Article Text

Abstract
Objective The ductular reaction (DR) involves mobilisation of reactive-appearing duct-like cells (RDC) along canals of Hering, and myofibroblastic (MF) differentiation of hepatic stellate cells (HSC) in the space of Disse. Perivascular cells in stem cell niches produce pleiotrophin (PTN) to inactivate the PTN receptor, protein tyrosine phosphatase receptor zeta-1 (PTPRZ1), thereby augmenting phosphoprotein-dependent signalling. We hypothesised that the DR is regulated by PTN/PTPRZ1 signalling.
Design PTN-GFP, PTN-knockout (KO), PTPRZ1-KO, and wild type (WT) mice were examined before and after bile duct ligation (BDL) for PTN, PTPRZ1 and the DR. RDC and HSC from WT, PTN-KO, and PTPRZ1-KO mice were also treated with PTN to determine effects on downstream signaling phosphoproteins, gene expression, growth, and migration. Liver biopsies from patients with DRs were also interrogated.
Results Although quiescent HSC and RDC lines expressed PTN and PTPRZ1 mRNAs, neither PTN nor PTPRZ1 protein was demonstrated in healthy liver. BDL induced PTN in MF-HSC and increased PTPRZ1 in MF-HSC and RDC. In WT mice, BDL triggered a DR characterised by periportal accumulation of collagen, RDC and MF-HSC. All aspects of this DR were increased in PTN-KO mice and suppressed in PTPRZ1-KO mice. In vitro studies revealed PTN-dependent accumulation of phosphoproteins that control cell-cell adhesion and migration, with resultant inhibition of cell migration. PTPRZ1-positive cells were prominent in the DRs of patients with ductal plate defects and adult cholestatic diseases.
Conclusions PTN, and its receptor, PTPRZ1, regulate the DR to liver injury by controlling the migration of resident cells in adult liver progenitor niches.
- CELL MIGRATION
- FIBROSIS
- CHOLESTATIC LIVER DISEASES
- IMMUNOHISTOCHEMISTRY
- STEM CELLS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Various types of liver injury promote a ductular reaction (DR) characterised by the periportal accumulation of small ductules, myofibroblasts and collagen matrix.
Pleiotrophin (PTN) is a heparin-binding growth factor that inhibits constitutive tyrosine phosphatase activity of its receptor, protein tyrosine phosphatase receptor Z1 (PTPRZ1) to regulate fate decisions in various stem/progenitor cells.
In the liver, PTN expression is induced in several conditions associated with liver progenitor accumulation, including bile duct ligation, partial hepatectomy and hepatocellular carcinoma.
Liver regeneration is inhibited in PTN-deficient mice after partial hepatectomy, suggesting that PTN promotes liver repair.
What are the new findings?
Cells in putative liver stem/progenitor niches, that is, the space of Disse and canals of Hering, express PTN and its receptor, PTPRZ1 and modulate PTN/PTPRZ1 expression during liver injury.
PTN-PTPRZ1 signalling controls the mobilisation of cells that normally reside in stem/progenitor niches of adult livers, thereby regulating the intensity of the DR.
PTN-PTPRZ1 interaction in liver cells modulates the tyrosine phosphorylation of factors that control cell-cell adhesion, cell-matrix interactions and cell migration.
PTPRZ1 is prominent in the DRs of von Meyenburg complexes, adult polycystic liver disease, primary biliary cirrhosis and primary sclerosing cholangitis.
How might it impact on clinical practice in the foreseeable future?
PTN/PTPRZ1 signalling might be manipulated therapeutically to optimise recovery from chronic cholestatic liver injury.
Inter-individual differences in PTN/PTPRZ1 pathway activity might be exploited as biomarkers for progressive liver fibrosis.
Introduction
Various types of liver injury promote a ductular reaction (DR) characterised by the periportal accumulation of small ductules comprised of reactive-appearing duct-like cells (RDC), myofibroblasts (MF) and collagen matrix. Neither the mechanisms driving this DR, nor its biological significance, are well understood.1 Bipotent liver progenitors that can differentiate into ductular cells reside along canals of Hering; vestiges of fetal ductal plates that persist around portal tracts in adult livers.2 Multipotent liver progenitors have also been identified in submucosal glands within the wall of the adult biliary tree.3 Mature hepatocyte and cholangiocytes can also cause RDC with properties of liver progenitors.4 ,5 It has been suggested that the DR reflects injury-related mobilisation of liver progenitors from varying sources.6 We evaluated the hypothesis that the DR is regulated by pleiotrophin (PTN) and its receptor, protein tyrosine phosphatase receptor zeta-1 (PTPRZ1).
PTN regulates fate decisions in various stem/progenitor cells.7–12 PTN expression has been demonstrated in two conditions associated with accumulation of liver progenitors; partial hepatectomy (PH) and hepatocellular carcinoma (HCC).13–16 Bile duct ligation (BDL), a well-established stimulus for the DR, also induces PTN.17 PTN appears to promote liver growth because regeneration after PH is inhibited in PTN-deficient mice.14 PTN is known to interact with various molecules, including syndecans, integrins, anaplastic lymphoma kinase (ALK) and PTPRZ1. Among these, PTPRZ1 is best characterised as a true receptor for PTN because PTN-PTPRZ1 interaction inhibits the constitutive tyrosine phosphatase activity of PTPRZ1, resulting in accumulation of phosphorylated tyrosine residues in ALK and other proteins that are ordinarily dephosphorylated by PTPRZ1.18 ,19 Prolonged PTN-dependent phospho-tyrosine-mediated tyrosine kinase activity modulates signalling in an array of kinase cascades, such as PI3-kinase,9 ,20 Akt,8 ,21 extracellular-signal regulated kinase (ERK)1/2,21 and PKCα/β,22 providing a mechanism by which PTN might globally impact cell fate. Cytoskeletal proteins that regulate migration, proliferation, and differentiation, are downstream effectors regulated by PTPRZ1.23–25
In certain stem cell niches, PTN is expressed by endothelial cells.11 Although resident liver pericytes (a.k.a., hepatic stellate cells (HSC)) produce PTN, it is not known if liver sinusoidal endothelial cells (LSEC) also produce this factor. Also unclear is how PTN expression by space of Disse-associated cells compares to that of other liver cell types. Nevertheless, PTN expression has been shown to increase as HSC become MF.17 ,26 This is intriguing because HSC-derived MF regulate the fate of liver progenitors, increase transiently after PH, and are necessary for post-PH liver regeneration.27 In mice, facultative progenitors for hepatocytes and cholangiocytes emerge within MF-HSC populations after acute PH and during chronic BDL-induced cholestatic liver injury.27 ,28 Populations of MF-HSC and progenitors expand during cirrhosis, an established risk factor for HCC.29 To the best of our knowledge, however, there have been no publications demonstrating PTPRZ1 expression in adult liver. In particular, the role of PTPRZ1 in the adult liver response to injury has not been well understood. Specific unknowns include which, if any, cell types involved in the DR express PTPRZ1 and whether or not the growth-promoting actions of PTN in adult liver require interaction with PTPRZ1.
To clarify the significance of PTN and its receptor, PRTPRZ1, in regulating cell fate decisions in adult liver, we subjected wild-type (WT), PTN-deficient and PTRPZ1-deficient mice to BDL and compared the intensity of the DR that was provoked by constant mechanical biliary obstruction. Studies in intact mice were complemented by analyses of isolated liver cells and human liver biopsies. The results demonstrate a previously unsuspected role for PTN-PTPRZ1 signalling in regulating the DR to liver injury.
Materials and methods
Human liver biopsies
Anonomised unstained liver sections from formalin-fixed paraffin embedded tissue blocks of individuals with normal liver (3), adult polycystic disease (n=1), von Meyenburg complexes (n=1), primary biliary cirrhosis (n=2) and primary sclerosing cholangitis (n=4) were obtained from the Duke Department of Pathology in accordance with our Duke IRB-approved protocol. Liver sections were processed for immunohistochemistry as detailed below.
Mice
PTN-GFP, PTN knockout (KO), and PTPRZ1 KO mice have been previously described.11 C57BL/6 and SV129 brown WT mice were used as strain-specific controls for PTN KO and PTPRZ1-KO mice, respectively, and were obtained from The Jackson Laboratory and Taconic, respectively.
To induce cholestatic injury, adult (10–12 weeks) mice (C57BL/6 WT (n=7), PTN-KO (n=9) mice, SV129 brown WT (n=6), and PTPRZ1-KO mice (n=8)) underwent BDL or sham surgery (n=3/group). After 14 days, animals were euthanised; blood and tissues were obtained as described.28 All studies were performed in accordance with a Duke University IACUC-approved protocol.
Cell culture
Primary cell isolation from mice
Primary HSC, LSEC, and hepatocytes were isolated from a 12-week-old mice.28 ,30
HSC and LSEC viability and purity were greater than 95% as previously described,28 ,30 HSCs were cultured for up to 9 days in standard Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, and 1% penicillin-streptomycin.
Hepatocytes were used immediately for RNA isolation. Cell viability exceeded 90% in all experiments as determined by Trypan-blue exclusion.
The murine ductular progenitor cell line, 603B, and the clonally-derived rat MF-HSC line, 8B, were maintained in culture as described.28
RNA isolation from cells/liver tissue and quantitative real-time RT-PCR
Total RNA was extracted from cells and whole liver tissue using commercially available kits (Zymo Research) and reversed transcribed to cDNA. 1.5% of the first-strand reaction was amplified using StepOne Plus Real-Time PCR Platform (ABI/Life Technologies) and specific oligonucleotide primers for target sequences. Target gene expression analysis was conducted according to the 2–ΔΔCt method with expression normalised to the housekeeping gene S9 as described.28 Primer sequences are listed in online supplementary table S1.
Cell viability assay
Cell viability was measured using the Cell Counting Kit-8 (CCK8; Dojindo Molecular Technologies).
Serum bilirubin and aspartate aminotransferase
Serum was collected at the time of sacrifice in all mice. Bilirubin and aspartate aminotransferase were measured using commercial kits (Biotron Inc) according to the manufacturer's instructions.
Hydroxyproline assay
Hepatic hydroxyproline content was quantified in flash frozen liver samples.28 Concentrations were calculated from a standard curve prepared with high-purity hydroxyproline (Sigma–Aldrich) and expressed as mg hydroxyproline per g of liver tissue.
Immunohistochemistry
Liver tissue was fixed in formalin and embedded in paraffin. Immunohistochemical staining was performed using the DAKO Envision System (DAKO Corporation) according to manufacturer's protocol as described.28 Primary and HRP-conjugated secondary antibodies are listed in online supplementary table S2. Double immunohistochemistry was performed using Vina Green, per manufacturer's recommendation (BioCare Medical).
Morphometry and tissue analysis
Quantification of bile infarcts, as well as morphometric analysis Sirius red-stained, α smooth muscle actin (αSMA)-stained, desmin-stained and elastin-stained liver sections from 20 randomly-selected fields at ×20 magnification per section per mouse was performed (MetaView, Universal Imaging Corp.28 Green fluorescent protein (GFP) staining was quantified by counting cells with stained nuclei in 10 randomly-chosen, ×20 fields per section per mouse.
Wound-healing assay
Wound-healing assays were performed by scraping cell monolayers. Cells were washed twice with PBS and then incubated with either vehicle (PBS) or recombinant human PTN 100 ng/mL (R&D Systems). A reference mark was created on the dish and a time 0 image was acquired. Subsequent images captured matched regions at the indicated times; the size of area lacking cells was quantified (Olympus DP2-BSW software).
Phosphotyrosine analysis
After PTN treatment (100 ng/mL), cultured cells were washed and lysed using SDS sample buffer. Samples were analysed by Western blotting using antiphosphotyrosine PY20 (1:1000; Upstate Laboratories). Immunoreactive proteins were visualised using horseradish peroxidase-catalysed chemiluminescence reaction (Amersham–Pharmacia).
To investigate global phosphotyrosine profiles in±PTN cell states, two independent approaches were utilised to increase coverage of potential downstream PTN dependent targets; a phosphotyrosine protein enrichment approach and a phosphotyrosine peptide enrichment approach (see online supplementary methods). Enriched phosphopeptides were then subjected to nanoscale capillary chromatography coupled with a high-resolution hybrid quadrupole-oribtrap mass spectrometer (QExactive Plus, Thermo). Spectra from liquid chromatography-mass spectrometry (LC-MS)/MS experiments were submitted to Mascot database searches against a forward/reverse SwissProt_Mouse database allowing for phosphorylation modifications on Ser, Thr and Tyr residues. Spectra were annotated at <1.0% false discovery rate with Scaffold. To define ambiguity in assignment of modification localisation, all spectra were processed through the Ascore algorithim and a per cent probability of correct localisation was assigned.31 ,32
Statistics
Data were expressed as mean±SEM. Statistical analysis was performed using Student's t test. Analysis was conducted using GraphPad Prism 4 software; p≤0.05 was considered to be statistically significant.
Results
Stromal cells are the predominant source of PTN in healthy liver and up-regulate PTN production during biliary obstruction
To localise PTN expression in adult liver, we used qRT-PCR to quantify PTN mRNAs in hepatocytes, HSC and LSEC freshly-isolated from healthy adult mice (figure 1A). PTN mRNA levels were similar in HSC and LSEC, and more than 60-fold lower in hepatocytes. Although PTN mRNA was easily demonstrated in freshly-isolated HSC or LSEC, we were unable to detect GFP marker in either cell type in healthy adult PTN-GFP transgenic mice, suggesting low PTN transcription in situ (figure 1B, C). Cholangiocytes of healthy PTN-GFP mice were also negative for GFP, but we observed low levels of PTN transcripts in 603B cells, a RDC line with features of a bipotent liver epithelial progenitor (figure 1A).

Adult liver stromal cells produce pleiotrophin (PTN). (A) PTN gene expression was assessed in primary hepatocytes (Hep), hepatic stellate cells (HSC), and liver sinusoidal endothelial cells (LSEC), as well as the ductular progenitor cell line, 603B by qRT-PCR. Results are normalised to S9 expression and expressed as fold-change over Hep (n=3–5 independent isolations). (B) Representative staining for PTN-driven GFP expression in PTN-GFP mice after sham operation or bile duct ligation (BDL) (magnification, ×10) and (C) Quantification of GFP(+) cells from 20 random fields. Results are expressed as fold-change over sham-operated controls. (D) Changes in PTN expression in during culture activation of primary HSC were analysed by qRT-PCR. Results are normalised to S9 and expressed as fold-change relative to day 0 (n=3–6 independent isolations). (E) PTN expression in whole liver from sham-operated or BDL mice was analysed by qRT-PCR. Results are normalised to S9 and expressed as fold-change over sham-operated control. (F) Double-staining demonstrating co-localisation of GFP (green) with either desmin (brown) or elastin (brown) in PTN-GFP mice after BDL (magnification: ×40; inset ×100). Double positive cells are indicated by black arrows. *p<0.05; **p<0.01; ***p<0.001.
Consistent with an earlier publication,17 we found that freshly isolated HSC dramatically upregulated PTN mRNA expression as they transdifferentiated into MF during culture. By culture day 7, PTN mRNA levels had increased more than 100-fold (figure 1D). BDL, an insult that provokes MF accumulation, also increased PTN mRNA levels and transcription (figure 1B, C, E). In PTN-GFP mice, GFP(+) cells were easily identified in periportal stroma 2 weeks after BDL, a marked change from the healthy adult liver. To better characterise PTN-producing cells in BDL livers, sections were costained for GFP and either desmin (a HSC marker) or elastin (to mark portal fibroblasts) (figure 1C, F). GFP colocalised with cells that expressed desmin and with cells that expressed elastin, demonstrating that MF derived from HSC and portal fibroblasts are sources of PTN during chronic cholestatic liver injury. In contrast, GFP staining was conspicuously absent in hepatocyte plates and in cholangiocytes lining intrahepatic bile ducts. Likewise, we were unable to demonstrate PTN transcription within any of the small periportal ductules that accumulated after BDL.
PTN depletion enhances the DR to liver injury
PTN regulates fate decisions in various types of progenitor cells;7–12 ,33 ,34 after PH, liver regeneration is acutely impaired in PTN-KO mice. Evidence that LSEC and HSC express PTN in healthy livers (figure 1A), and that PTN-producing cells localise periportally in injured livers (figure 1B, F), suggests that PTN is enriched in the space of Disse and near canals of Hering, two putative progenitor niches in adult livers. To clarify the significance of PTN induction in chronically injured livers, we compared hepatic responses to BDL in PTN-KO mice and age/gender-matched WT controls. Livers of healthy PTN-KO mice and WT mice appeared normal (see online supplementary figure S1). As expected, BDL provoked cholestatic liver injury (see online supplementary figure S2) and stimulated periportal accumulation of small keratin (Krt)19(+) ductules and fibrous matrix. Most of the RDC demonstrated nuclear staining for Sox9, a marker of biliary progenitors.35 Many hepatocytic cells in periportal areas were also Sox9-positive (see online supplementary figure S3). The DR to BDL and liver injury were enhanced in PTN-KO mice (see figure 2A, B and online supplementary figure S2). Periportal areas of PTN-KO livers accumulated many more small Krt19-stained ductules and Sox9-postive RDC compared to WT livers after BDL. This portal tract expansion was associated with induction of Krt19 and Sox9 mRNA expression and greater accumulation of Sirius red-stained fibrils (see figure 2B, C and online supplementary figure S3). Increased matrix deposition in the PTN-KO mice post-BDL was confirmed by qRT-PCR analysis of collagen1α1 (Col1α1) mRNA levels and by hepatic hydroxyproline content (figure 2C). Cholestasis was also worse in the PTN-KO group; serum bilirubin level in PTN-KO mice were double that of controls 2 weeks after BDL (figure 2A). The aggregate findings suggest that induction of PTN after biliary obstruction controls the fate of cells involved in the DR, and that loss of PTN, as in the PTN-KO mouse, is associated with amplification of the DR, enhanced cholestatic liver injury and worse fibrosis.

Pleiotrophin (PTN) deficiency enhances the ductular reaction during cholestatic injury. (A) Representative H&E staining of liver sections from wild-type and PTN deficient mice (n=7–9 mice per group; magnification ×4). Differences in portal tract size were evaluated by morphometry and graphed as mean±SEM relative to sham-operated control. Serum bilirubin levels in sham-operated or bile duct ligation (BDL) wild type (WT) and PTN-knockout (KO) mice. Values are expressed as mean±SEM relative to WT sham-operated controls (mg/dL). (B) Representative immunohistochemistry (magnification, ×20), corresponding quantification of immunostained images and qRT-PCR analysis of Krt19 mRNA in whole liver RNA is shown in panels to the right. Results are expressed relative to sham-operated WT mice. (C) Representative sections of Sirius red stained liver (magnification, ×4) with Col1α1 qRT-PCR analysis and quantification of hepatic hydroxyproline in sham-operated or BDL WT and PTN-KO mice. Results are expressed relative to WT sham-operated mice. *p<0.05; ** p<0.01; ***p<0.001.
PTN inhibits migration of liver MF and ductular cells
To more directly assess the effects of PTN on ductular cells and MF, we treated cell lines (603B RDC and 8B MF-HSC) with recombinant PTN. Although PTN had no effect on viability or proliferation of either cell type (see online supplementary figure S4), it dramatically inhibited RDC and liver MF migration in a scratch wound assay (figure 3A, B). Further study of primary HSC-derived MF from PTN-KO mice confirmed the negative effects of PTN on cell migration (figure 3C). Primary cultures of HSC from PTN-KO mice demonstrated significantly greater migratory activity than cultures of HSC from WT mice, and this effect was abrogated by providing PTN to PTN-depleted HSC. Because migration is a component of epithelial-to-mesenchymal transition (EMT),36 we examined whether or not concentrations of PTN that inhibited cell migration influenced expression of EMT-related genes. PTN decreased expression of the pro-EMT transcription factor, SNAIL, in 603B RDC and liver MF (figure 3D). PTN treatment also decreased mRNA expression of several Hedgehog-regulated genes (figure 3E). This was accompanied by decreased mesenchymal marker expression (eg, Col1α1, αSMA and vimentin mRNA), and increased expression of genes that block EMT and promote HSC quiescence (eg, BMP7, E-cadherin and PPARγ). Therefore, increases in PTN that normally accompany biliary obstruction are likely to constrain the EMT-related migration of MF and RDC involved in the DR.

Pleiotrophin (PTN) inhibits liver cell migration and epithelial-to-mesenchymal transition (EMT). Cell migration was assessed by wound-healing assay in the murine ductular progenitor line 603B (A), rat hepatic stellate cells (HSC) line 8B (B), and primary HSC isolated from either wild type (WT) or PTN- knockout (KO) mice (C) in the presence of either PBS (untreated, UT) or PTN (PTN; 100 ng/mL) then quantified after 48 h (magnification, ×10). Leading edge of the repairing cell monolayers is indicated by yellow bars. Results are expressed as mean±SEM of three independent measurements. (D) SNAIL gene expression by qRT-PCR in 603B and 8B treated with either PBS (UT) or PTN (100 ng/mL) for 48 h. Results are normalised to S9 and expressed as fold-change relative to UT control for each cell type. (E) Changes in expression of either mesenchymal or epithelial (boxed) genes were assessed by qRT-PCR analysis in myofibroblastic-hepatic stellate cells (MF-HSC) treated with either PBS (UT) or PTN (100 ng/mL) for 48 h. Results are normalised to S9 and expressed as fold-change relative to UT control. *p<0.05; **p<0.01.
Cells that localise in hepatic progenitor niches express the PTN receptor, PTPRZ1
PTN is a heparin-binding protein that interacts with matrix-associated molecules, including syndecans and integrins, thereby maintaining pockets of PTN-enriched matrix. Since we found that PTN-expressing cells localised in the space of Disse and near canals of Hering in adult livers, we used qRT-PCR and immunostaining to determine if cells that typically reside in these locations express PTPRZ1. Quiescent (Q)-HSC and LSEC freshly-isolated from healthy adult livers, as well as the RDC line, 603B, expressed PTPRZ1 transcripts (figure 4A), although immunohistochemistry was unable to demonstrate PTPRZ1 protein in livers of healthy adult mice (figure 4B). After BDL, qRT-PCR analysis of whole liver RNA demonstrated significant PTPRZ1 mRNA induction (figure 4A). PTPRZ1 protein was readily identified in RDC and sinusoidal cells after BDL. PTPRZ1(+) RDC localised in intralobular bile ducts and in small ductules that accumulated periportally during biliary obstruction. Double staining for PTPRZ1 and a marker of HSC (desmin) confirmed perisinusoidal desmin(+) cells also expressed PTRPZ1 after BDL (figure 4B). Thus, injured livers accumulate PTPRZ1(+) cells in locations that are enriched with the PTPRZ1 ligand, PTN.
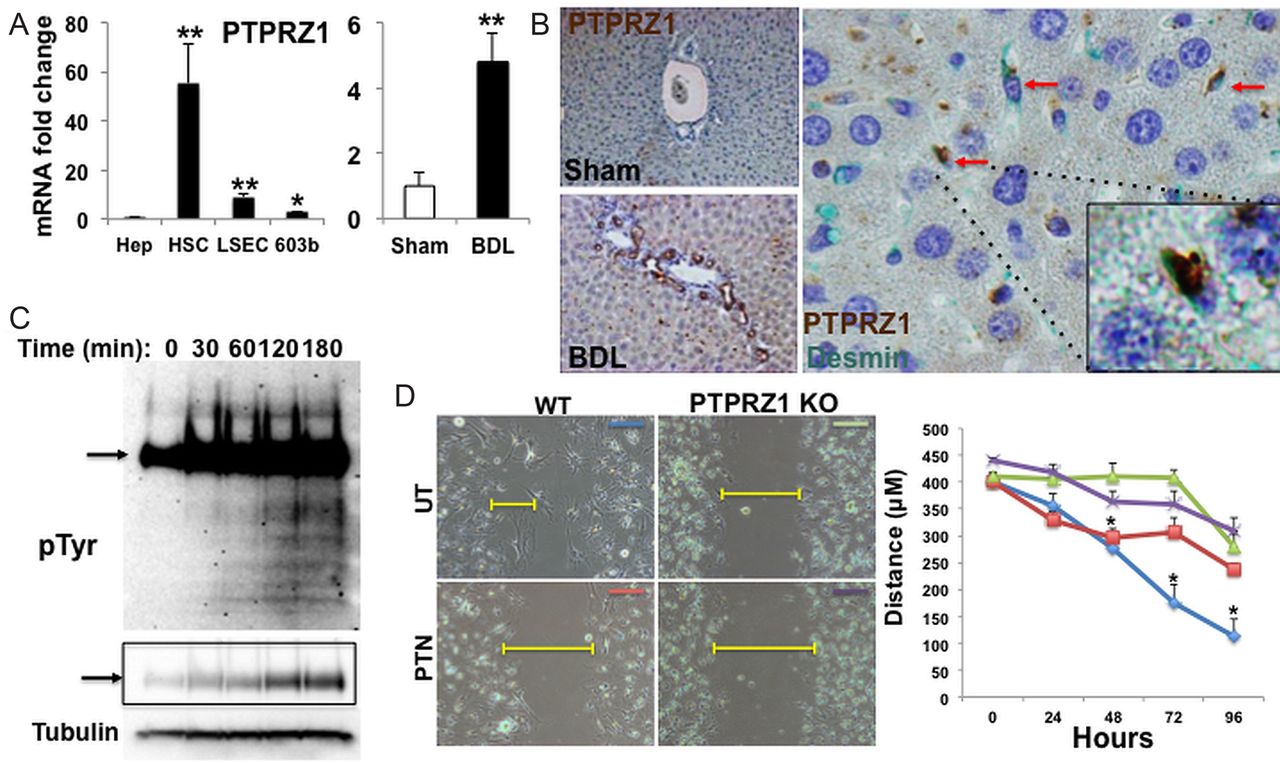
Protein tyrosine phosphatase receptor zeta-1 (PTPRZ1) signalling is required for pleiotrophin (PTN)-mediated inhibition of cell migration. (A) PTPRZ1 gene expression in different types of liver cells, and whole liver from either sham-operated or bile duct ligation (BDL) mice, was analysed by qRT-PCR. Results are normalised to S9 and expressed as fold-change relative to primary hepatocytes or sham-operated controls respectively. (B) Representative liver sections for PTPRZ1 immuno-staining (brown) from either sham-operated or BDL mice (magnification, ×10). Double immunostaining reveals co-localisation (red arrows) of PTPRZ1 (brown) and desmin (green) with a double-stained cell highlighted (inset, magnification ×100). (C) Changes in total tyrosine phosphorylation over time in 603B treated with PTN (100 ng/mL) were visualised by Western analysis using anti-PY20 antibody. Phosphoprotein expression (arrow) at decreased exposure is highlighted in boxed region. Tubulin was used as protein loading control. (D) Wound-healing assay in primary hepatic stellate cells ( isolated from either WT or PTPRZ1- knockout (KO) mice and treated either PBS (UT) or PTN (100 ng/mL). Leading edge of the repairing cell monolayers is indicated by yellow bars. Cellular migration was quantified and represented graphically with results expressed as mean±SEM of three independent measurements. *p<0.05; ***p<0.01.
PTN interacts with PTPRZ1 to regulate migration of cells that reside in liver progenitor niches
In PTN-sensitive cells, PTN binding to PTPRZ1 inhibits the constitutive tyrosine-phosphatase activity of the receptor, resulting in accumulation of tyrosine-phosphorylated target proteins.18 ,19 To determine if PTN-PTRZ1 interaction influenced the phenotype of PTPRZ1(+) liver cells, we treated the RDC line and MF-HSC with PTN and evaluated time-dependent changes in downstream phosphoprotein mediators. Western blot demonstrated a rapid increase and progressive accumulation of protein phosphotyrosine after PTN treatment, demonstrating functional signalling in these cells (figure 4C). To begin to evaluate the pathways affected by PTN treatment, we stimulated cells for 3 h and interrogated downstream phosphorylation events using two orthogonal enrichment approaches (see Materials and Methods section). High-resolution mass spectrometry analysis revealed increased phosphorylation of several proteins known to modulate intercellular connections and cellular migration (table 1). To determine if PTPRZ1 is involved in antimigratory effects of PTN (figure 3A–C), we isolated primary HSC from PTPRZ1-KO mice and evaluated HSC migration in the scratch wound assay. Compared to HSC from WT mice, PTPRZ1-deficient HSC migrated poorly and PTN did not further inhibit their migration (figure 4D). The aggregate findings show that PTN inhibits constitutive PTPRZ1 activity in liver cells. Further, the results demonstrate that PTPRZ1 normally promotes liver cell migration, because either addition of PTN (figure 3A–C) or loss of PTPRZ1 inhibits migration (figure 4D), while loss of PTN stimulates migration of PTPRZ1(+) liver cells (figure 3C). Thus, the negative effects of PTN on cell migration require interaction with PTPRZ1.
PTN/PTPRZ1 signalling alters the phosphorylation state of key regulators of cell migration
PTPRZ1 deletion prevents the DR during liver injury
To determine the relative significance of PTPRZ1 in controlling cell migration in intact liver tissue, we subjected PTPRZ1-KO mice and age/gender-matched controls to BDL. PTPRZ1 deletion severely attenuated the DR and liver injury post-BDL, significantly reducing portal tract expansion, bile infarcts, accumulation of Krt19(+) and Sox9(+) RDC, periportal fibrosis and hepatic hydroxyproline content (see figure 5 and online supplementary figures S2 and S3). Moreover, loss of PTPRZ1 blocked induction of mRNAs encoding several Hedgehog-regulated genes (eg, Gli1, Gli2, Ptc; see online supplementary figure S5), as well as accumulation of αSMA transcripts and αSMA(+) cells after BDL (see online supplementary figure S6A). Similarly, in primary HSC from PTPRZ1-KO mice, culture-related induction of αSMA gene expression was also significantly reduced compared to HSC from controls (see online supplementary figure S6B). Finally, incubation with PTN had no effect on αSMA expression in HSC-derived MF from PTPRZ1-KO mice, but reduced αSMA expression by more than 80% in HSC-derived MF from WT controls (see online supplementary figure S6B). The aggregate data demonstrate that PTPRZ1 activity promotes a mesenchymal/migratory phenotype in liver cells expressing that receptor, and that these effects are blocked by PTN-PTPRZ1 interaction.

Protein tyrosine phosphatase receptor zeta-1 (PTPRZ1) deficiency inhibits the ductular reaction during cholestatic injury. (A) Representative sections of H&E stained livers (magnification, ×10) from wild type (WT) or PTPRZ1-deficient mice after sham-operation or bile duct ligation (BDL) are shown. Portal tract diameter was evaluated by morphometry with results expressed as mean±SEM relative to WT sham-operated mice. (B) Representative immunohistochemistry (magnification, ×20), corresponding quantification of immunostained images and qRT-PCR analysis of Krt19 mRNA in whole liver RNA is shown in panels to the right. Results are expressed relative to sham-operated WT mice. (C) Representative Sirius red-stained sections in WT or PTPRZ1-deficient mice after sham-operation or BDL (magnification, ×20) with associated morphometric analysis and quantification of hepatic hydroxyproline content to assess fibrosis. Results are displayed as mean±SEM relative to sham-operated WT mice. *p<0.05 **p<0.01.
Since macrophages are known to regulate the fate of RDC involved in the DR,37 we assessed whether deleting PTN or PTPRZ1 influenced macrophage accumulation and/or polarisation. Hepatic macrophage accumulation and polarisation were similar in PTN-KO mice and WT mice before and after BDL. However, both parameters were significantly inhibited in BDL PTPRZ1-KO mice (see online supplementary figures S7–S9). The proliferative activity of RDC and hepatocytes were also uniquely suppressed in PTPRZ1-KO mice post-BDL (see online supplementary figure S10). The aggregate data, therefore, reveal previously unsuspected roles for PTPRZ1 in regulating various aspects of the DR. The findings also support the concept that PTPRZ1 activity is controlled by multiple factors in injured livers because simply deleting PTN was not sufficient to disrupt all of PTPRZ1's actions in BDL mice.
Human livers with ductal plate abnormalities accumulate PTPRZ1(+) ductular cells
Our findings in mice demonstrate that PTN-PTPRZ1 interactions regulate the retention/migration of cells in hepatic progenitor niches during cholestatic liver injury. To determine if similar mechanisms might operate in humans, we screened three representative healthy human livers, two livers with hepatic ductal plate abnormalities and livers from six other patients with adult-onset chronic cholestatic liver disease for evidence of PTN-PTPRZ1 axis activity. Although PTN-producing cells are not reliably localised by immunostaining because PTN is a secreted heparin-binding protein, our studies of mouse livers (figures 4 and 5) indicated that immunohistochemistry would be useful for localising PTPRZ1-expressing cells in human livers. As in healthy adult mice (figure 4B), in healthy adult humans, PTPRZ1(+) liver cells were rarely detected (figure 6A, H). As in murine livers with the DR (figure 4B), in a human liver with adult polycystic disease (figure 6B), a human liver with von Meyenburg complexes (figure 6C–E), livers from two patients with primary biliary cirrhosis (figure 6F, H) and four patients with primary sclerosing cholangitis (figure 6G, H), atypical ductular structures strongly expressed PTPRZ1. For example, numbers of PTPRZ1-positive cells in primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC) livers were increased more than 10-fold relative to healthy livers (figure 6H). Moreover, as in mice, PTPRZ1-expressing cells in humans included immature liver epithelial cells and HSC-derived MF because PTPRZ1 colocalised with Krt7 (figure 6D), a marker of immature biliary epithelia, and desmin, a marker of HSC (figure 6E). PTPRZ1-positive ductular structures were also prominent in mdr2−/− mice, a murine model of primary sclerosing cholangitis that also accumulates atypical ductular structures (see online supplementary figure S11). The aggregate mouse and human data support the concept that PTN-PTPRZ1 interactions regulate the DR.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Protein tyrosine phosphatase receptor zeta-1 (PTPRZ1)(+) cells accumulate during the ductular reaction in humans. (A) PTPRZ1 is not detectable in normal adult human liver (NHL). Patients with ductal plate abnormalities, for example, adult polycystic liver disease (B) and von Meyenburg complexes (C), demonstrated a marked increase in PTPRZ1 expression, particularly in ductular cells. (D, E) PTPRZ1 (brown) localises in (D) Krt7(+) ductular cells (green) and (E) Desmin(+) stromal cells (green) in representative liver sections from patients with von Meyenburg complexes (magnification ×10; inset ×100). PTPRZ1-positive ductular cells were also evident in liver sections from representative patients with primary biliary cirrhosis (F; magnification ×100), primary sclerosing cholangitis (G; magnification ×100) with ductular reactions. Arrows indicate PTPRZ1 positive cells in atypical ductular structures. (H) Quantification of PTPRZ1-positive ductular cells in liver sections from PBC patients (n=2) and PSC patients (n=4) relative to NHL (n=3 individuals).
Discussion
Our findings revealed that PTN-PTPRZ1 signalling controls the retention of cells that normally reside in stem/progenitor niches of adult livers. Specifically, we showed LSEC, HSC, and RDC produce PTN ligand and express its receptor, PTPRZ1. Additionally, we demonstrated that HSC and RDC modulate their expression of the ligand and receptor during liver injury. By comparing responses to liver injury and studying cultured cells from mice deficient in either PTN or PTPRZ1, we also characterised how ligand-receptor interaction changes the phenotypes of niche cells: PTN signalling via PTPRZ1 inhibits cell migration and acquisition of other mesenchymal cell characteristics, while PTPRZ1 alone promotes a migratory/mesenchymal phenotype when unopposed by PTN. The aggregate findings, therefore, indicate that localised increases in PTN retain PTPRZ1-expressing cells within liver stem/progenitor niches. Conversely, PTPRZ1(+) liver cells migrate out of their niches when the niche microenvironment becomes relatively depleted of PTN. In addition to identifying a regulatory mechanism for the DR to liver injury, this discovery reveals a previously unsuspected similarity between liver and haematopoietic stem/progenitor cells, as Himburg et al11 demonstrated that PTN production by blood vessel-associated cells retained PTPRZ1(+), multipotent haematopoietic stem cells within the bone marrow stem cell niche.
A diverse array of signalling programmes have been shown to regulate cell migration, but how these pathways are integrated to achieve tissue repair is poorly understood. Integrins, for example, are transmembrane receptors that mediate cell–cell and cell–matrix interactions.38 Ductular cells and MF in injured livers express certain integrins (eg, αvβ6 or αvβ1, respectively) that regulate biliary fibrosis, consistent with an important role for these receptors in modulating the DR.39 ,40 Paracrine signalling involving the Hedgehog pathway,28 ,41 as well as vascular endothelial growth factor (VEGF) and one of its receptors, VEGFR2, have also been shown to control the DR.42 The present study demonstrates previously unsuspected roles for PTN and PTPRZ1 in DR regulation. Further research is needed to clarify if and how all of these systems interact to orchestrate liver repair. The present work provides novel evidence for interactions between the Hedgehog and PTN-PTPRZ1 pathways. Other cross talk is conceivable based on emerging evidence in the field of cancer cell biology. For example, the PTN/PTPRZ1 pathway was discovered to regulate human cancer cell migration via phosphorylation-dependent changes in αvβ3 activity.43 ,44 Importantly, we identified several known regulators of cell migration as potential mediators of PTN-PTPRZ1 signalling. PTN-PTPRZ1 interaction inhibits constitutive tyrosine phosphatase activity of PTPRZ1, resulting in tyrosine phosphorylation of target substrates,10 ,23 ,45 that in turn activate downstream nodal effectors, including PI3-kinase,9 ,20 Akt,8 ,21 ERK1/2,21 and PKCα/β.22 We demonstrated that treating PTPRZ1-positive ductular progenitor cells with PTN caused time-dependent accumulation of many tyrosine-phosphorylated proteins, including talin, tensin, paxillin, afadin and supervillin. These latter proteins control cell migration, and our results demonstrate that PTN-PTPRZ1 interactions directly regulate the migratory activity of cultured HSC and liver progenitor cell (LPC).
In non-liver cells, talin, paxillin, and tensin control stability of focal adhesions,46 ,47 and supervillin regulates podosome turnover and function, endosomal recycling of integrins, and ERK signalling.32 ,48 Therefore, migration of PTPRZ1-expressing liver cells is likely to be impacted by PTN-dependent phosphorylation of these proteins. Evidence that PTN-PTPRZ1 signalling modulates phosphorylation of afadin in liver cells is also relevant because afadin is a key component of adherens junctions49 ,50 and phosphorylation results in afadin translocation from adherens junctions into the nucleus.51 This translocation step is regulated by PI3 K/Akt, the latter being activated by PTN/PTPRZ1 signalling.20 Evidence that deleting either PTN or PTPRZ1 dramatically altered the extent of neoductule formation (ie, the DR) in livers of adult mice with extrahepatic biliary obstruction supports the concept that PTPRZ1-dependent phosphorylation of afadin may also influence adherens junction stability. This could impact the construction/deconstruction of liver epithelia because adherens junctions normally exist between adjacent ductular cells,52 and connect HSC to each other.53 ,54
Maintenance of intercellular connections that tether neighbouring epithelial cells is a pre-requisite for epithelial integrity. Disassembly of epithelial cell-cell contacts is one of the earliest events in the EMT, and reassembly of these connections is the end point of mesenchymal-to-epithelial transition (MET).55 Hence, the aggregate data in our study favour the possibility that subpopulations of cells in liver stem/progenitor niches undergo EMT/MET during adult liver repair and suggest that PTN-PTPRZ1 signalling helps to control that process. Although it has been debated whether EMT/MET occurs during regeneration of injured adult livers,56 ,57 it is generally accepted that this process is necessary for organogenesis during development.55 Indeed, proper morphogenesis of the intrahepatic biliary tree requires perivascular stromal cell-derived signals that instruct neighbouring liver progenitors to undergo EMT/MET, thereby forming the tubular structures of the primitive ductal plate.2 We showed that PTPRZ1 is strongly expressed by immature ductular cells lining dysmorphic ducts in von Meyenburg complexes and in adult polycystic liver disease. Both of those conditions are believed to reflect ductal plate defects.58 Given that RDC involved in the adult DR that accompanies primary biliary cirrhosis and primary sclerosing cholangitis also express PTPRZ1, and PTPRZ1-initiated signals modify proteins that regulate cellular adherence and migration, formation of neoductules during the adult DR may recapitulate some of the processes involved during development of the intrahepatic biliary tree, including EMT/MET.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
GAM and AT contributed equally.
Correction notice This article has been corrected since it published online first. The OA licence has now been added.
Contributors GAM designed and performed experiments, analysed data, and wrote the manuscript. AT designed and performed experiments, analysed data and wrote the manuscript. MS-S, MVM, designed and performed experiments. SSC designed and performed experiments and wrote the manuscript. LK, ES, JWT, and MM-S designed and performed experiments. HAH, CAM, CDG, KSG, RTP, and JPC analysed data. AMD designed experiments, supervised research, analysed data and wrote the manuscript.
Funding This work was supported in part by grants R37 AA010154 and R01 DK077794 (AMD) and Howard Hughes Medical Institute Medical Research Fellowship (AT).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.