK33 (aa261–514). The amplified fragments were then digested with BamH1/Xho1 and ligated into the vector of DUALGFP-CCM (Vector Biolabs) for Ad-STK33-N and Ad-STK33-C construction. The construct was verified by DNA sequence analysis. The N- or C-terminus of STK33 protein fused with Flag tag was expressed. The high titres of custom-made adenoviral N-terminus of Ad-STK33 (Ad-STK33-N, aa1–260), adenoviral C-terminus of Ad-STK33 (Ad-STK33-C, aa261–514), and the control adenoviral green fluorescent protein (GFP) (Ad-GFP) were purchased from Vector Biolabs. Adenovirus (1×108 pfu) containing the above construct was injected into the tail vein of 8-week-old mouse every 7 days, and protein expression was detected at 0, 1, 2, 3, 4, 5, 6 and 7 days after injection. Adenovirus was first injected on the day of tumour cell implantation in the nude mice group and on the day of tamoxifen (TAM) treatment in the TAM-inducible AlbERT2-STK33 tumour burst model group.
Statistical analysis
Overall survival (in months) was defined as the interval between the date of surgery and the date of death or the last follow-up. Overall survivals were estimated using the Kaplan–Meier method, and the difference in survival was evaluated using the log-rank test. p values of <0.05 and <0.01 were considered significant and very significant, respectively. All calculations were performed using R V.2.9.0 software (http://www.r-project.org).
Results
Expression of STK33 correlates with HCC progression and shortened survival
We detected the expression of STK33 protein in 251 patients with HCC (patients’ clinical data in online supplementary table S1) using immunohistochemical analysis. Compared with normal liver tissue, hepatic tissues from patients with HCC expressed higher levels of STK33. Furthermore, STK33 expression was detected in 14.29% (5/35) of stage I HCC samples, 27.78% (20/72) of stage II HCC samples, 76.4% (68/89) of stage III HCC samples, and 87.27% (48/55) of stage IV HCC samples. Notably, STK33 staining was much stronger in higher-grade HCC than in lower-grade HCC samples (figure 1A, B). Clear nuclear localisation of STK33 was observed. These results were confirmed using real-time PCR analysis, in which more STK33 mRNA was detected in higher-grade HCC tissues than in lower-grade HCC and adjacent normal tissue samples (figure 1C). We also examined the expression of STK33 in HCC cell lines with western blotting using STK33 antibodies. Compared with normal liver tissue lysate, elevated STK33 expression was observed in all seven HCC cell lines. The immortalised hepatocyte cell line, L02, also showed weak upregulation of STK33 (figure 1D). These results were also confirmed with real-time quantitative PCR analysis (figure 1E).

Increased expression of STK33 in hepatocellular carcinoma (HCC). (A) The level of STK33 in normal tissues compared with stage I–IV HCC tissues detected through immunostaining with the STK33 antibody. Representative pictures are shown. Bar=50 μm. (B) Positive percentage of STK33 expression in stage I–IV HCC. (C) The expression of STK33 was verified by quantitative reverse transcription PCR in normal tissues and HCC samples. Each box represents the 5th and 95th centiles, the middle line represents the mean value, and the whiskers represent the SD. (D) STK33 protein expression in normal liver, immortalised L02 cells and HCC cell lines detected through immunoblotting analysis using antibody to STK33; tubulin was used as a loading control. (E) STK33 mRNA levels in normal livers, immortalised L02 cells and HCC cell lines detected through quantitative reverse transcription PCR analysis. (F) Kaplan–Meier analyses were performed according to immunoreactivity of STK33 in HCC patients. The staining score of STK33 is described in and Materials and methods section: 0–2 and ≥3 were considered negative (STK33−) and positive (STK33+) staining, respectively. Overall (left) and disease-free survival (right) of patients who had high STK33 expression was evidently shorter. Results represent at least three separate experiments. Error bar ±SD. *p<0.5, **p<0.01.
STK33 was previously reported to be critical for the survival of KRAS-dependent cancer cells.8–11 To explore whether overexpression of STK33 proteins is associated with Ras mutations in HCC, we detected Ras mutations in tissue samples of 251 patients with HCC. However, we found only 11 samples in total with STK33 mutations, of which six were found from STK33+ samples and five were found from STK33− samples (see online supplementary table S2). It is consistent with previous studies showing that somatic mutations of the Ras gene are uncommon.12–17 Among the HCC cell lines used in this study, only HepG2 was found with one N-RasQ61L mutation, while all other six HCC cell lines had the wild-type Ras gene.
We further evaluated whether STK33 expression correlated with survival in 251 patients with HCC. We observed that upregulation of STK33 predicted shorter overall survival and disease-free survival of patients with HCC (figure 1F). Multivariate survival analysis using the Cox proportional hazards model further indicated that upregulation of STK33 correlated with a higher HR and poor clinical outcomes (overall survival, p=0.005, HR 4.327; disease-free survival, p=0.012, HR 2.853) (table 1). These results indicate that expression of STK33 was associated with HCC progression and shortened survival.
Multivariate Cox regression analysis of STK33 expression in HCC
STK33 enhances HCC cell proliferation and tumour growth
To investigate the biological role of STK33 in HCC cells, we performed gain- or loss-of-function studies. We overexpressed STK33 in L02 and HepG2 cells, which expressed lower endogenous STK33 (figure 2A). We subsequently examined the role of STK33 in HCC cell proliferation using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The results showed a significant increase in the growth curve, indicating that cell proliferation was enhanced in vitro after transfection with STK33 in both L02 and HepG2 cells (figure 2B). We also examined whether STK33 could enhance tumour growth in vivo. When cells were injected subcutaneously into athymic nude mice, overexpression of STK33 resulted in dramatically increased tumour volumes compared with vector controls for both L02 and HepG2 cells in vivo (figure 2C). Using STK33 short hairpin (sh)RNA, we then knocked down STK33 expression in HKCL2 and SMMC7721 cells, which have abundant endogenous STK33 protein (figure 2D). Compared with control shRNA (Ctrl shRNA), the cells treated with STK33 shRNA1 and STK33 shRNA2 grew more slowly in vitro in both HKCL2 and SMMC7721 cells with MTT assay (figure 2E). The mice inoculated with HKCL2/STK33 shRNA and SMMC7721/STK33 shRNA cells exhibited dramatically reduced tumour volumes compared with those receiving HKCL2/Ctrl shRNA and SMMC7721/Ctrl shRNA cells (figure 2F). These results, both in vitro and in vivo, show that STK33 potently promoted HCC cell proliferation and tumour growth.
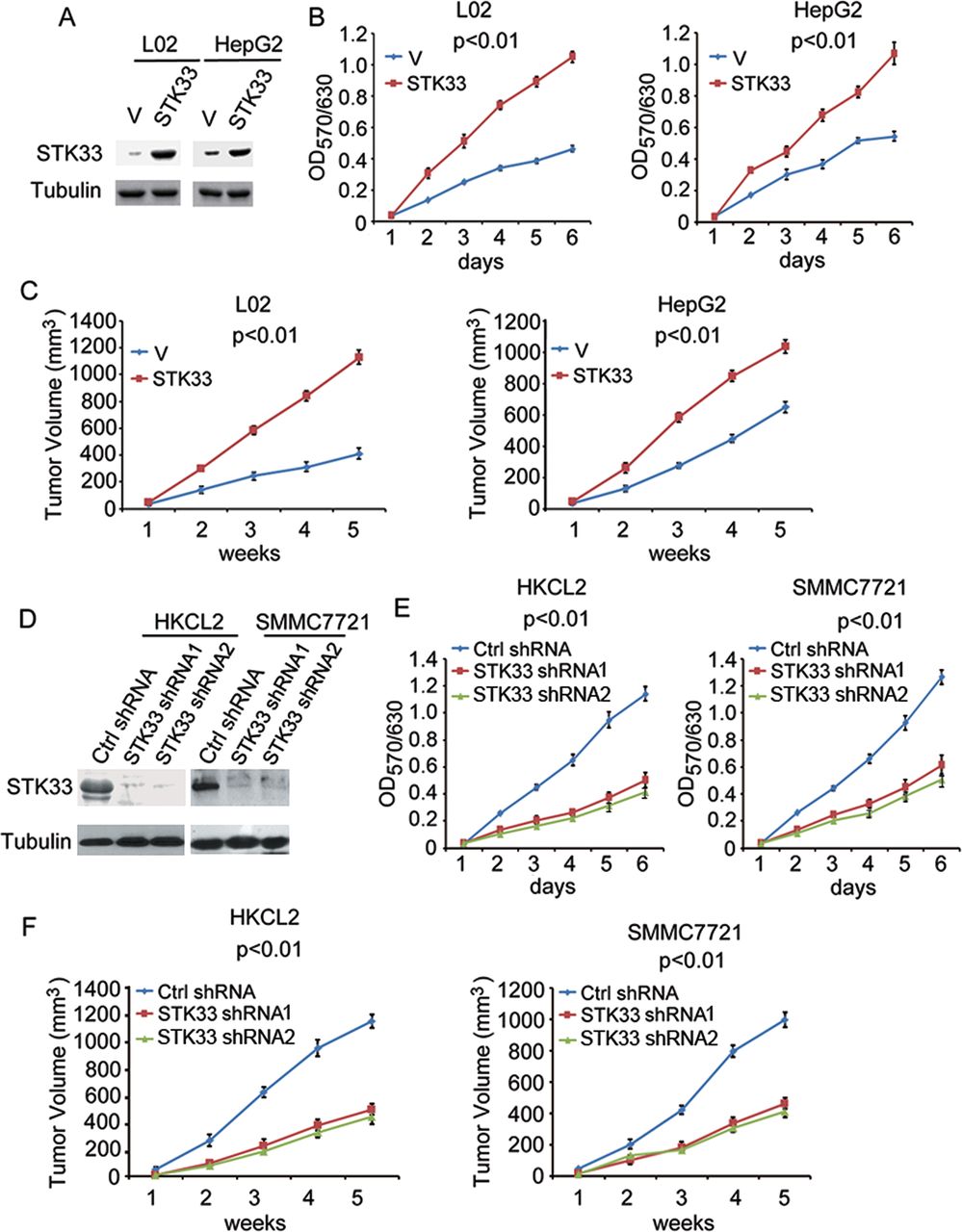
STK33 promotes hepatocellular carcinoma cell proliferation and tumour growth. (A) L02 and HepG2 cells transfected with plain vector (V) or plasmid encoding STK33 (STK33). Cell lysates were immunoblotted using the STK33 antibody; tubulin was used as the loading control. (B) In vitro growth of L02/V, HepG2/V (V) and L02/STK33, HepG2/STK33 (STK33) cells, measured using the MTT assay. (C) Average tumour volume in athymic nude mice subcutaneously inoculated with L02/V, HepG2/V or L02/STK33, HepG2/STK33 cells. (D) HKCL2 and SMMC7721 cells transfected with control shRNA (Ctrl shRNA) or STK33 shRNA1 and STK33 shRNA2. STK33 levels were detected through immunostaining using STK33 antibody, with tubulin as the loading control. (E) In vitro growth of HKCL2/Control shRNA, SMMC7721/Control shRNA (Ctrl shRNA) HKCL2/STK33 shRNA1, SMMC7721/STK33 shRNA1 (STK33 shRNA1), and HKCL2/STK33 shRNA2, SMMC7721/STK33 shRNA2 (STK33 shRNA2) cells, measured using the MTT assay. (F) Average tumour volume in athymic nude mice subcutaneously inoculated with HKCL2/Ctrl shRNA, SMMC7721 control shRNA (Ctrl shRNA) or HKCL2/STK33 shRNAs, SMMC7721/STK33 shRNAs (STK33 shRNA1 and STK33 shRNA2). For (A), (B), (D) and (E), results represent at least three separate experiments. For (C) and (F), n=10 mice/group. Error bar ±SD.
STK33 accelerates mouse hepatocyte proliferation and hepatocellular tumour growth
To investigate whether STK33 affected hepatocyte proliferation in vivo, we generated inducible liver-specific STK33 transgenic mice, STK33Tgflox/flox, Alb-ERT2-Cre (figure 3A). We treated STK33Tgflox/flox, Alb-ERT2-Cre mice with TAM and examined the livers at day 7 after TAM injection. Overexpression of STK33 was demonstrated by western blot analysis, which did not occur in controls, STK33Tgflox/flox, Alb-ERT2-Cre mice treated with corn oil and STK33Tgflox/flox mice treated with TAM (see online supplementary figure S1A). STK33 overexpression was also confirmed by immunohistochemical staining of a paraffin-embedded section 7 days after TAM or corn oil injection (see online supplementary figure S1B).

Increased cell proliferation and tumour generation after STK33 overexpression in mice liver. (A) Scheme of STK33 overexpression using STK33Tgflox/flox, Alb-ERT2-Cre mice. (B) Liver weight to body weight ratios. (C, D) Immunohistochemical analysis for BrdU (C) and Ki67 (D) shows the differences in hepatocyte proliferation between STK33Tgflox/flox, Alb-ERT2-Cre mice treated with either tamoxifen (TAM) or corn oil and wild-type littermate control mice treated with TAM. Quantification of BrdU- and Ki67-positive cells is on the right. (E) Time/dose curve for incidence of visible tumours in STK33Tgflox/flox, Alb-ERT2-Cre mice treated with TAM. Tumours were first found 8 months after TAM treatment. (F) Incidence of visible tumours in STK33Tgflox/flox, Alb-ERT2-Cre mice treated with either corn oil or TAM and STK33 floxed littermate control mice treated with TAM. Mice were killed 10 months after TAM treatment. The data shown are the mean of three independent experiments. n=10 mice/group for (B)–(E). Bar=50 μm. Error bar represents ±SD. *p<0.05, **p<0.01.
Short-term (7 days) overexpression of STK33 resulted in a significant increase in liver to body weight ratio (figure 3B). Hepatocyte proliferation was analysed by immunohistochemistry for BrdU and Ki67 antigens. We also found a significantly higher number of BrdU and Ki67 antigens in mice with STK33 overexpression compared with controls, STK33Tgflox/flox, Alb-ERT2-Cre mice treated with corn oil and wild-type mice treated with TAM, which was detected at day 7 after TAM or corn oil injection (figure 3C, D). To determine the effect of STK33 overexpression on hepatic tumour progression, we treated STK33Tgflox/flox, Alb-ERT2-Cre mice with TAM and examined the liver tissue after injection. Tumour burst was first found at 8 months after TAM treatment in 2 of 12 (16.67%) STK33Tgflox/flox, Alb-ERT2-Cre mice (figure 3E). All 12 (100%) STK33Tgflox/flox, Alb-ERT2-Cre mice were found to have liver tumours at 10 month after TAM treatment, compared with none in controls, STK33Tgflox/flox, Alb-ERT2-Cre mice treated with corn oil or STK33Tgflox/flox mice treated with TAM (figure 3F).
STK33 knockout mice are resistant to DEN-induced hepatocarcinogenesis
To further investigate the role of STK33 in liver tumorigenesis, STK33KOflox/flox mice were crossed with Alb-Cre-ERT2 transgenic mice to generate a temporal and conditional knockout of the STK33 gene (figure 4A). To confirm hepatocyte-specific disruption of STK33, mice were treated with one dose of vehicle or TAM 24 h before liver, and extrahepatic tissues were analysed. STK33 truncation was only observed in livers from TAM-treated STK33KOflox/flox, Alb-ERT2-Cre mice but not from STK33KOflox/flox or vehicle-treated mice (figure 4B). A decrease in STK33 mRNA level was only achieved in livers from TAM-treated STK33KOflox/flox, Alb-ERT2-Cre mice; STK33 mRNA was not changed in extrahepatic tissues of TAM-treated STK33KOflox/flox, Alb-ERT2-Cre mice (figure 4C). Western blot analysis of liver extracts revealed diminished STK33 protein levels in TAM-treated STK33KOflox/flox, Alb-ERT2-Cre mice (figure 4D). These data indicate that an efficient inducible liver-specific knockout of STK33 is achieved using the Alb-Cre-ERT2 system.

Conditional and temporal knockout of STK33 in hepatocytes. (A) Scheme of the model of STK33 conditional knockout in liver. (B) PCR diagnostic for activated ERT2-Cre-mediated knockout of exon 1 of STK33 allele in genomic DNA isolated from kidney and liver of STK33KOflox/flox or STK33KOflox/flox, Alb-ERT2-Cre mice treated with vehicle control (CO) or tamoxifen (TAM) for 24 h. (C) Quantitative PCR analysis measuring STK33 mRNA level in liver, lung, kidney and skeletal muscle from STK33KOflox/flox or STK33KOflox/flox, Alb-ERT2-Cre mice treated with TAM and killed 24 h after treatment (n=8–10). ***p<0.001 vs STK33fl/fl group. (D) Western blot of STK33 for whole liver extracts from STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre mice treated with TAM and killed 24 h after treatment.
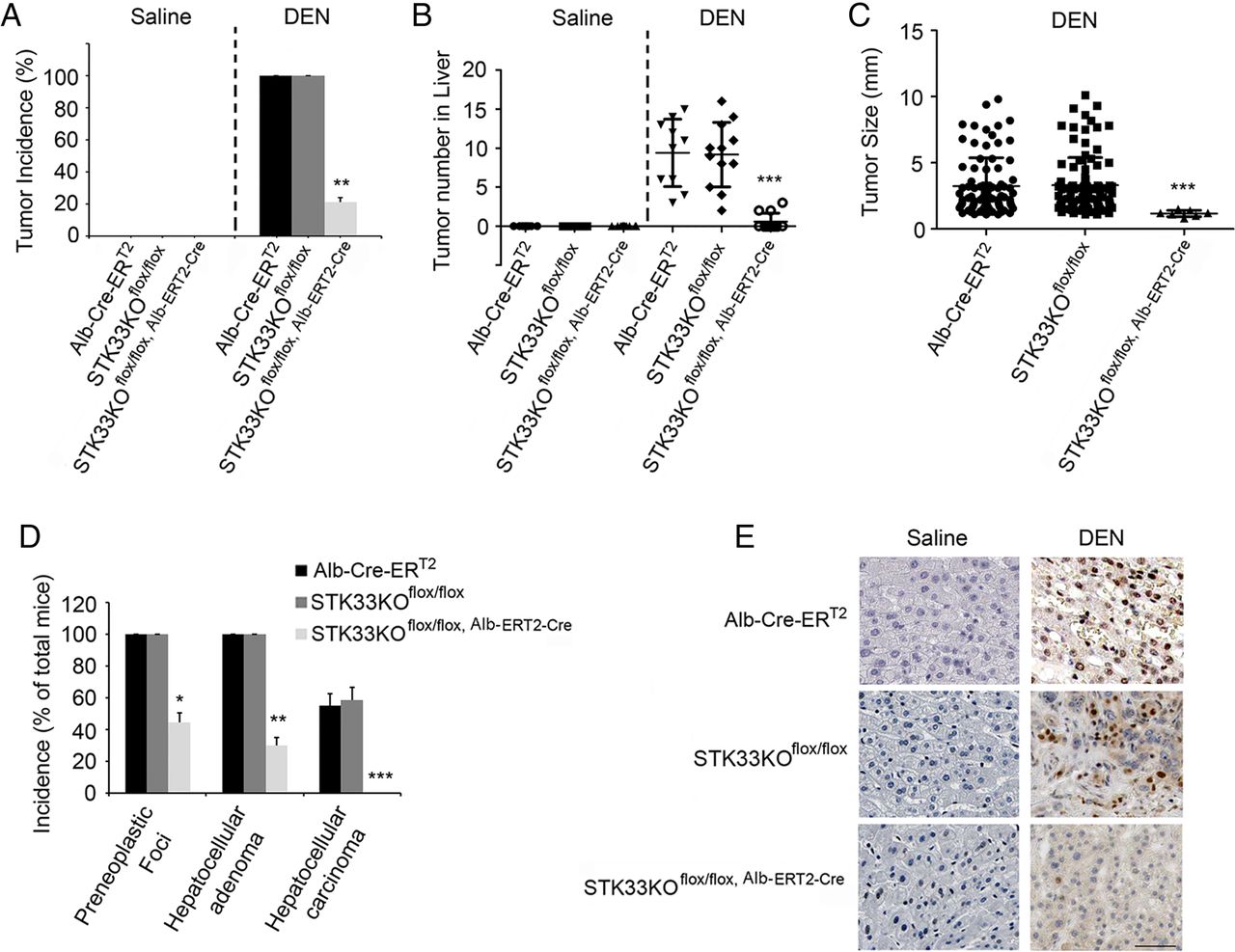
To investigate the direct role of STK33 in liver tumorigenesis, the susceptibility of Alb-ERT2-Cre, STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre mice to DEN-induced liver cancer was determined. After TAM treatment, injection of 50 μg/g DEN resulted in 100% incidence of visible tumours in Alb-ERT2-Cre mice and STK33KOflox/flox mice, but only 25% incidence in STK33KOflox/flox, Alb-ERT2-Cre mice (figure 5A); the number and size of tumours were also decreased in STK33KOflox/flox, Alb-ERT2-Cre mice (figure 5B, C). Histological analysis demonstrated that STK33KOflox/flox, Alb-ERT2-Cre mice had a 41% incidence of preneoplastic foci and 25% adenomas but no carcinomas, whereas 100% of Alb-ERT2-Cre mice and STK33KOflox/flox mice had both preneoplastic foci and adenomas, with 56% and 58% of them developing carcinomas, respectively (figure 5D). Ki67 immunostaining showed a significant increase in hepatocyte proliferation in DEN-treated Alb-ERT2-Cre mice and STK33KOflox/flox mice livers, which was markedly reduced in STK33KOflox/flox, Alb-ERT2-Cre mice livers (figure 5E).

STK33KOflox/flox, Alb-ERT2-Cre mice are resistant to diethylnitrosamine (DEN)-induced liver tumorigenesis. (A) Incidence of visible tumours in tamoxifen (TAM)-pretreated Alb-ERT2-Cre, STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre mice treated with 50 μg/g DEN at the age of 20 days and killed 10 months later. (B) The number of tumours counted on the surface of liver lobes. (C) Tumour size measured on the surface of liver lobes. (D) Incidence of preneoplastic foci, hepatocellular adenoma and hepatocellular carcinoma in DEN-treated Alb-ERT2-Cre, STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre mice. Data are expressed as percentage of total number of mice. (E) Representative images of Ki67 staining from DEN-induced tumours in Alb-ERT2-Cre, STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre mice. For (A)–(E), mice were treated with 50 μg/g DEN at age 20 days and killed 10 months later. n=10 for Alb-ERT2-Cre group, n = 12 for STK33KOflox/flox and STK33KOflox/flox, Alb-ERT2-Cre groups. *p<0.05, **p<0.01, ***p<0.001.
STK33 binds to c-Myc, enhances c-Myc transcription activity and rescues c-Myc depletion in HCC cells
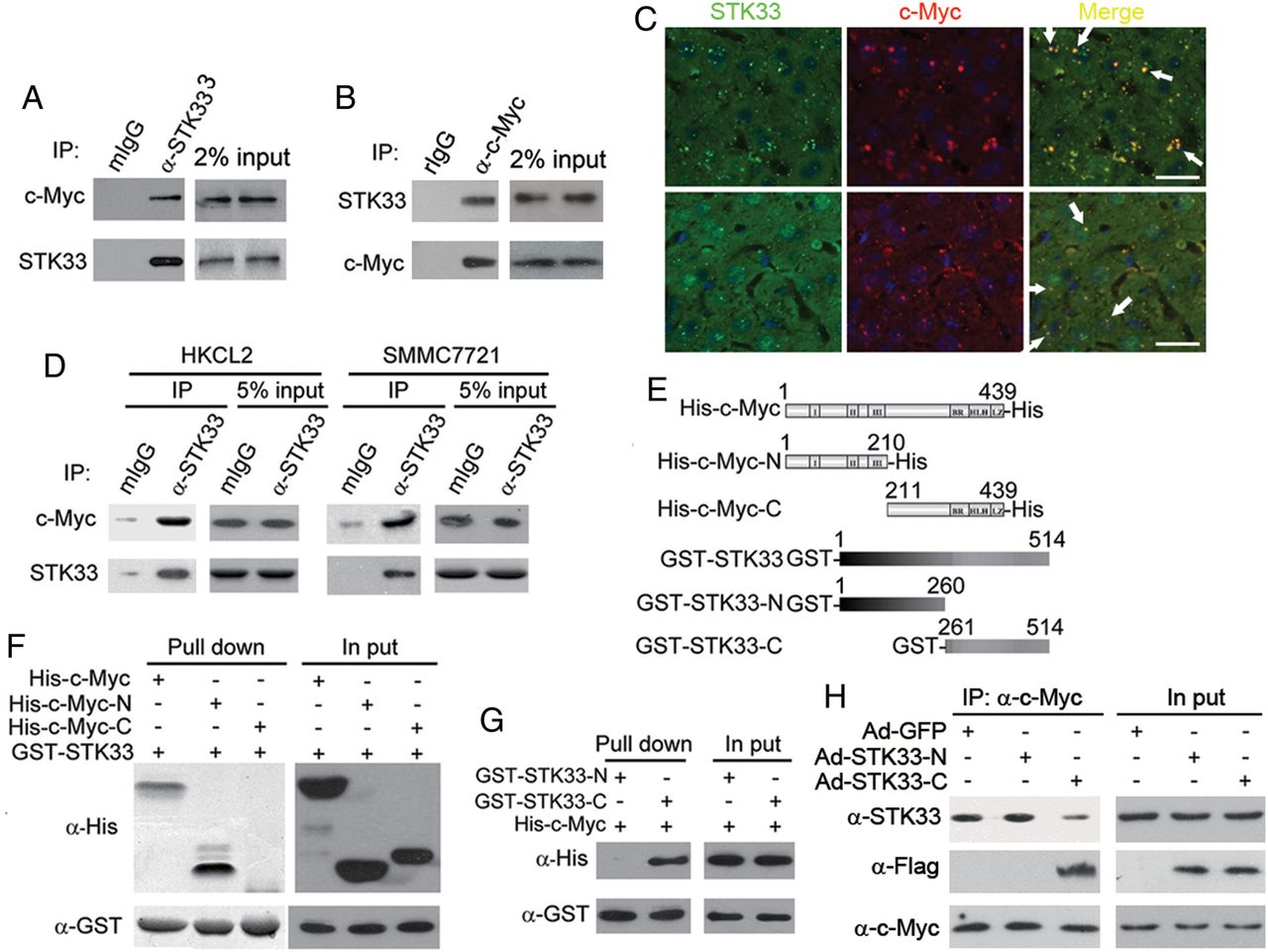
We investigated the mechanism by which STK33 regulates HCC tumour cell and liver cell proliferation. Using a coimmunoprecipitation (co-IP) assay in primary HCC sample lysates, we found that STK33 was coimmunoprecipitated with endogenous c-Myc (figure 6A, B). To confirm the interaction of STK33 and c-Myc, we stained the two proteins in human HCC samples and DEN-induced liver tumour sample and found that STK33 colocalised with c-Myc (figure 6C). We also verified, using the co-IP assay, that STK33 could bind to c-Myc in HKCL2 and SMMC7721 cells (figure 6D). We subsequently examined whether STK33 interacted with c-Myc directly using purified recombinant proteins, in which a glutathione S-transferase (GST) tag was fused to full-length STK33, N-terminus (aa1–260, GST-STK33-N) or C-terminus (aa261–514, GST-STK33-C) of STK33, and a His tag was fused to the full-length c-Myc (His-c-Myc), N-terminus (aa1–210, His-c-Myc-N) or C-terminus (aa211–439, His-c-Myc-C) of c-Myc (figure 6E). We found that the N-terminus but not the C-terminus of c-Myc bound directly to GST-STK33 (figure 6F), and that the C-terminus but not the N-terminus of STK33 bound to His-c-Myc (figure 6G).

The C-terminus of STK33 binds directly to c-Myc. (A) Endogenous STK33 bound to c-Myc in hepatocellular carcinoma (HCC) samples. Endogenous STK33 was immunoprecipitated from the HCC tumour lysates using mouse STK33 antibody, followed by immunoblotting with antibodies to STK33 and c-Myc. Mouse IgG (mIgG) was used as an immunoprecipitation-negative control antibody. Sample loading controls are shown (2% input). (B) Endogenous STK33 bound to c-Myc in HCC samples. Endogenous c-Myc was immunoprecipitated from the HCC tumour lysates using rabbit c-Myc antibody, followed by immunoblotting with antibodies to c-Myc and STK33. Rabbit IgG (rIgG) was used as an immunoprecipitation-negative control antibody. Sample loading controls are shown (2% input). (C) Colocalisation of STK33 and c-Myc by immunofluorescent staining with STK33 antibody and c-Myc antibody in human HCC samples (upper) and diethylnitrosamine (DEN)-induce mouse liver tumour samples (lower). DAPI (4’,6-diamidino-2-phenylindole; blue) was used to indicate the nucleus. (D) STK33 binds to c-Myc in HKCL and SMMC7721 cells. STK33 was immunoprecipitated from the cell lysates followed by immunoblotting using antibodies to STK33 and c-Myc. mIgG was used as an immunoprecipitation-negative control antibody. (E) Scheme for c-Myc and STK33 mutants. (F) STK33 directly binding to c-Myc. Isolated His-fused c-myc protein (His-c-Myc), N-terminus of c-Myc (His-c-Myc-N), C-terminus of c-Myc (His-c-Myc-C), and GST-fused STK33 protein (GST-STK33) were incubated with glutathione beads as indicated. After extensive washing, they were immunoblotted with the His antibody (α-His); GST-STK33 was detected with GST antibody (α-GST). Sample loading controls are shown (2% input). (G) c-Myc bound to C-terminus of STK33. Isolated His-fused c-myc protein (His-c-Myc), GST-fused N-terminus of STK33 (GST-STK33-N) and C-terminus of STK33 (GST-STK33-C) were incubated with glutathione beads as indicated. After extensive washing, they were immunoblotted with the His antibody (α-His); GST-STK33 was detected with GST antibody (α-GST). Sample loading controls are shown (2% input). (H) C-terminus of STK33 blocked endogenous STK33/c-Myc association. c-Myc antibodies were used to immunoprecipitate c-Myc from SMMC7721 cell lysates, STK33 antibodies were used to detected endogenous STK33, and Flag antibodies were used to detect STK33-N or STK33-C.
We evaluated whether STK33 affected c-Myc transcription activity. Using two c-Myc response promoter luciferase report genes pTERT-Luc and M4mintK-Luc, we found that, when overexpressing STK33 in L02 or HepG2 cells, both reporters were enhanced (see online supplementary figure S2A, B), whereas after knockdown of STK33 by shRNA in HKCL2 and SMMC7721 cells, both pTERT-Luc and M4mintK-Luc luciferase values were reduced (see online supplemental figure S2C, D).
We then investigated whether overexpression of STK33 could rescue c-Myc depletion. Using shRNA, we knocked down endogenous c-Myc in HepG2 cells (see online supplementary figure S3A) and found (using the MTT assay) that the knockdown of c-Myc inhibited HepG2 cell proliferation in vitro. Moreover, this inhibitive effect was rescued by overexpression of STK33 (see online supplementary figure S3B). Furthermore, the in vivo tumour growth was dramatically rescued by overexpression of STK33 in c-Myc knockdown HepG2 cells (see online supplementary figure S3C).
The C-terminus of STK33 blocked STK33/c-Myc association and inhibited HCC cell proliferation and liver tumour growth
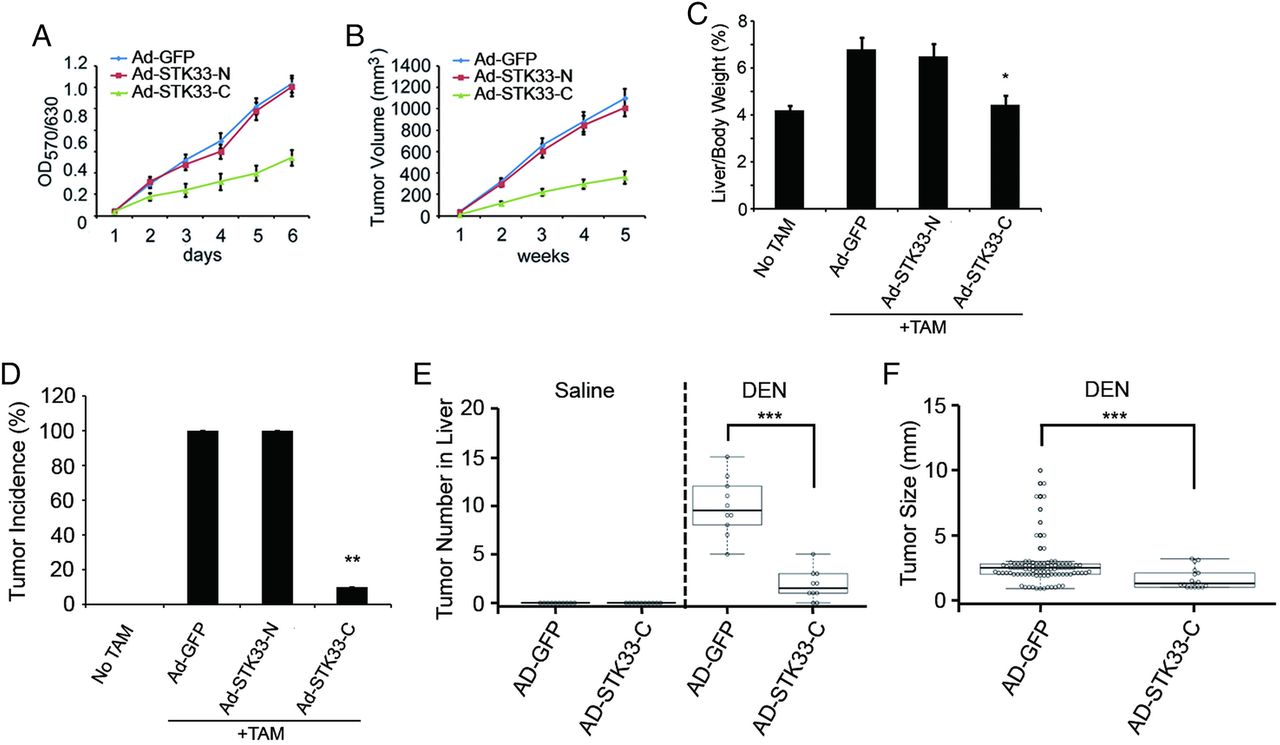
We generated adenovirus encoding the N-terminus of STK33 (Ad-STK33-N, aa1–260) or the C-terminus of STK33 (Ad-STK33-C, aa261–514) and infected SMMC7721 cells with adenovirus control vector (Ad-GFP), Ad-STK33-N or Ad-STK33-C. Through co-IP assay using antibody to c-Myc, we found that less STK33 coimmunopricipatitated with c-Myc when cells were treated with Ad-STK33-C, compared with those treated with Ad-GFP vector or Ad-STK33-N. This indicates that STK33-C competed with endogenous STK33 for binding to c-Myc (figure 6H). We then explored whether STK33-C affected c-Myc transcription activity and HCC cell proliferation through blocking STK33/c-Myc association. In fact, c-Myc-targeted reporter gene transcription was inhibited by Ad-STK33-C (see online supplementary figure S4A, B). The proliferation of SMMC7721 cells were dramatically inhibited by Ad-STK33-C, but not by Ad-GFP vector or by Ad-STK33-N measured using the MTT assay (figure 7A). Nude mice inoculated subcutaneously with SMMC7721/Ad-STK33-C cells exhibited dramatically reduced tumour volumes compared with mice receiving SMMC7721/Ad-GFP andSMMC7721/Ad-STK33-N cells (figure 7B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The C-terminus of STK33 inhibited hepatocellular carcinoma cell proliferation and liver tumour growth. (A) Ectopic expression of STK33-C inhibited in vitro growth of SMMC7721 cells, measured using the MTT assay. (B) Ectopic expression of STK33-C inhibited average tumour volume in athymic nude mice subcutaneously inoculated with SMMC7721 cells. (C) STK33-C decreased liver weight/body weight ratios, detected at day 7 after adenovirus injection. Tamoxifen (TAM) induced STK33Tgflox/flox, Alb-ERT2-Cre mice treated with adenovirus control vector (Ad-GFP), Ad-STK33-N and Ad-STK33-C. (D) Liver tumour burst incidence in normal mice, TAM-induced STK33Tgflox/flox, Alb-ERT2-Cre mice treated with Ad-GFP, Ad-STK33-N and Ad-STK33-C. (E) The number of tumours counted on the surface of liver lobes. (F) Tumour size measured on the surface of liver lobes. For (C)–(F), C57BL/6 mice were treated with 50 μg/g diethylnitrosamine (DEN) at the age of 20 days and killed 10 months later. Results represent at least three separate experiments. n=10/group (B–F). Error bar represents ±SD. *p<0.05, **p<0.01, ***p<0.001.
Using the TAM-inducible STK33Tgflox/flox, Alb-ERT2-Cre mice model, we injected mice with Ad-GFP, Ad-STK33-N or Ad-STK33-C once every 7 days. Expression of STK33-N and STK33-C is shown in online supplementary figure S5A–C. We found in mice treated with Ad-STK33-C a significantly decreased liver/body weight ratio at day 7 after adenovirus injection (figure 7C). Furthermore, Ad-STK33-C inhibited liver tumour burst in STK33Tgflox/flox, Alb-ERT2-Cre mice (figure 7D).
To further investigate the role of STK33 in liver tumorigenesis, the DEN-induced liver cancer model was used. We injected DEN (50 μg/g) into C57BL/6 mice and found that STK33 expression was upregulated in DEN-induced tumour tissues, compared with liver tissues from PBS-treated mice (see online supplementary figure S6A, B). Expression of c-Myc was also upregulated in DEN-induced liver tumours. However, expression of KRAS was not found to be significantly changed at either the protein or mRNA level (see online supplementary figure S6A, B). When mice were injected with Ad-STK33-C once every 7 days (see online supplementary figure S5D, E), both the number and size of tumours were decreased (figure 7E, F). These results indicate that the C-terminus of STK33 inhibits liver tumour growth.
STK33 kinase inhibitor BRD-8899 failed to inhibit STK33-induced cell proliferation and migration
To explore the role of kinase activation of STK33 in HCC cells, we used a STK33 kinase inhibitor, BRD-8899.9 We found that BRD-8899 failed to inhibit the proliferation and migration of HepG2/STK33 cells (see online supplementary figure S7A, B). It suggests that the kinase activation effect of STK33 is not necessary in HCC cell proliferation and migration.
Discussion
In this study, we found that STK33 expression was frequently upregulated in patients with HCC (56.2%), particularly at an advanced stage (80.56%). A positive correlation between STK33 expression and advanced clinicopathological features was also demonstrated. Functional analysis by gain- or loss-of-function studies showed that STK33 overexpression enhanced HCC cell proliferation, while STK33 knockdown using shRNA inhibited HCC cell proliferation both in vitro and in vivo. In the mechanism study, we found that STK33 could bind to c-Myc directly and promote c-Myc transcription activity. As such, we created adenovirus coding the C-terminus of STK33 (aa261–514) and found that the C-terminus of STK33 blocked STK33/c-Myc association, decreased c-Myc transcription activity, and inhibited HCC cell proliferation both in vitro and in vivo.
We developed models of overexpression and knockout of STK33 using a TAM-inducible albumin promoter driving transgenic technology. Our study clearly indicates that overexpression of STK33 promoted tumour burst in the liver by 100%, whereas the C-terminus of STK33 could inhibit this effect. Furthermore, the C-terminus of STK33 could dramatically decrease the number and size of tumours induced by DEN. Mice with hepatocyte-specific STK33 knockout were largely resistant to DEN-induced hepatocarcinogenesis, demonstrating that STK33 is essential in hepatocellular proliferation and tumorigenesis. These results indicate that STK33 is a potential target for liver tumour therapy.
c-Myc has been studied for more than 30 years; it contains an N-terminal transcriptional regulatory domain and a C-terminal HLH-Zip domain.18–24 The N-terminal domain has been reported to form complexes with many factors including TRRAP, GCN5 and TBP to regulate transcription. The C-terminal domain dimerises with Max and bends DNA through binding motifs (5′-CACGTG-3′) termed E boxes, and then recruits complexes that modify chromatin.25–28 c-Myc is thought to contribute to hepatocyte proliferation, liver regeneration and tumorigenesis in response to hepatic injury and carcinogen exposure. Recently, Qu et al29 demonstrated that Myc is essential in hepatocellular proliferation and tumorigenesis. It is interesting that, in our study, HCC cell proliferation and liver tumour growth were affected by blockage of STK33/c-Myc association, whereas the STK33 kinase inhibitor, BRD-8899, did not inhibit HCC cell proliferation, which is consistent with results from previous studies.10 These results suggest that it is the STK33/c-Myc association rather than the kinase activity of STK33 that is important in HCC cell proliferation. The STK33/c-Myc association could work well as a target for HCC therapeutic medicine scanning.
The three human Ras genes (H-Ras, N-Ras and K-Ras) encode four proteins that function as small GTP-binding proteins, H-Ras, N-Ras, K-Ras4A and K-Ras4B. Single amino acid substitutions at N-Ras codon 12, H-Ras codon 13 or K-Ras codon 61, that unmask Ras-transforming potential, create mutant proteins that are insensitive to GAP (Ras p120 GTPase activation protein) stimulation.30 ,31 Consequently, these oncogenic Ras mutant proteins are locked in the active, GTP-bound state, leading to constitutive, deregulated activation of Ras function. The present study examined tumour tissues from 251 patients with HCC for somatic mutations in codons 12, 13 and 61 of the KRAS, HRAS or NRAS genes, which are known hot spots in various malignancies. However, the results showed that only 11 of the 251 patients (4.38%) had somatic Ras mutations, indicating that Ras gene mutations do not appear to be related to the pathogenesis of most HCCs. There have been several reports with small sample sizes regarding Ras gene mutations in HCC. Most have reported that somatic mutations of the Ras gene in HCCs are uncommon, which is similar to our study. Taketomi et al12 found only one patient with a RAS point mutation among 61 patients with HCC. Tsuda et al13 found only two tumour samples with Ras point mutations from 30 patients with HCC. Tada et al14 found that there were no point mutations in any of 12 patients with HCC in codons 12, 13 or 61 of the Ras genes. Leon and Kew15 found that only one of 12 patients with HCC had a RAS point mutation. Ogata et al16 found that only two of 19 patients with HCCs had a RAS mutation. Although STK33 was reported to be critical for the survival of KRAS-dependent haematopoietic cancer cell lines and some other cancer cells, the interaction of STK33 and Ras did not seem to play crucial roles in HCC.
Both c-Myc and Ras were considered to be oncogenes. Ample studies have also proved that c-Myc has potent oncogenicity, which can be further enhanced by collaborations with other oncogenes such as Bcl-2 and activated Ras. Studies on the collaborations of c-Myc with Ras in oncogenicity have disclosed their underlying mechanisms in tumour biology, including ‘immortalisation’ and ‘transformation’, through a cycD1–CDK4 complex.32 It would be interesting to explore whether STK33 is involved in the collaborations of c-Myc with Ras in oncogenesis.
Acknowledgments
This study was supported by National Natural Science Foundation of China (Nos 81472284, 81101578, 81372262 and 81172020), Shanghai ‘Rising-Star’ Science Foundation for Youths (No 12QA1404800), the Program for Excellent Young Scholars of SMMU, the Charitable Project on Cancer Research of Shanghai, the State Key Project on Infectious Diseases of China (No 2012ZX10002-016), and the Technology Committee of Shanghai Municipal Government (No 09411960200).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
TY, BS and JZ contributed equally.
Contributors TY, BS, JZ, G-SY, HZ and W-FY performed all the experiments and analysed the results. FS, M-CW and J-HL designed the experiments and wrote the manuscript.
Competing interests None.
Patient consent Obtained.
Ethics approval Clinical Research Ethics Committee of Second Military Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.