Article Text

Abstract
Objective Using whole genome sequencing, we identified gene amplification of solute carrier family 12 member 5 (SLC12A5) located at 20q13.12 in colorectal cancer (CRC). We analysed its amplification, overexpression, biological effects and prognostic significance in CRC.
Design SLC12A5 amplification status was evaluated by fluorescence in situ hybridisation (FISH). The effects of SLC12A5 re-expression or knockdown were determined in proliferation, apoptosis, invasion and metastasis assays. SLC12A5 target genes and related pathways were identified by reporter activity and cDNA microarray analyses. Clinical impact of SLC12A5 overexpression was assessed in 195 patients with CRC.
Results Amplification of SLC12A5 was verified in 78 out of 191 (40.8%) patients with primary CRC by FISH, which was positively correlated with its protein overexpression (p<0.001). Biofunctional investigation of SLC12A5 revealed that SLC12A5 significantly increased cell proliferation, G1-S cell cycle transition, invasion/migration abilities, but suppressed apoptosis in vitro and promoted xenograft tumour growth as well as lung metastasis in vivo. The antiapoptosis effect by SLC12A5 was mediated through inhibiting apoptosis-inducing factor and endonuclease G-dependent apoptotic signalling pathway; and the pro-metastasis role was by regulating key elements of the matrix architecture, including matrix metallopeptidase and fibronectin. After a median follow-up of 50.16 months, multivariate analysis revealed that patients with SLC12A5 protein overexpression had a significant decrease in overall survival. Kaplan–Meier survival curves showed that SLC12A5 overexpression was significantly associated with shortened survival in patients with CRC.
Conclusions SLC12A5 plays a pivotal oncogenic role in colorectal carcinogenesis; its overexpression is an independent prognostic factor of patients with CRC.
- COLORECTAL CANCER
- GENE EXPRESSION
- MOLECULAR ONCOLOGY
- TUMOUR MARKERS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
SLC12A5, a gene located at 20q13.12, belongs to the SLC12 gene family and encodes the protein of solute carrier family 12, member 5.
Gene amplification is one of the most common mechanisms which contributes to overexpression of genes favouring tumour development.
What are the new findings?
SLC12A5 amplification is identified for the first time in colorectal cancer (CRC) by whole genome sequencing. We have further verified SLC12A5 amplification by fluorescence in situ hybridisation in 78 out of 191 (40.8%) patients with primary CRC.
SLC12A5 plays strong tumourigenic role through inhibiting apoptosis and promoting metastasis evidenced by in vitro and in vivo tumorigenicity experiments.
SLC12A5 inhibits apoptosis through suppressing apoptosis-inducing factor/endonuclease G-dependent apoptotic signalling and promotes metastasis through regulating key elements of the matrix architecture.
SLC12A5 overexpression mediated by amplification is associated with poor prognosis of patients with CRC.
How might it impact on clinical practice in the foreseeable future?
Detection of SLC12A5 overexpression may serve as a new biomarker for the prognosis of patients with CRC.
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in men and the second in women worldwide.1 In Asian countries, the incidence of CRC has been rising rapidly.2 Approximately 50%–60% of patients diagnosed with CRC develop metastases, which is the main cause of mortality in patients with CRC.3 ,4 The pathogenic mechanisms underlying CRC development appear to be complex and heterogeneous. Emerging evidences indicate that CRC development is a multistep process with the accumulation of genetic and epigenetic alterations.5 Copy number aberrations (CNAs), including chromosome gains and losses, or localised amplifications and deletions, are frequently found in human CRC and are major causes of aberrant activation of oncogenes and inactivation of tumour suppressor genes.6–10 Some CNAs, such as copy number loss of chromosome 18q12.2, copy number gain at the chromosomal region between 11q13.3 and 11q22.3, gene amplification of MPL1/PZR, are closely related to clinical outcome or metastatic progression.11–16 It is of great importance to identify and functionally characterise novel genes with CNAs that are associated with CRC.
It is well documented that CNAs on chromosome 20 are involved in human cancers. 20q13.12 is one of the most frequently amplified regions in CRC, as well as in other human cancers, which contains several putative oncogenes.17–21 Our recent study using single-cell sequencing demonstrated an activating point mutation of solute carrier family 12 member 5 (SLC12A5), a gene located at 20q13.12, in CRC.22 Targeted capture sequencing in a larger cohort of patients with CRC revealed that the mutation prevalence of SLC12A5 is only 3.8% (7 out of 182 cases).23 We found that SLC12A5 was widely expressed in CRC tissues, while silenced or downregulated in their adjacent mucosal tissues. We, therefore, hypothesised that amplification of SLC12A5 could be an important mechanism for its activation. Indeed, using whole-genome sequencing, we identified the frequent amplification of SLC12A5 in human CRC. This was confirmed by an analysis of CRC genomic data of The Cancer Genome Atlas (TCGA), showing the frequent amplification of SLC12A5 in CRC cases (https://www.synapse.org/#!Synapse:syn300013).
SLC12A5, also known as potassium chloride cotransporter 2, belongs to the SLC12 gene family and encodes the protein of solute carrier family 12, member 5.24 SLC12A5 protein is an integral membrane KCl cotransporter that maintains chloride homeostasis in neurons.25 ,26 However, the role of SLC12A5 amplification in CRC has not yet been explored. In this study, we evaluated the amplification status of SLC12A5, characterised the functional mechanisms and the clinical implication of SLC12A5 in colorectal carcinogenesis.
Materials and methods
Tissue samples
CRC tissue microarrays (TMAs) were constructed according to a method described previously.27 The first set of TMA was generated from formalin-fixed, paraffin-embedded archive tissues of 191 patients with CRC, which were collected at The Prince of Wales Hospital, Hong Kong, from 2002 to 2010. The second set of TMAs was prepared from 195 CRC tissues, which were obtained from the Sun Yat-Sen University and Guangdong Provincial People's Hospital, Guangzhou, China, between January 2000 and November 2006. The patient's demographic and clinicopathological features were shown in table 1 and online supplementary table S1. The TNM status of CRCs was assessed according to the criteria of the sixth edition of the TNM classification of the International Union Against Cancer (2002).28 Patients were being regularly followed up and the median follow-up duration since the time of diagnosis was 50.16 months (range 7–60 months). In addition, 15 paired primary tumour and adjacent non-tumour tissues from patients with CRC were obtained during operation prior to any therapeutic intervention at the Prince of Wales Hospital in Hong Kong. All subjects provided informed consent for obtaining the study specimens.
Relationship between SLC12A5 expression and clinicopathological features in 195 colorectal cancer cases
Fluorescence in situ hybridisation
The bacterial artificial chromosome clone RP11-465L10 at 20q13.12 containing SLC12A5 gene was labelled with Sperctrum-Red (Vysis, Downers Grove, Illinois, USA). Chromosome 20 centromere probe labelled with Spectrum-Green (Vysis) was used as control. For cell line fluorescence in situ hybridisation (FISH), metaphase cells were collected after colcemid treatment, followed by hypotonic treatment and fixation, then were spread on slides. For tissue FISH, paraffin-embedding TMA sections were pretreated by de-waxing, gradient hydration and Proteinase K digestion. The Probes hybridisation procedure was performed according to the previous method.29
Statistical analysis
The results were expressed as mean±SD. Statistical analysis was performed using the SPSS statistical software package (standard V.16.0). The Pearson correlation coefficient was used to evaluate the correlation between SLC12A5 gene amplification and expression in the clinical samples. The χ2 test was used for comparison of patient characteristics and distributions of expression and covariates by vital status. Crude relative risks (RRs) of death associated with SLC12A5 expression and other predictor variables were estimated by univariate Cox proportional hazards regression model first. Multivariate Cox model was constructed to estimate the adjusted RR for SLC12A5 expression. Overall survival in relation to expression was evaluated by the Kaplan–Meier survival curve and the log-rank test. Mann–Whitney U test or Student's t test was performed to compare the variables of two groups. The difference in cell viability and tumour growth rate between the two groups of nude mice was determined by repeated-measures analysis of variance. p Values <0.05 were taken as statistical significance. Additional detailed description of the materials and methods can be found in the online supplementary materials and methods section.
Results
Amplification of SLC12A5 is identified in CRCs
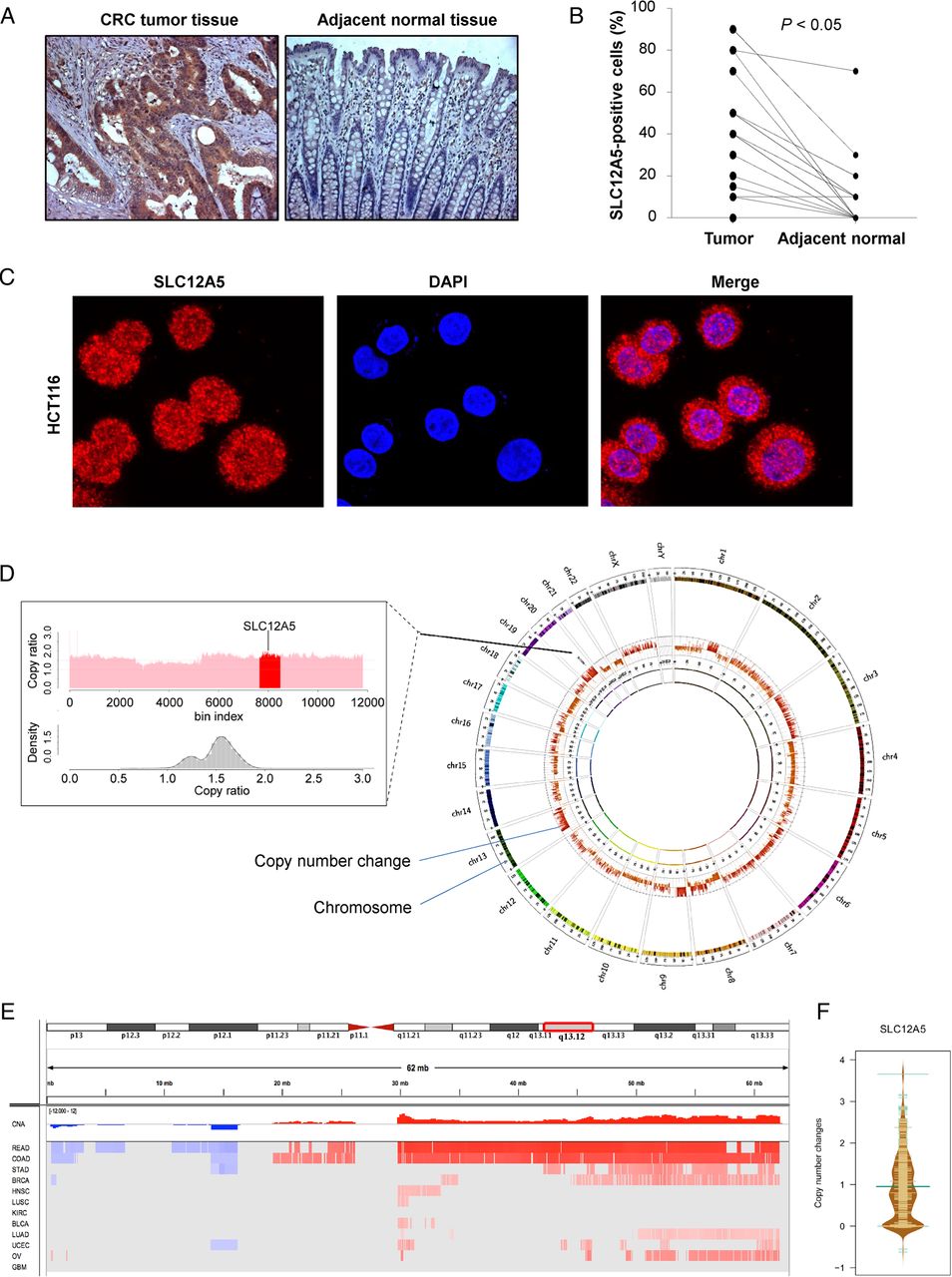
We evaluated SLC12A5 protein expression in 15 pairs of colorectal tumours and adjacent normal tissues by immunohistochemistry. SLC12A5 protein expression was distributed in both nuclear and cytoplasm of CRC tumour cells in 93% of CRCs (14/15), but not in normal colon tissue cells (figure 1A). The protein expression level of SLC12A5 was significantly higher in primary CRCs as compared with their adjacent normal tissues (p<0.05) (figure 1B). We performed immunofluorescence and confocal microscopy of colon cancer cell line HCT116 and confirmed that the SLC12A5 protein expression was distributed in both nuclear and cytoplasm (figure 1C). As activating mutation of SLC12A5 was only detected in 3.8% (7 out of 182 cases) CRCs by targeted capture sequencing,23 other mechanisms such as gene amplification may contribute to the enhanced expression of SLC12A5.

SLC12A5 is amplified in colorectal cancer (CRC). (A) Representative images of SLC12A5 protein expression in CRC tumour tissues and their adjacent tissues by immunohistochemistry. (B) The protein expression level of SLC12A5 was significantly higher in primary CRC tumour as compared with their adjacent tissue (p<0.05). (C) SLC12A5 was expressed in both nuclear and cytoplasm in colon cancer cell line HCT116 by immunofluorescence and confocal microscopy. (D) The right panel shows the genomic profiles of CRC by whole genome sequencing, where the copy number alterations were indicated with red bars. The left panel shows the visualisation of copy ratio particularly for the whole chromosome 20. 20q13.12 is highlighted with red colour, which harbours the amplified gene SLC12A5. (E) Significant genomic alterations of 20q13.12 across 12 human cancer types in The Cancer Genome Atlas (TCGA) cohort. Red colour indicates copy number gain, while blue colour refers to copy number loss. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; KIRC, kidney renal clear cell carcinoma; COAD, colon adenocarcinoma; DAPI, 4’,6-diamidino-2-phenylindole; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; LAML, acute myeloid leukaemia; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; READ, rectum adenocarcinoma; OV, ovarian serous cystadenocarcinoma; UCEC, uterine corpus endometrioid carcinoma. (F) Copy number changes of SLC12A5 in 575 colorectal cancers from TCGA.
By whole-genome sequencing analyses, we identified SCL12A5 amplification in CRC (figure 1D), which was confirmed by array comparative genomic hybridisation showing copy number gain of SLC12A5 (see online supplementary table S2). To further confirm our identification, we evaluated the copy number variation from TCGA data (https://www.synapse.org/#!Synapse:syn300013). In TCGA cohort of 575 CRCs, SLC12A5 was amplified in both colon cancer (q value=8.99E-07, frequency=72.6%) and rectal cancer (q value=0.003, frequency=88.9%), signifying its common occurrence in CRC (figure 1E, F).
Amplification of SLC12A5 contributes to its overexpression in CRC tumour tissues
To examine the relationship between DNA amplification of SLC12A5 and its overexpression, the DNA copy number change of SLC12A5 was investigated by FISH in a TMA containing 191 CRC cases. Amplification of SLC12A5 was not observed in normal metaphase lymphocytes, but detected in colon cancer cell line Caco-2 (figure 2A). Importantly, 40.8% (78/191) of primary CRCs exhibited amplification of SLC12A5, while there was no obvious copy number gain in adjacent normal tissues (figure 2B). The protein expression of SLC12A5 was investigated in the same TMA by immunohistochemistry (figure 2C). Pearson correlation analysis indicated that amplification of SLC12A5 in CRC tissues was positively correlated with its protein overexpression (p<0.001) (figure 2D). These data suggest that upregulation of SLC12A5 protein in CRC is at least in part contributed by DNA amplification.
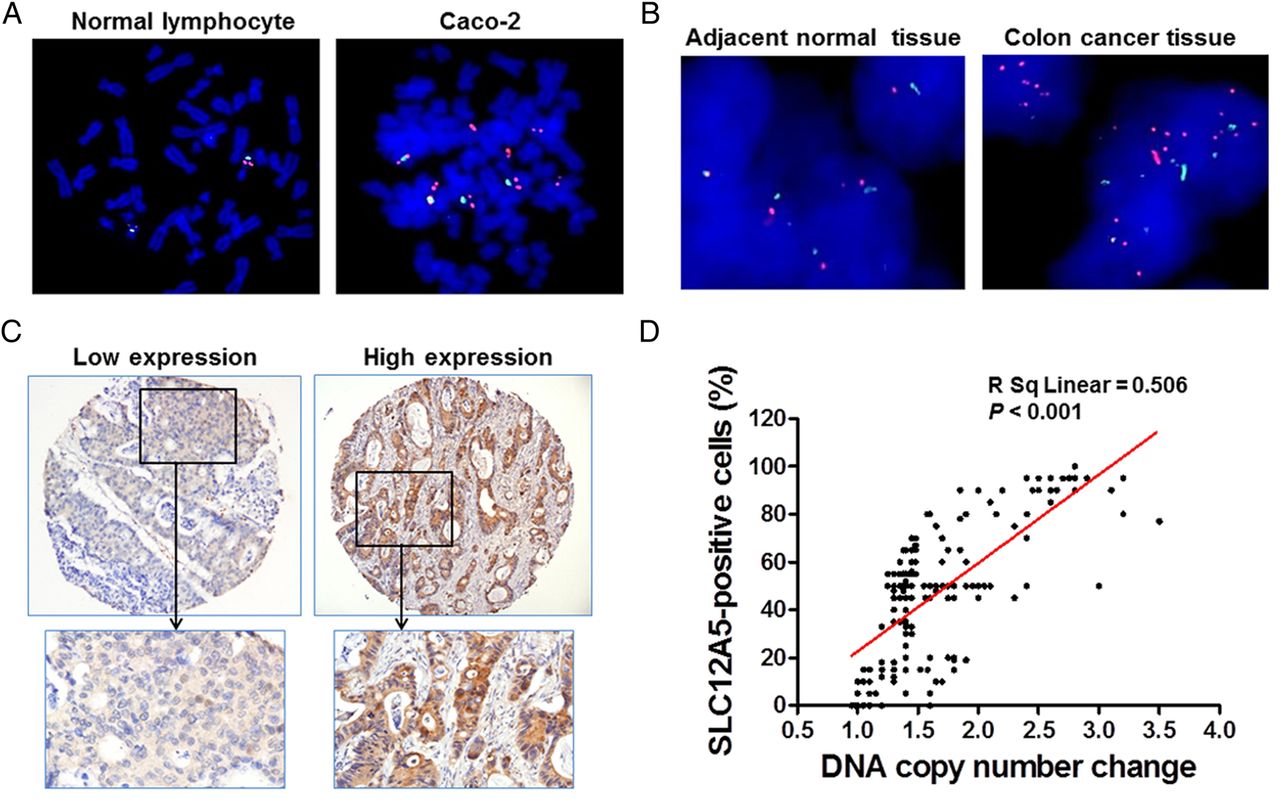
Amplification and overexpression of SLC12A5 in colorectal cancers (CRCs). (A) Fluorescence in situ hybridisation (FISH) was performed in normal metaphase lymphocytes and colon cancer cell line Caco-2. The red signal indicates the location of SLC12A5 and the green signal refers to centromere of chromosome 20. This result implied that the probe of SLC12A5 was correctly located on chromosome 20. Amplification of SLC12A5 was observed in Caco-2. (B) Representative image of SLC12A5 gene amplification detected by FISH in primary CRC tissues, but not detected in their adjacent tissues. (C) Representative staining of high and low expression levels of SLC12A5 in CRC tissue microarray slides by immunohistochemistry. (D) The correlation between copy number changes of SLC12A5 and protein overexpression (R2=0.506, p<0.001).
SLC12A5 promotes CRC cell growth in vitro and in vivo
To assess the potential function of SLC12A5, a series of in vitro biological experiments were performed with gain-of-function or loss-of-function of SLC12A5. SW480 and SW116 cells were stably transfected with different doses of SLC12A5 plasmids. Ectopic expression of SLC12A5 in these cells (figure 3A) caused a significant increase in the number of viable cells in a dose-dependent fashion in SW480 and SW1116 (p<0.01) compared with empty vector transfected cells, respectively (figure 3B). In keeping with this, the number of colonies increased significantly in a dose-dependent manner when transfected with SLC12A5 plasmids compared with empty vector-transfected SW480 and SW1116, respectively (figure 3C, D). On the other hand, we examined whether SLC12A5 is required for the tumourigenic phenotypes of CRC cells by silencing SLC12A5 expression with siRNA. The introduction of SLC12A5-specific siRNA (siSLC12A5) into HCT116 dramatically decreased the expression of SLC12A5 relative to control cells transfected with scrambled siRNA (figure 3E). SLC12A5 depletion by siRNA reversed the tumourigenic phenotype by inhibiting the cell growth rate compared with control cells (p<0.001) (figure 3F).

SLC12A5 promotes colorectal cancer cell growth. (A) Expression levels of SLC12A5 were increased after transfection with different doses of SLC12A5 plasmids (0.8 and 1.6 µg) in colon cancer cell lines SW480 and SW1116. (B) Ectopic expression of SLC12A5 significantly enhanced cell viability in a dose-dependent manner in both cell lines. (**p<0.01, ***p<0.001) (C) Empty vector and different doses of SLC12A5 plasmids (0.8 and 1.6 µg) were used to transfect cells in 24-well plates. Compared with empty vector-transfected cells, colony numbers significantly increased with transfection of 0.8 and 1.6 µg SLC12A5 plasmids in SW480 in a dose-dependent manner. (D) The number of colonies increased in a dose-dependent manner when transfected with SLC12A5 plasmids in SW1116. (E) Knockdown efficiency of SLC12A5 in HCT116 cells by transient transfection of siSLC12A5 was examined by real-time PCR (left panel) and western blot (right panel), respectively. (F) Knockdown of SLC12A5 significantly inhibited cell viability in HCT116. (G) Knockdown efficiency of SLC12A5 in HCT116 cells by stable transfection of shSLC12A5 was demonstrated by real-time PCR (left panel) and western blot (right panel). (H) The tumour growth curve of HCT116 stably transduced with shSLC12A5 in nude mice was significantly dampened compared with HCT116 transduced with Control. (I) A representative picture of tumour formation in nude mice subcutaneously inoculated with shSLC12A5- or Control-HCT116 (left panel). Histogram represents mean of the tumour weight from the shSLC12A5 and Control groups (right panel).
To further investigate the in vivo tumourigenic ability of SLC12A5, we constructed a lentiviral vector carrying shSLC12A5 to stably knock down the expression of SLC12A5 in HCT116 (figure 3G). The Control-HCT116 and shSLC12A5-HCT116 cells were injected subcutaneously into the dorsal flank of nude mice. Tumours induced by shSLC12A5-HCT116 cells showed significantly longer latency and smaller mean tumour volume than tumours induced by control cells (p<0.001) (figure 3H). At the end of experiments, xenograft tumours were isolated and the mean tumour weight of the shSLC12A5-HCT116 group was reduced by more than 70% as compared with controls (p<0.05) (figure 3I). Collectively, these data indicate that SLC12A5 possesses strong tumourigenicity in CRC both in vitro and in vivo.
SLC12A5 inhibits apoptosis in colon cancer cells
To explore the molecular basis involved in SLC12A5-enhanced tumour growth, the effect of SLC12A5 on apoptosis was assessed quantitatively by flow cytometry after staining with Annexin V and 7-amino-actinomycin. We found that overexpression of SLC12A5 significantly inhibited early apoptosis in SW480 and SW1116 (p<0.001) (figure 4A), as compared with control vector-transfected cells. To further clarify whether knockdown of SLC12A5 could reverse this phenotype, siRNAs against SLC12A5 were transfected into HCT116 cells. The percentage of early apoptotic cells in siSLC12A5 HCT116 cells was significantly increased, as compared with the control cells (p<0.01) (figure 4B).

SLC12A5 reduces apoptosis in colorectal cancer cell lines. (A) Overexpression of SLC12A5 significantly inhibited early apoptosis in SW480 and SW1116 (B) Knockdown of SLC12A5 significantly increased the proportion of apoptotic cells in HCT116 cells by flow cytometry analyses following Annexin V and 7-amino-actinomycin (7-AAD) staining (p<0.01). (C) Protein expression of key apoptosis-related genes was evaluated by western blot in HCT116 cells transfected with siSLC12A5 or Control. After silencing SLC12A5 expression, no changes of cleaved Caspase 3, 7 nor cleaved poly(ADP-ribose) polymerase (PARP) were observed. SLC12A5 silencing increased the protein expression of p53, p53 upregulated modulator of apoptosis (PUMA), Bax and Bak and downregulated the inhibitor of apoptosis protein Survivin. Knockdown of SLC12A5 enhanced the nuclear protein levels of apoptosis-inducing factor (AIF) and endonuclease G (EndoG). (D) TUNEL-stained sections of mouse xenograft tumours displayed apoptotic cells; representative images of AIF and EndoG immunohistochemistry-stained sections of mouse xenograft tumours. (E) Schematic diagram for the proposed mechanisms of SLC12A5 exerting antiapoptotic function via caspase-independent signalling pathway. (F) The number of cell distribution was determined by flow cytometry. Values are mean±SD. (G) Protein expression of cyclin D1 and cyclin-dependent kinase (CDK4) was determined by western blot.
SLC12A5 exerts an antiapoptotic function through the caspase-independent pathway
To define the molecular basis of cell death after SLC12A5 depletion, we assessed the key apoptosis regulators. Activation of caspases and the subsequent cleavage of poly (ADP-ribose) polymerase (PARP) are classic molecular hallmarks of apoptosis.30 However, no changes of cleaved Caspase 3, 7 or cleaved PARP were observed after silencing SLC12A5 expression (figure 4C), indicating that SLC12A5 may inhibit apoptosis via a caspase-independent pathway. Apoptosis-inducing factor (AIF) and endonuclease G (EndoG) are known for mediating cell death independent of caspases by translocating from mitochondria into the nucleus where they induce DNA cleavage.30 We, therefore, examined the nuclear protein expression of AIF and EndoG. We found that knockdown of SLC12A5 enhanced the nuclear protein levels of AIF and EndoG (figure 4C). The mitochondrial release of AIF and EndoG is mediated by pro-apoptotic Bax and Bak and their upstream regulators, p53 and p53 upregulated modulator of apoptosis (PUMA), which can influence the mitochondrial membrane permeabilisation.31 Concordantly, SLC12A5 silencing increased the protein expression of pro-apoptotic factors p53, PUMA, Bax and Bak (figure 4C). Emerging evidence supported the fact that the inhibitor of apoptosis protein Survivin decreases the amount of AIF in the nucleus after apoptosis induction, thus protecting cells from death.32 ,33As shown in figure 4C, Survivin was significantly downregulated after knockdown of SLC12A5. To further confirm the antiapoptotic effect of SL12A5 in vivo, we performed TUNEL staining on the paraffin slides of mouse xenograft tumours. We found that TUNEL-positive apoptosis cells were significantly more in the shSLC12A5-HCT116 tumours compared with the control-HCT116 tumours (p<0.01) (figure 4D). Consistently, the nuclear protein expression of AIF and EndoG was significantly increased in the shSLC12A5-HCT116 tumours compared with the control-HCT116 tumours (figure 4D), which is consistent with the in vitro study by western blot (figure 4C). These results collectively suggest that SLC12A5 inhibits apoptosis by suppressing the activation of AIF–EndoG-dependent apoptotic pathway (figure 4E).
SLC12A5 promotes G1 to S phase transition in colon cancer cells
To characterise the oncogenic mechanism of SLC12A5 in colon cancer cell growth, we further investigated the role of SLC12A5 in cell cycle progression. We found that ectopic expression of SLC12A5 in SW480 (SLC12A5-SW480) significantly decreased the number of cells in G1 phase (p<0.01), but increased the number of cells in S phase (p<0.01) compared with control vector-transfected cells 12 h after serum stimulation (figure 4F). Conversely, knockdown SLC12A5 in HCT116 cells by siSLC12A5 arrested the cell cycle at the G1–S transition (p<0.01) (figure 4F). Western blot analysis showed that the protein expression of two master G1–S checkpoint regulators cyclin D1 and cyclin-dependent kinase (CDK4) was increased in SLC12A5-SW480 compared with the vector-transfected cells, while knockdown SLC12A5 in HCT116 had the opposite effect (figure 4G), confirming the role of SLC12A5 on promoting cell growth by regulating cell cycle progression in colon cancer cells.
SLC12A5 inhibits p53 and p21 reporter activity
To gain insights into the downstream signalling pathways modulated by SLC12A5 in CRC tumourigenesis, we examined the functional effect of SLC12A5 in several important cancer pathways, including p53, p21, activator protein-1 (AP-1), nuclear factor (NF)-κB and Wnt/β-catenin (TOPFlash), by luciferase reporter activity assay. Ectopic expression of SLC12A5 significantly suppressed p53 and p21 luciferase reporter activities in HCT116 (figure 5A), whereas no significant activity changes were observed in AP-1, NFκB and Wnt/β-catenin pathway reporters (figure 5A). To further confirm the importance of the p53 and p21 pathways mediated by SLC12A5 in colon cancer cells, we assessed the ability of abolishing SLC12A5 in the reporter activities of p53 and p21. Knockdown SLC12A5 significantly enhanced p53 and p21 reporter activities (figure 5B). Western blot results further validated that ectopic expression of SLC12A5 reduced the protein level of p53 and p21, while knockdown of SLC12A5 upregulated p53 and p21 in HCT116 cells (figure 5C).

SLC12A5 suppressed p53 and p21 luciferase reporter activity. (A) To screen for SLC12A5 target signalling pathways, a serial of promoter-luciferase assays (p53-luc, p21-luc, activator protein 1 (AP1)-luc, nuclear factor κB (NF κB)-luc, and Wnt/β-catenin) were performed in SLC12A5 stably transfected p53 wildtype (WT) HCT116 cells compared with vector control cells. Luciferase activities were determined by dual luciferase assay system at 48 h post-transfection. Ectopic expression of SLC12A5 suppressed p53 and p21 luciferase reporter activity in p53 WT HCT116. (B) Knockdown of SLC12A5 increased p53 and p21 luciferase reporter activity in p53 WT HCT116. (C) The western blot results confirmed the findings of luciferase reporter activity assays in p53 WT HCT116. (D) Ectopic expression of SLC12A5 was determined in p53 knockout (KO) HCT116 cells by real-time PCR. (E) Ectopic expression of SLC12A5 significantly promoted cell growth in p53 KO HCT116 cells. (F) SLC12A5 in HCT116 cells suppressed p21-luciferase reporter activity in both p53 KO HCT116 and SW116 cells.
As SLC12A5 promoted cell proliferation and inhibited cell apoptosis in p53-mutated cells SW480 and SW1116, which implies the tumour-promoting function of SLC12A5 could be independent of p53. To confirm this hypothesis, we transfected SLC12A5 in p53 knockout (KO) HCT116 cells (figure 5D). Ectopic expression of SLC12A5 significantly enhanced cell growth in p53 KO HCT116 cells (figure 5E). We then determined the above five luciferase reporters in p53 KO HCT116 and SW1116. As shown in figure 5F, only p21 reporter activity was significantly downregulated. These data indicated that SLC12A5 suppressed p21 activity irrespective of p53 status.
SLC12A5 promotes tumour invasion and metastasis in vitro and in vivo
We also investigated the effects of SLC12A5 on metastasis using in vitro migration and Matrigel invasion assays and in vivo metastasis assay. The migration and Matrigel invasion assays showed that both the migration and the invasive capability of SLC12A5-SW1116 cells were greater than those of Vector-SW116 cells (figure 6A). By contrast, knockdown SLC12A5 expression by siRNA in HCT116 cells eliminated the migratory and invasive ability of the HCT116 cells (figure 6B). These results indicate that SLC12A5 increases cell invasion and metastasis, which we further validated in vivo.

SLC12A5 promotes tumour metastasis in vitro and in vivo. (A) Ectopic expression of SLC12A5 promoted cell invasion and migration in SW1116 cells. (B) Knockdown of SLC12A5 suppressed cell invasion and migration in HCT116 cells. (C) Representative images of lungs with or without metastasis (left panel). H&E staining of lung tissues from nude mice injected with Control-HCT116 or shSLC12A5-HCT116 cells (200x magnification, middle panel). Incidence of lung metastasis (right panel). (D) Validation of the key metastasis-related genes identified by cDNA expression array by RT-PCR. (E) Western blot further confirmed the deregulation of metastasis-related genes. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HGF, hepatocyte growth factor; MMP, matrix metallopeptidase.
The in vivo metastasis assay was performed by injecting HCT116 cells transduced with shSLC12A5 or Control-shRNA into nude mice through the lateral tail vein to examine the cells’ lung metastasis ability. Six weeks following injection, the incidence of lung metastases (5/5, 100%) was significantly higher in mice injected with Control-HCT116 cells than in mice injected with shSLC12A5-HCT116 cells (1/5, 20%) (p<0.01) (figure 6C). Metastatic nodules in the lungs were confirmed histologically (figure 6C).
SLC12A5 regulates extracellular matrix remodelling genes
To gain further insight into the mechanisms by which SLC12A5 promotes CRC cell invasion and metastasis, SLC12A5-modulated downstream target genes were characterised by Human Tumour Metastasis RT2 Profiler PCR array in HCT116 cells transfected with siSLC12A5 or control siRNA. The results showed that knockdown of SLC12A5 in HCT116 cells upregulated nine antimetastasis genes, including metastasis suppressor (MTSS1), p53, NME/NM23 nucleoside diphosphate kinase (NME1), fibronectin 1 (FN1), neurofibromin 2, FAT tumour suppressor homologue 1 (FAT1), breast cancer metastasis suppressor 1 (BRMS1), tissue inhibitor of metalloproteinase 2 (TIMP2), cathepsin K (CTSK) and downregulated four pro-metastasis genes, namely matrix metallopeptidase 2 (MMP2), hepatocyte growth factor (HGF), MMP 13 and MMP10, as compared with Control-HCT116 cells, with more than a twofold change at mRNA levels (see online supplementary table S3). Selected downstream effectors of SLC12A5 were validated in two more colon cancer cell lines by overexpression of SLC12A5. Consistent with the array data, ectopic expression of SLC12A5 in SW480 and SW1116 downregulated MTSS1 and FN1, but upregulated MMP2, HGF and MMP13 as determined by RT-PCR and western blots (figure 6D, E). Taken together, our findings strongly suggest that SLC12A5 promote invasion and metastasis by regulating extracellular matrix-remodelling genes.
Overexpression of SLC12A5 is associated with poor prognosis of patients with CRC
We further evaluated the clinicopathological and prognostic significance of SLC12A5 in patients with CRC. As shown in table 1, overexpression of SLC12A5 was positively associated with a more advanced TNM stage (p<0.05), which is consistent with the role of SLC12A5 on tumour metastasis. Kaplan–Meier survival analysis showed that patients with CRC with SLC12A5 overexpression had significantly shorter survival compared with those with SLC12A5 low expression (p=0.009, log-rank test) (figure 7). In univariate Cox regression analysis, SLC12A5 overexpression was associated with an increased risk of cancer-related death (RR, 2.38; 95% CI 1.21 to 4.66; p=0.012) (table 2). After the adjustment for potential confounding factors, multivariate Cox regression analysis showed that SLC12A5 overexpression was an independent predictor of poorer survival of patients with CRC (RR, 2.30; 95% CI 1.16 to 4.60; p=0.018) (table 2).
Cox regression analysis of potential poor prognostic factors for patients with colorectal cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical significance of SLC12A5 in colorectal cancers (CRCs). Kaplan–Meier survival analysis according to SLC12A5 expression in195 patients with CRC. Patients with CRC with SLC12A5 overexpression had poorer survival than others. The difference is statistically significant based on the log-rank test (p=0.009).
Discussion
In this study, amplification of SLC12A5 was detected in 40.8% (78/191) of primary CRCs by FISH, but not in their adjacent tissues. Compared with the low frequency of SLC12A5 point mutation (3.8%) reported in our recent study based on targeted capture sequencing,23 amplification of SLC12A5 is a common event in colorectal tumourigenesis. SLC12A5 was located on chromosome 20q13.12, which is one of the most frequently amplified regions in CRC21 and in other cancer types such as breast, ovarian and gastric cancers.19 ,20 The 20q13.12 amplicon encodes 11 genes, none of which have been unequivocally described with oncogenic function in CRC yet.21 Gene amplification is thought to promote overexpression of genes favouring tumour development.34 ,35 Importantly, we revealed that the amplification of SLC12A5 was positively associated with its protein overexpression in the 191 CRC cases (p<0.001), suggesting that increased SLC12A5 copy number contributes to the up-regulation of SLC12A5, which might play an important oncogenic role in CRC. In keeping with our finding, SLC12A5 was upregulated in cervical cancer.36
A series of in vitro and in vivo functional experiments revealed that SLC12A5 possessed a strong tumourigenic function in CRC. SLC12A5 enhanced tumour cell survival, which is attributable to its antiapoptotic and pro-proliferative ability. Defective apoptosis is one of the hallmarks of cancer. Although caspase activation is considered a cardinal feature of apoptosis,37 an alternative pathway of apoptosis has been described that does not require caspase activation, which is characterised by the nuclear translocation of AIF and EndoG.30 ,38 ,39 In this study, knockdown of SLC12A5 induced apoptosis without caspase activation, suggesting that SLC12A5 modulated apoptosis via a caspase-independent pathway. In line with this hypothesis, we found that silencing SLC12A5 enhanced the nuclear protein levels of AIF and EndoG (figure 4). The release of AIF and EndoG is regulated by mitochondrial outer membrane permeability, which is modulated by the relative abundance of the pro-apoptotic Bax and Bak.31 After being activated by p53, PUMA interacts with antiapoptotic Bcl-2 family members, thereby freeing pro-apoptotic molecules, Bax and Bak.30 Consistent with the antiapoptotic role of SLC12A5, nuclear translocation of AIF and EndoG by silencing SLC12A5 was paralleled with the upregulation of p53, PUMA, Bax and Bak. Bax and Bak can form complexes at the mitochondrial outer membrane to facilitate the release of AIF and EndoG into nucleus.40 On the other hand, the inhibitor of apoptosis protein Survivin retrains AIF in mitochondria and suppresses AIF activity, thus further enhancing apoptotic resistance.32 Collectively, our data support that SLC12A5 inhibits caspase-independent apoptosis by deregulating AIF and EndoG in CRC (figure 4E). Cell cycle analysis revealed a decrease in G1 phase cells and an increase in S phase cells in SLC12A5-transfected cells, and a concomitant increase of protein expression of the master G1–S transition regulators cyclin D1 and CDK4. These results indicated that SLC12A5 could facilitate DNA synthesis and promote G1–S transition, contributing to its oncogenic effect in CRC.
Moreover, SLC12A5 inhibited p53 and p21 luciferase reporter activities in HCT116, which is p53 wild type. p53 and p21 were implicated in apoptosis,30 and the inhibition of these factors may contribute to the antiapoptotic effect of SLC12A5. Consistent with this effect, it has been reported that supplementation of KCl in extracellular medium was most effective in preventing hypoxia-induced apoptosis.41 It is further revealed that KCl supplementation downregulated a distinct p53-regulated cellular sub-network of genes involved in regulation of DNA replication.41 On the other hand, SLC12A5 also exhibited tumour-promoting effect and suppressed p21 reporter activity in p53 KO and mutated CRC cells, indicating that SLC12A5 promotes tumourigenesis by inhibiting p21 reporter activity regardless of p53 status. In line with this, the carboxyl terminus of p21 was reported to activate cell apoptosis without caspase involvement in a p53-independent manner, which is associated with the release of AIF and EndoG from mitochondria.42
Consistent with its tumourigenic function, SLC12A5 also induces tumour invasion and metastasis. Ectopic expression of SLC12A5 in colon cancer cell lines promoted cell migration and invasion in vitro. Moreover, in a cancer metastasis mouse model, depletion of SLC12A5 by shRNA in HCT116 cells significantly inhibited lung metastasis in vivo. The molecular mechanisms by which SLC12A5 exerts its pro-invasive and pro-metastasis functions in CRC were evaluated by cDNA microarray and immunoblotting. We revealed that the enhanced cell invasion and migration by SLC12A5 were mediated via downregulation of key antimetastasis genes, including MTSS1, p53, NME1, FN1, NF2, FAT1, BRMS1, TIMP2 and CTSK, and upregulation of the important pro-metastasis genes HGF, MMP10, MMP13 and MMP2 (see online supplementary table S3). Of which, MTSS1 plays a role in governing the metastatic nature of breast cancer cells, as well as a prognostic indicator of disease-free survival in patients with breast cancer.43 FN1 is a glycoprotein and is known to be involved in matrix remodelling, cell adhesion and migration processes, which affect cell motility by regulating actin polymerisation. The inhibitory effects of FN1 on cancer cell migration and metastasis have been well documented.44 ,45 MMP10, MMP13 and MMP2 are members of extracellular proteinases with key functions in the formation and remodelling of tumour invasion.46 ,47 HGF stimulates tumour cell migration by binding specifically to its receptor c-met in CRC.44 ,46–48 Taken together, SLC12A5 promoted tumour metastasis through mediating key elements of the matrix architecture such as MMP and fibronectin.
A clinical association evaluation showed that overexpression of SLC12A5 was associated significantly with advanced tumour stage (p<0.001). Patients with CRC vary greatly in clinical outcome, depending on the growth status and aggressiveness of tumours. At present, the most important clinical prognostic indicator of disease outcome is TNM staging. Nevertheless, some patients with CRC with low TNM stages are still afflicted with disease recurrence. Therefore, additional prognostic biomarkers are needed to provide better risk assessment. In this regard, we further examined the influence of SLC12A5 overexpression on clinical outcomes of 195 patients with CRC. Both univariate and multivariate cox regression analyses showing high expression levels of SLC12A5 were correlated significantly with shortened overall survival, suggesting that SLC12A5 overexpression could be regarded as an independent new prognostic marker for CRC.
In conclusion, amplification of SLC12A5 is a common event in CRC. Overexpression of SLC12A5 plays a pivotal oncogenic role in colorectal carcinogenesis by inhibiting apoptosis through mediating AIF-dependent and EndoG-dependent apoptotic signalling pathway and promoting metastasis by regulating key elements of the matrix architecture. SLC12A5 may serve as an independent prognostic biomarker for patients with CRC.
Acknowledgments
The authors thank Professor Bert Vogelstein for kindly providing HCT116 p53 knockout cell line.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors LX, XL, MC, JC and WK performed the experiments; MC, JT and K-FT provided the samples; X-YG provided technical support; LX, XL, XL, WKKW and JY analysed the data; LX and JY wrote the paper; JJYS commented on the study; JY and FKLC designed and supervised the research.
Funding This project was supported by China 863 Program (2012AA02A506), China 973 Program (2013CB531401), Shenzhen Technology and Innovation Project Fund, Shenzhen (JSGG20130412171021059), Shenzhen Municipal Science and Technology R & D fund (JCYJ20120619152326450) and Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics Committee of the Chinese University of Hong Kong and the Clinical Research Ethics Committee of the Sun Yat-sen University of Medical Sciences.
Provenance and peer review Not commissioned; externally peer reviewed.