Article Text

Abstract
Objective The generation of acinar and ductal cells from human pluripotent stem cells (PSCs) is a poorly studied process, although various diseases arise from this compartment.
Design We designed a straightforward approach to direct human PSCs towards pancreatic organoids resembling acinar and ductal progeny.
Results Extensive phenotyping of the organoids not only shows the appropriate marker profile but also ultrastructural, global gene expression and functional hallmarks of the human pancreas in the dish. Upon orthotopic transplantation into immunodeficient mice, these organoids form normal pancreatic ducts and acinar tissue resembling fetal human pancreas without evidence of tumour formation or transformation. Finally, we implemented this unique phenotyping tool as a model to study the pancreatic facets of cystic fibrosis (CF). For the first time, we provide evidence that in vitro, but also in our xenograft transplantation assay, pancreatic commitment occurs generally unhindered in CF. Importantly, cystic fibrosis transmembrane conductance regulator (CFTR) activation in mutated pancreatic organoids not only mirrors the CF phenotype in functional assays but also at a global expression level. We also conducted a scalable proof-of-concept screen in CF pancreatic organoids using a set of CFTR correctors and activators, and established an mRNA-mediated gene therapy approach in CF organoids.
Conclusions Taken together, our platform provides novel opportunities to model pancreatic disease and development, screen for disease-rescuing agents and to test therapeutic procedures.
- PANCREAS
- CYSTIC FIBROSIS
- STEM CELLS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Human pluripotent stem cells (PSCs) present a powerful tool for developmental studies and regenerative medicine.
Directed differentiation of PSCs towards pancreatic cell fates requires the formation of PDX1/NKX6.1-positive progenitor cells.
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) perturb fluid transport causing chronic airway infections or biliary cirrhosis with variable phenotypes while the pancreas is one of the first organs affected.
What are the new findings?
Efficient generation of high yields of pancreatic progenitors from several human pluripotent stem cell lines.
PSC-derived pancreatic progenitors form pancreatic organoids that comprise acinar/ductal-like progeny and resemble human pancreas upon orthotopic transplantation in mice.
Induced PSCs from cystic fibrosis patients display normal pancreatic commitment in vitro and in vivo at least until a fetal developmental stage.
Pancreatic organoids from patients with cystic fibrosis recapitulate defective CFTR function in vitro, allowing subsequent drug screening but also mRNA-mediated gene supplementation.
How might it impact on clinical practice in the foreseeable future?
Our system provides a novel approach to model human pancreatic development and disease.
Humanised platform for (organ-specific and patient-specific) drug screening and testing of therapeutic options in vitro and in vivo.
Introduction
Given their capacity to differentiate into every cell type of the human body, human-induced pluripotent stem cells (hiPSCs) provide a unique platform for developmental studies and regenerative medicine.1–4 The generation of pancreatic progenitor (PP) cells from PSCs follows the sequential induction of virtually pure definitive endoderm (DE), foregut endoderm (GTE) and pancreatic endoderm (PE, figure 1A).5–7 Over the last decade, a series of studies have aimed at improving pancreatic differentiation protocols.5–7 While most studies focused on the generation of PDX1-positive PE,8 ,9 true PP cells should coexpress both NKX6.1 and PDX1.10 While the exocrine and endocrine lineages develop, NKX6.1 is still expressed in β cells, but becomes mutually exclusive with the expression of Ptf1a driving the exocrine lineage.11 Thus, the presence of NKX6.1 is one of the key distinguishing features of these two lineages and hence can be used to monitor the emergence of true progenitors. Despite recent progress in differentiating PSCs towards endocrine pancreatic progeny,12 ,13 the generation of ductal and exocrine-like cells has not yet been adequately achieved, apart from a recent study modelling human pancreatic cancer.14 Three-dimensional organoid models generated from PSCs can faithfully model the in vivo situation,13 ,15 ,16 and disease-specific iPSCs allow the generation of distinct human disease models.12 ,13 ,15 Nevertheless, the generation of human pancreas in mice upon xenotransplantation of non-transformed organoids has not been achieved to date. However, this would open up entirely new research avenues. In addition, inherited pancreatic diseases would benefit from in vivo gene supplementation strategies as recently shown for a lung disease with specifically modified mRNA, pre-evaluated in organoids.17 ,18 To tackle this unmet need in the pancreatic field, we describe herein a new PSC-based organoid system that was used to model pancreatic aspects of cystic fibrosis (CF).

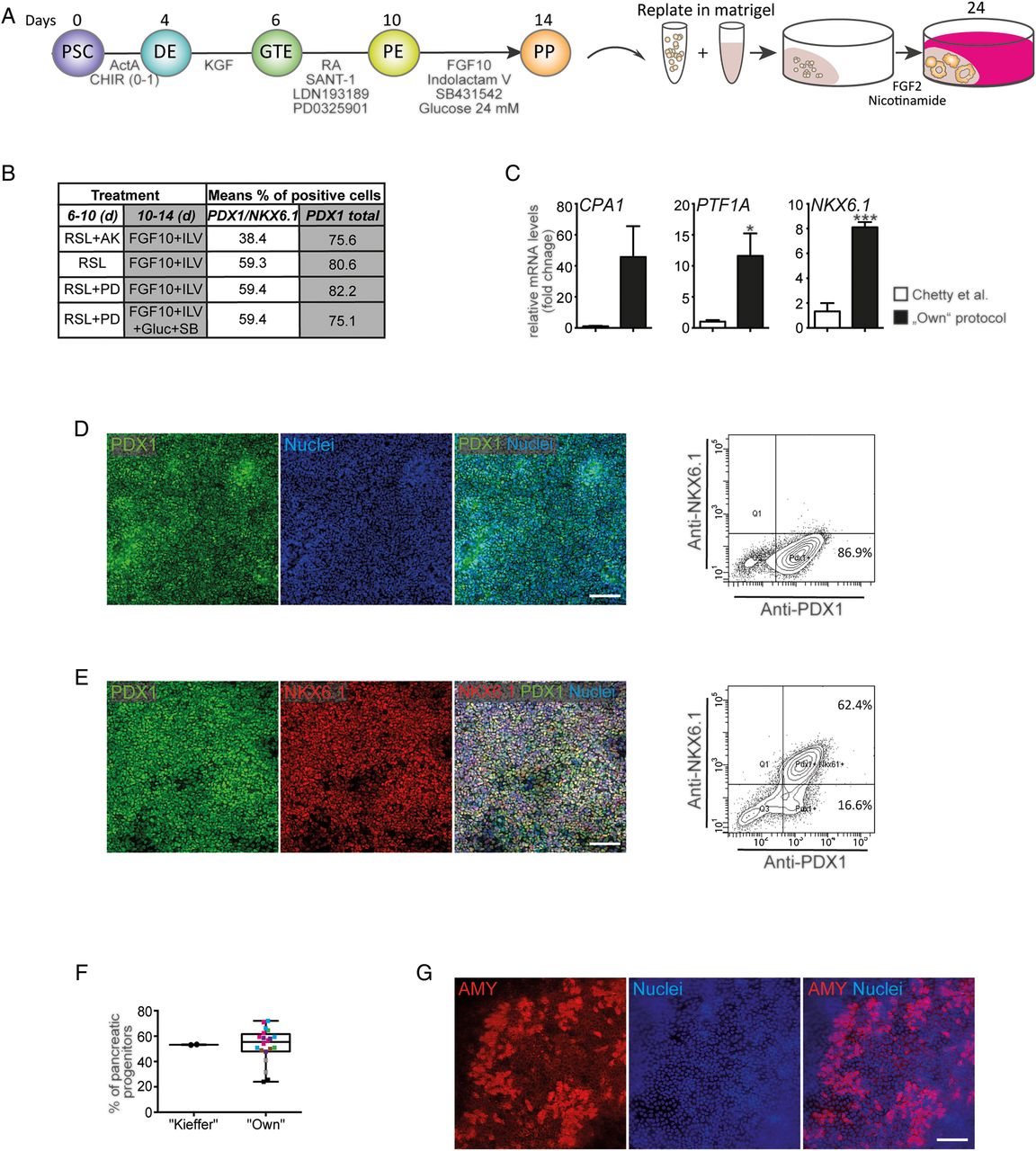
Pancreatic progenitor cells derived from human pluripotent stem cells (PSCs). (A) Schematic outline of the developed protocol. Growth factors, small molecules and timing are indicated. DE, definitive endoderm; GTE, gut-tube endoderm; PE, pancreatic endoderm; PP, pancreatic progenitor. (B) Overview of fluorescence-activated cell sorting (FACS) results during protocol optimisation to generate true PPs. A, Activin A; K, keratinocyte growth factor (KGF); L, LDN-193189; PD, PD0325901; R, retinoid acid; S, SANT-1 . (C) qPCR at the PP stage comparing a previously described protocol53 with our optimised protocol (first vs last row in B, fold change, PP markers as indicated). Data are presented as mean±SEM, n=3 experiments, statistical significance was determined by two-tailed t-test, *p<0.05, ***p<0.001. (D and E) Immunostaining (left) and representative FACS plots (right) for the indicated markers ((D) day 10; (E) day 14). (F) FACS-based quantification of PDX1/NKX6.1-double positive cells generated after 13 days of PP differentiation from HUES8 cells using the ‘Kieffer’13 protocol or our ‘own’ protocol (see A) in seven different PSC lines (indicated by different colours). Dots represent independent experiments. Boxes show the median, and the 25th and 75th percentiles. The whiskers of the graph show the largest and smallest values. (G) Amylase-positive cells derived after 24 days of pancreatic differentiation in monolayer culture. If not stated otherwise, all data were obtained using HUES8 cells. Scale bars: 100 µm.
CF is an inherited disease caused by either nonsense or missense mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, resulting in complete absence of the protein, a misfolded polypeptide that is degraded by the unfolded protein response, or a dysfunctional protein.19 CFTR encodes a chloride channel gated by cyclic AMP-dependent phosphorylation that is necessary for electrolyte and fluid homeostasis of epithelia in various organs including the lung, liver, intestine and pancreas. Dysfunction of the CFTR leads to the production of hyperviscous mucus causing chronic airway infections or biliary cirrhosis with variable phenotypes.20 Although the pancreas is one of the first organs affected, knowledge about the pathophysiology of the pancreas during CF is limited. Briefly, distinct CFTR genotypes have been shown to not only increase the probability of developing either pancreatitis or perinatal exocrine insufficiency21 but also pancreatic cancer.22 Moreover, exocrine insufficiency drives a complex and poorly understood cascade of events leading to endocrine exhaustion.23 Additionally, the expression of the CFTR gene during early pancreatic development suggests that CFTR mutations could have a developmental impact;24 however, current CF animal models recapitulate only limited aspects of the human disease sparing the pancreas, and in vitro studies have been hampered by the lack of primary human pancreatic progeny. Moreover, with increasing CF patient survival pancreatic manifestations of CF are becoming progressively more clinically relevant but the underlying pathomechanisms remain to be explored. Thus, innovative model systems for pancreatic CF and other pancreatic disorders are clearly warranted.
Materials and methods
Differentiation of human PSCs into PP cells
For differentiation, human PSCs were grown to 95% confluence on growth factor-reduced matrigel (BD, 354 230) and FTDA medium25 was refreshed 3 hours before initiating differentiation. The backbone medium for the first 6 days of differentiation was BE1: MCDB131 (Invitrogen) with 0.8 g/L cell culture tested glucose (Sigma), 1.174 g/L sodium bicarbonate (Sigma), 0.1% fatty acid free (FAF) BSA (A7030, Sigma), 2 mM l-glutamine. Later differentiation was performed in BE3 as the backbone medium: MCDB131 with 0.44 g/L glucose, 1.754 g/L sodium bicarbonate, 2% FAF-BSA, 2 mM l-glutamine, 44 mg/L l-ascorbic acid, 0.5× insulin-transferrin-selenium-ethanolamine (ITS-X). Cells in differentiation were cultured at 37°C in a 5% CO2 incubator with daily media change. For the first day of differentiation, cells were washed with phosphate buffered saline (PBS) (Sigma) and incubated with BE1 supplemented with 3 µM GSK3β-inhibitor (CHIR99021) (Axon MedChem) and 100 ng/mL Activin A. The next day the medium was replaced by BE1 with 100 ng/mL Activin A. After 3 days, media was changed to BE1 with 50 ng/mL KFG (Peprotech) for 2 days. From day 6 until day 10, cells were cultured in BE3 medium containing 0.25 µM SANT-1 (Sigma), 2 µM retinoic acid (Sigma), 200 nM LDN-193189 (Sigma) and 500 nM PD0325901 (Calbiochem). At days 10–14, the cells received BE3 supplemented with 50 ng/mL fibroblast growth factor (FGF10) (Peprotech), 330 nM Indolactam V (Stem Cell Technologies), 10 µM SB431542 (Axon MedChem) and additional 16 mM glucose. An outline of the differentiation protocol can also be found in figure 1A.
3D pancreatic organoid culture
At the PP stage (day 12 of differentiation), cells were washed with PBS, incubated with TrypLE at 37°C for 5–6 min and carefully resuspended in DMEM/F12 resulting in clumps of 3–10 cells. After centrifugation at 400 g for 5 min, the pellet was washed in BE3 medium, centrifuged again and resuspended in precooled day 12 medium (detailed above) supplemented with 10 µM Rock inhibitor. The cell suspension was mixed on ice with growth factor reduced matrigel at a 1:3 ratio and 25 µL were transferred to a 48-well plate (NunclonΔSurface). Following incubation for 10 min at 37°C, the solidified drop of matrigel was overlayed with 200 µL of day 12 medium (see above) containing 10 µM Rock inhibitor (always added for the first 4 days in three-dimensional (3D) culture), which was replaced the next day. On day 14, differentiation was continued with 10 ng/mL FGF2 and 10 µM Rock inhibitor in BE3 medium. From day 18 on, organoids were cultured in BE3 with 10 ng/mL FGF2 and 10 mM nicotinamide (NA) (Sigma) (referred to as ‘FN’). Medium was changed every 2–3 days. Alternatively, organoids were generated by replating PPs in suspension in ultra-low-attachment plates (Corning). To prevent aggregation, cell clusters were triturated after 1 hour and again after 2 days. For changing media, half of the fluid was pipetted off and replaced with fresh differentiation media. Medium conditions were identical to the matrigel-based culture. For suspension cultures, another medium (referred to as ‘FEPC’) was tested (based on conditions published for mouse embryonic PPs26) composed of DMEM/F12, 10% knockout serum replacement (KOSR) and 0.1 mM β-mercaptoethanol supplemented with 50 ng/mL FGF10, 25 ng/mL epidermal growth factor (EGF) (novoprotein), 2 µM CHIR99021 and 16 nM phorbol myristate acetate (Sigma). For passaging, matrigel was scraped off, pipetted in order to mechanically dissociate the organoids into small clumps, collected in a 15 mL falcon, and further processed as described above. Organoids were passaged every 10–14 days and cultured at 5% CO2 and 37°C.
Functional CFTR assay
Organoids were split 1:4 and 8 µL of the cell/matrigel suspension was transferred to the inner border of each well of a 96-well plate (NunclonΔSurface), solidified for 5 min and 100 µL culture medium was added. Six days after seeding, organoids were incubated with 20 µM forskolin (FSK) and 100 µM 3-isobutyl-1-methylxanthine (IBMX) (or 1:500 dimethyl sulfoxide (DMSO) (all from Sigma) as negative control) in FN medium for 2 hours at 37°C. The forskolin-induced swelling assay was performed as described before in ref 27. Pictures were captured before and immediately after treatment using a Keyence Biozero BZ-9000 microscope. Images were manually analysed and spheres were also encircled manually. To quantify the surface area increase relative to the organoid area before CFTR induction (0 hour), we used the area measurement application of the BZII-analyser software (Keyence). The mean area increase (of at least 30 organoids) was calculated per well followed by summarising the results of three individual wells per condition.
CFTR corrector screen
For screening CFTR modulators, organoids (cultivated in matrigel for 6 days) were preincubated with the respective compound, or combinations thereof, diluted to 10 µM (DMSO, 1:500 was used as control) in FN medium for 24 hours (except for P4 and P9, which were applied during forskolin (FSK) treatment only), and the functional CFTR assay was subsequently carried out as described above. The effect of selected hit compounds was quantified by measuring the mean area increase after CFTR induction in all organoids (8–12 organoids per condition) using ImageJ. All CFTR corrector and potentiator compounds, specified in online supplementary table S1, were provided by the Cystic Fibrosis Foundation Therapeutics.
Supplemental material
Results
Genuine PP cells from human PSCs
Recent studies have described a complex sequential arrangement of growth factors to generate high numbers of PPs.12 ,13 Therefore, we implemented, fine-tuned and specifically tailored a previously described protocol for our requirements: (i) by limited use of growth factors in exchange with cost-effective small molecules instead, (ii) a PP suitable for acinar/ductal lineage commitment and (iii) broad applicability across various human PSC lines has been generated (figure 1A). The growth factors and small molecules used included the sonic hedgehog antagonist SANT1,28 LDN193189 as an inhibitor of bone morphogenetic protein (BMP) signalling, the mitogen-activated protein kinase (MEK) inhibitor PD0325901, FGF2 and FGF10,29 SB431542 to inhibit transforming growth factor-β (TGF-β)-signalling,30 NA and Indolactam V to promote PP expansion and exocrine specification.31 ,32 Details of our pilot tests are outlined in the online supplementary results in supplementary figures S1–3 and figure 1B, C. Briefly, high yields of PDX1-positive PE were achieved by retinoic acid treatment, sonic hedgehog (SHH) and BMP inhibition, and inhibition of MEK/ERK-signalling (figure 1D). Similarly, we optimised the commitment towards PDX1/NKX6.1-positive PPs (figure 1B, C,E, F and online supplementary figures S2 and S3) with a combination of Indolactam V,31 FGF10 and SB431542 in high-glucose, serum-free media. This protocol robustly allowed the generation of up to 70% PDX1/NKX6.1-positive PPs across several human PSC lines (figures 1E, F, 4C, F and online supplementary figure S8). This combination also resulted in the strongest increase in PP-marker gene expression (figure 1C) while non-pancreatic lineage marker gene expression was lowest under these conditions (see online supplementary figure S3C). Cell death was virtually absent at various intermediate stages, while PPs remained proliferative (see online supplementary figure S4). To provide an objective rating of the quality of our protocol, we applied a protocol published in a recent landmark study,13 which aimed to develop β cells from PDX1/NKX6.1-positive PPs, to HUES8 cells and found similar, if not superior, results with our protocol across seven different human PSC lines (figure 1F). Upon subsequent monolayer differentiation (using FGF2 and then FGF2+NA) to test our novel PP population for its exocrine differentiation capacity, we observed the generation of some amylase (AMY)-positive cell clusters (figure 1G).
Pancreatic organoids efficiently generate acinar-like and ductal-like progeny
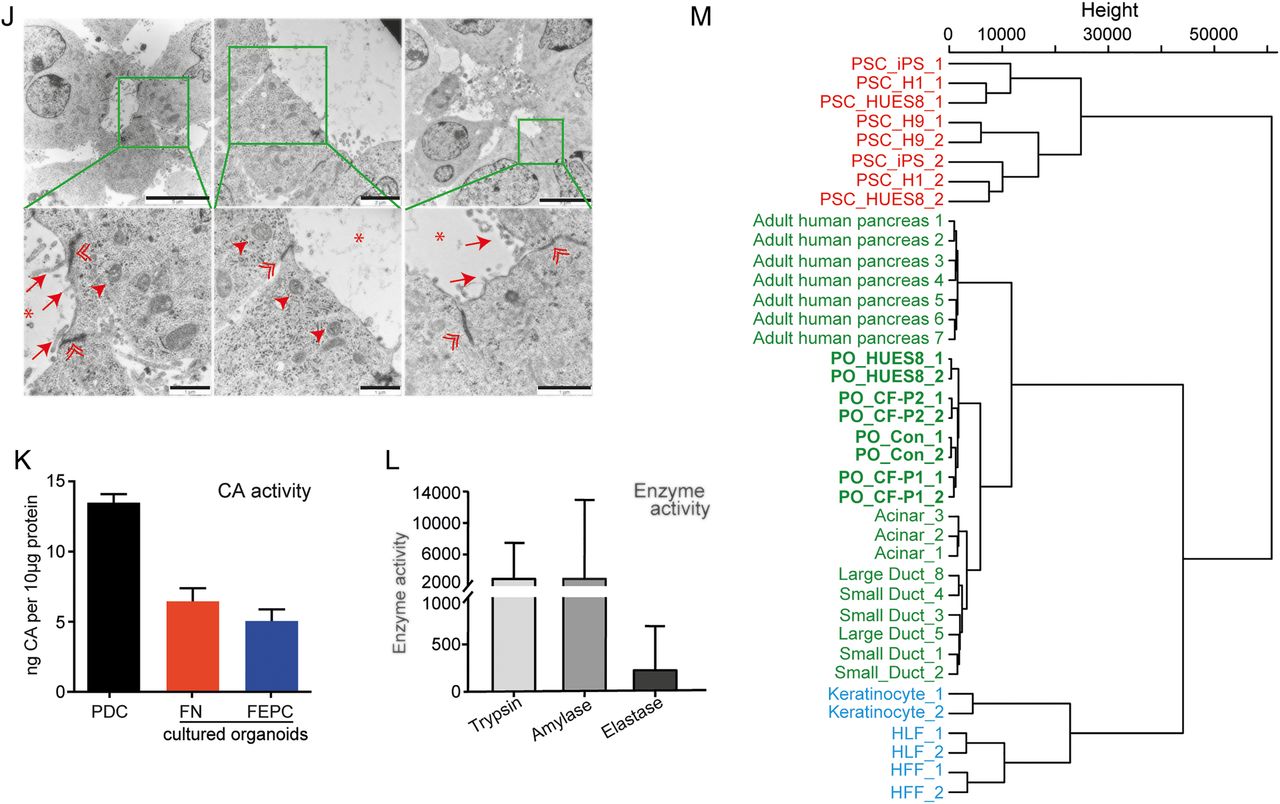
It has been shown that human PSC-derived organoids emphasise maturation and functionality across several cell types.33 ,34 In order to direct lineage differentiation of PPs in favour of pancreatic maturation, 3D culture conditions were employed (figure 1A, right). Whether reseeded into suspension or embedded into matrigel, PPs rapidly formed cyst-like structures further referred to as pancreatic organoids (POs) (figure 2A and online supplementary figures S5A and S9A). Of note, organoids arose from cell clusters comprising on average 4–5 cells (figure 5A and online supplementary figure S5A). Intriguingly, POs could be expanded for >6 passages and regrew upon subsequent freeze-thaw cycles (not shown). Maturated organoids expressed pancreatic exocrine markers such as AMY or chymotrypsin C (CTRC), ductal markers such as SOX9 and cytokeratin 19 (CK19) and also CFTR, as well as the epithelial marker E-cadherin and the progenitor marker NKX6.1 (figure 2B–G). While we observed heterogeneity in pancreatic maturation across all organoids, POs with a relevant architecture of acinar/ductal progeny contained 34%±15% acinar and 61%±19% ductal cells. Interestingly, organoids arising from reaggregated, FACS-purified PPs derived from an NKX6.1-GFP reporter embryonic stem cell (ESC) line displayed similar morphology, further substantiating the pronounced pancreatic fate of the organoids (see online supplementary figure S5B–E). In particular, POs exhibiting a hollow central lumen displayed a basal lamina and signs of apical-basal polarity as shown by staining for the apical marker ZO1 and basal marker laminin-α5 (figure 2H). We could not detect relevant amounts of apoptosis (cleaved CASP3) and each organoid contained a few Ki67-positive cells indicating proliferation (figure 2I). Ultrastructural analysis revealed typical ductal morphology with microvilli (arrow) and tight junctions (double arrowhead) but also acinar-like cells as identified by secretory granula (arrowhead) and lumen formation (asterisk, figure 2J).

Pancreatic organoids containing acinar-like /duct-like cells. (A) Brightfield view of pancreatic organoids (POs) in FN (left) or FEPC (right) medium (for details, see online supplementary figure S9). Scale bars: 50 µm. (B–G) Immunostaining of POs for the indicated markers. CTRC, chymotrypsin C; ECADH, E-cadherin. (E) Acinar-like cells staining positive for amylase (AMY) are mostly excluded from NKX6.1-positive regions (marked by arrowhead). (F) SOX9-positive cells mainly do not express NKX6.1 (indicated by arrowhead). (H) Analysis of polarity in organoids derived in matrigel-based culture conditions showing basolateral expression of laminin-α5 (LAM) and apical localisation of the tight junction protein ZO1. (I) Staining for Ki67 and cleaved caspase 3 (CASP3) indicating proliferation and very few apoptotic cells (marked by asterisk) in organoids derived in matrigel. Scale bars: (B, H, I) 20 µm, (C–G) 25 µm. (J) Electron microscopy of POs harbouring duct-like and acinar-like structures. Marked inlets are depicted in the lower row and show microvilli (arrows), tight junctions (double arrowheads) and secretory granules (arrowheads); asterisks mark the lumen. Scale bars: 5 µm (upper row), 2 µm (upper row middle) or 1 µm (bottom row). (K) Carbonic anhydrase (CA) assay reflecting enzymatic activity of POs compared with primary pancreatic ductal cells (PDC) depicted as ng CA/10 µg protein (means of two triplicate measurements±SEM are shown). (L) Digestive enzyme activity in POs generated in suspension culture (FN medium). Trypsin and elastase activity is given as RFU/min/µg protein and amylase activity is indicated in mU calculated against purified enzyme (porcine amylase). Measurements were performed in triplicates and results are represented as mean±SD. (M) Hierarchical clustering shows in-house generated data (ie, HUES8, Con-iPSCs and CF-P1/P2-derived POs) clustered with acinar, small/large ductal cells (E-MTAB-463) and adult human pancreas (GSE72492) and are clearly separated from pluripotent stem cells (PSCs) (GSE56130, GSE63101) and other somatic cells (ie, human keratinocytes, GSE63101; human foreskin/lung fibroblasts (HFF/HLF), GSE55820). If not indicated otherwise, all data were obtained in human ES cell (HUES8)-derived POs.

Continued.
POs are functional and globally cluster with human pancreas
To functionally assess our PO cultures, we applied an enzymatic assay to measure carbonic anhydrase (CA) activity in duct-like cells. CA is a key enzyme expressed in pancreatic ducts to catalyse the reaction CO2+H2O=HCO3−+H+. Indeed, organoids showed CA activity in a similar range as freshly isolated primary ductal cells (figure 2K). Additionally, enzymatic activity for key acinar enzymes such as AMY, trypsin and elastase could be detected (figure 2L). Similar to the aforementioned heterogeneous pancreatic marker expression profile, enzymatic activity also varied across individual organoids.
Next, we performed global gene expression analysis of PSC-derived POs and applied unsupervised clustering analysis in an attempt to globally assess the maturation stage of our cultures. Accordingly, transcriptome comparison with reference data sets from the literature35–37 revealed clustering of POs close to human adult pancreatic tissue but also with acinar and ductal cells derived from human pancreas. In contrast, POs clustered away from somatic cells such as keratinocytes and fibroblasts and human PSC (figure 2M). In addition, pancreas-specific gene signatures confirmed such clustering (see online supplementary figure S6 and table S4). Thus, POs faithfully recapitulate human pancreatic tissue based on protein markers, gene expression profiling and function. Taken together, these results would suggest that our system of POs is suitable for drawing conclusions on human disease.
Plucked human hair to generate CF-specific induced PSCs
Next, we aimed to generate a disease model for CF as an application for this PO model system. Clonal iPSC lines from two patients with CF and healthy donors were generated via reprogramming of keratinocytes derived from plucked human hair (figure 3A).38 ,39 Expanded clones displayed hallmarks of pluripotency as judged by immunostaining and mRNA expression of pluripotency markers such as OCT3/4, NANOG, SSEA4 and SOX2 (figure 3B–D). Gene expression profiling also showed clustering of CF lines close to HUES8 human embryonic stem cells but also together with iPSC samples from public databases. Of note, all pluripotent cell lines were located apart from somatic counterparts (figure 3E). Next, we confirmed the genotype of the two patients with CF by targeted sequencing of the CFTR gene in cultured iPSCs (figure 3F, G). Cells were derived from two unrelated individuals affected by (classical) CF. The first patient (CF-P1, figure 3F) was homozygous for the mutation c.3276C>A (p.Y1092*), and the second patient (CF-P2, figure 3G) was compound heterozygous for the mutations c.1521_1523delCTT (p.F508del) and c.3773dupT (p.L1258Ffs*7). All three mutations are well-known mutations and lead to a complete or near-complete loss of function of the CFTR protein (class I or II). Further details are provided in the online supplementary materials and methods section.
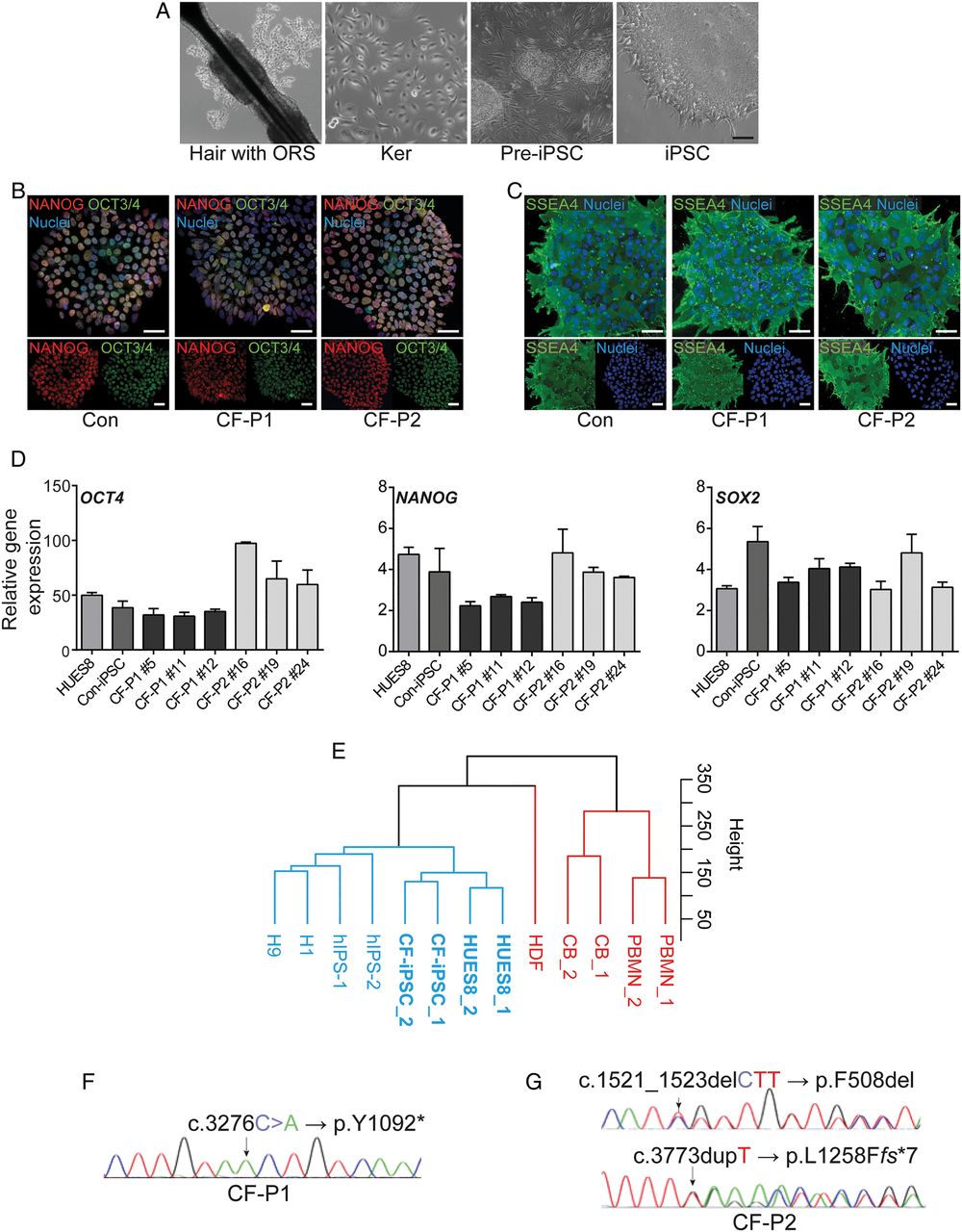
Modelling pancreatic cystic fibrosis (CF) with disease-specific human-induced pluripotent stem cells (iPSCs). (A) Reprogramming outline of plucked human hair (with outer root sheath (ORS)) keratinocytes (ker) from a patient with CF towards iPSCs. (B–D) Representative immunostaining (B, C) and qPCR analysis (D) for indicated pluripotency markers in a control-iPSC line and two CF-iPSC lines derived from independent CF individuals. Scale bars: 50 µm. Error bars represent mean±SEM, n=3. (E) Hierarchical clustering shows a high similarity between CF-iPSCs and pluripotent cells, including human embryonic stem cells (HUES8, own cultures; H1, GSM1040172; H9, GSM1040173) and human iPSCs established elsewhere (hiPSC_1, GSM1040179; hiPSC_2, GSM1040180). All pluripotent cell types are labelled in blue. CF-iPSCs show a high dissimilarity with and clustered away from somatic cell types such as peripheral blood monocytes (PBMN_1, GSM1040233; PBMN_2, GSM1040234), cord blood cells CB_1, GSM104023; CB_2, GSM1040232) and human dermal fibroblasts (HDF, GSM1040229). All somatic cell types are labelled in red. (F and G) DNA chromatogram of patient CF-P1 (F) and of patient CF-P2 (G) depicting the mutated nucleotides (arrows) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene locus. Note the two different mutations at different positions of the CFTR gene in CF-P2.
PSCs from patients with CF display normal commitment towards PPs
To demonstrate the eligibility of our organoid system to model pancreatic development and human disease, we focused on the pancreatic aspects of CF. It is assumed that mucus accumulating within the ductal lumen of tubular structures leads to obstruction and subsequent acinar degeneration. Although this sequence of events appears logical, it remains unclear whether mutated CFTR alters human pancreatic development, as suggested by its early expression and the pronounced perinatal damage in case of severe mutations.21 As such, we first assessed DE formation via surface marker cell cytometry for CXCR4 and c-KIT and immunostaining for SOX17 and FOXA2. Interestingly, commitment towards DE occurred in CF-iPSC lines at the same purity as in control lines (figure 4A, D and online supplementary figure S7). Similarly, formation of PDX1-positive PE was uniform across all genotypes and subclones upon directed pancreatic differentiation (figure 4B, E and online supplementary figure S8A). Applying our novel protocol to generate NKX6.1/PDX1-double positive PPs (figure 1A) also revealed no genotype-linked differences (figure 4C, F and online supplementary figure S8B, C).

Differentiation of cystic fibrosis (CF)-mutated human-induced pluripotent stem cells towards pancreatic progenitors. (A–C) Immunostaining (left) and representative FACS plots (right) for the definitive endoderm (DE) markers SOX17, CXCR4 and c-KIT, PDX1 and PDX1/NKX6.1-positive cells at day 4 (A), day 10 (B) and day 14 (C). Genotypes are indicated. Scale bars: 100 µm. (D–F) Quantification of several FACS experiments at (D) DE, (E) pancreatic endoderm and (F) pancreatic progenitor stage detected using indicated marker combinations. Data are shown as mean±SEM from three independent experiments. Individual experiments were grouped according to genotype (wildtype vs CF) and statistically compared via t-test.
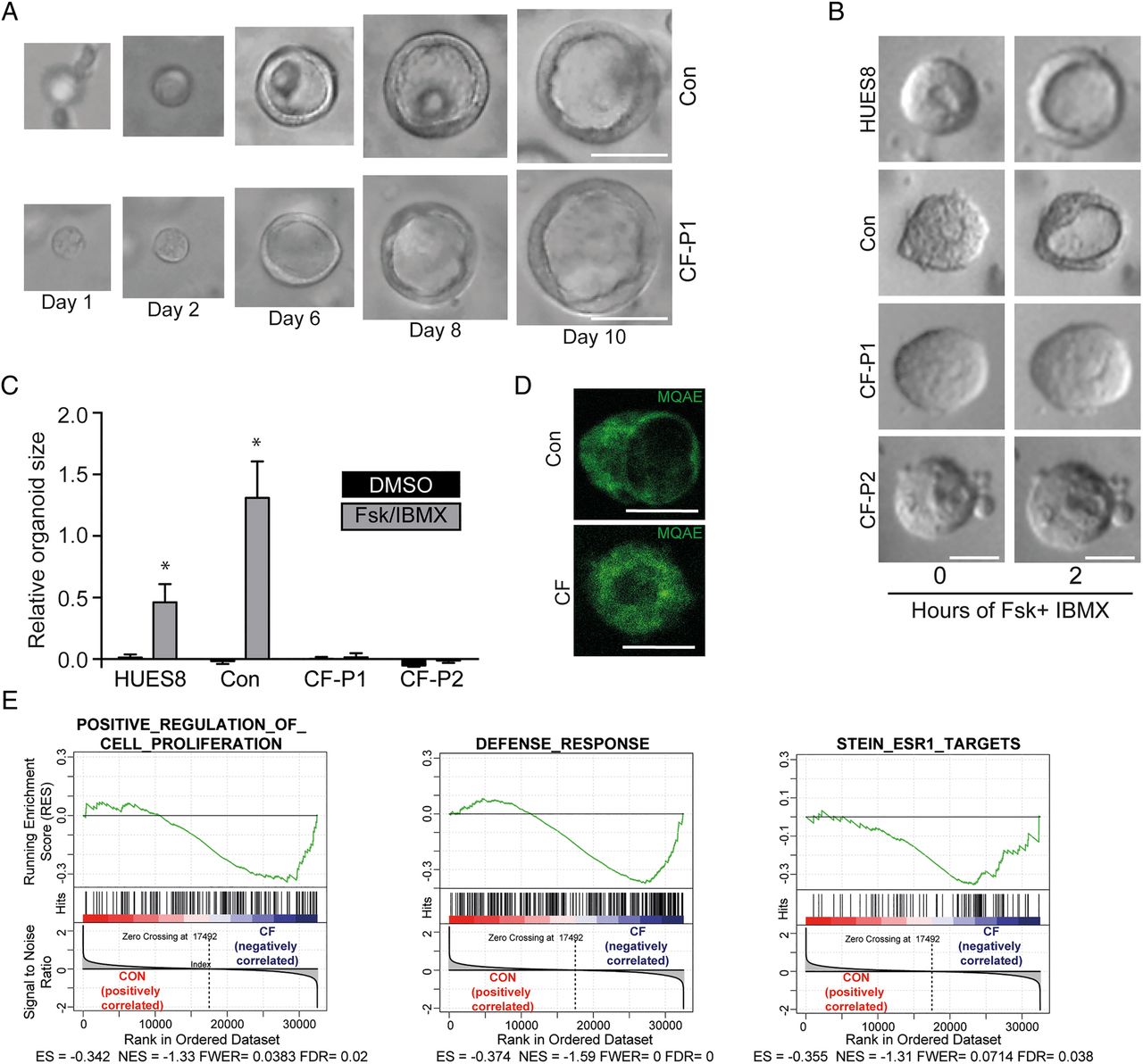
POs display a CF phenotype
We hypothesised that in POs a CF phenotype might become more traceable compared with the in vivo situation. Intriguingly, CF-POs developed without gross abnormalities in size and morphology, suggesting that this commitment step towards acinar-like/duct-like cells is unaltered in CF (figure 5A and online supplementary figure S9A). Also, we did not observe obvious changes in polarity, proliferation and apoptosis but also lineage fate within wild-type (WT) and CF POs as assessed via qPCR and immunostaining (see online supplementary figure 9B–D). Forskolin and IBMX activate CFTR and lead to subsequent fluid secretion into the organoid lumen;27 however, this effect should be disturbed in CF POs. Indeed, while in WT organoids swelling was pronounced, CF organoids remained unchanged (figure 5B, C). To further test CFTR function in both patient and control organoids, we used MQAE (N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide), a fluorescent dye quenched by the presence of chloride but not affected by other anions or pH changes, to visualise functional alterations in the respective organoid genotypes. Interestingly, the expected differences in chloride content within the organoid lumen could be observed (figure 5D).

Generation of pancreatic organoids (POs) from cystic fibrosis (CF)-mutated patients and phenotypic rescue. (A) Time course of growing POs derived from control (Con) and CF- human-induced pluripotent stem cells (iPSCs) (CF-P1). (B) Images of POs before and after 2 hours treatment with forskolin and IBMX. (C) Corresponding quantification from forskolin-induced PO swelling (B) in each indicated genotype. Error bars represent mean area increase±SEM of three individual wells, statistical significance was determined by two-tailed t-test, *p<0.05. (D) Luminal (N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide) (MQAE) fluorescence (Cl−-sensitive dye) is quenched in wild-type (WT) organoids indicating an increase in intraluminal chloride secretion after cystic fibrosis transmembrane conductance regulator (CFTR) activation and subsequent challenge with a chloride ion-rich solution, which is impaired in CF organoids. (E) Gene set enrichment analysis of three gene signatures were performed on WT ESC/iPSC (CON) and CF samples. Genes involved in cell proliferation, immune response and oestrogen receptor 1 (ESR1) signalling are negatively correlated in CFTR-mutated pancreatic organoids as previously described for airway cells.54 CON: HUES8 (WT ESC line, n=2) and Con1 (WT iPSC) line, n=2), CF: CF-P1 and CF-P2, n=2 each. ES, enrichment score; FDR, false discovery rate; FWER, family-wise error rate; NES, normalised enrichment score. (F and G) Small-scale rescue screen using indicated compounds in two independent CF patient-derived iPSC-POs. Organoids were preincubated with compounds before applying FSK/IBMX. Green (CF-P1) and red (CF-P2) boxes mark most pronounced rescue, which is represented as quantification of 8–12 organoids in (G). Dotted lines indicate relative organoid size upon treatment with DMSO as a solvent control. All scale bars: 50 µm.
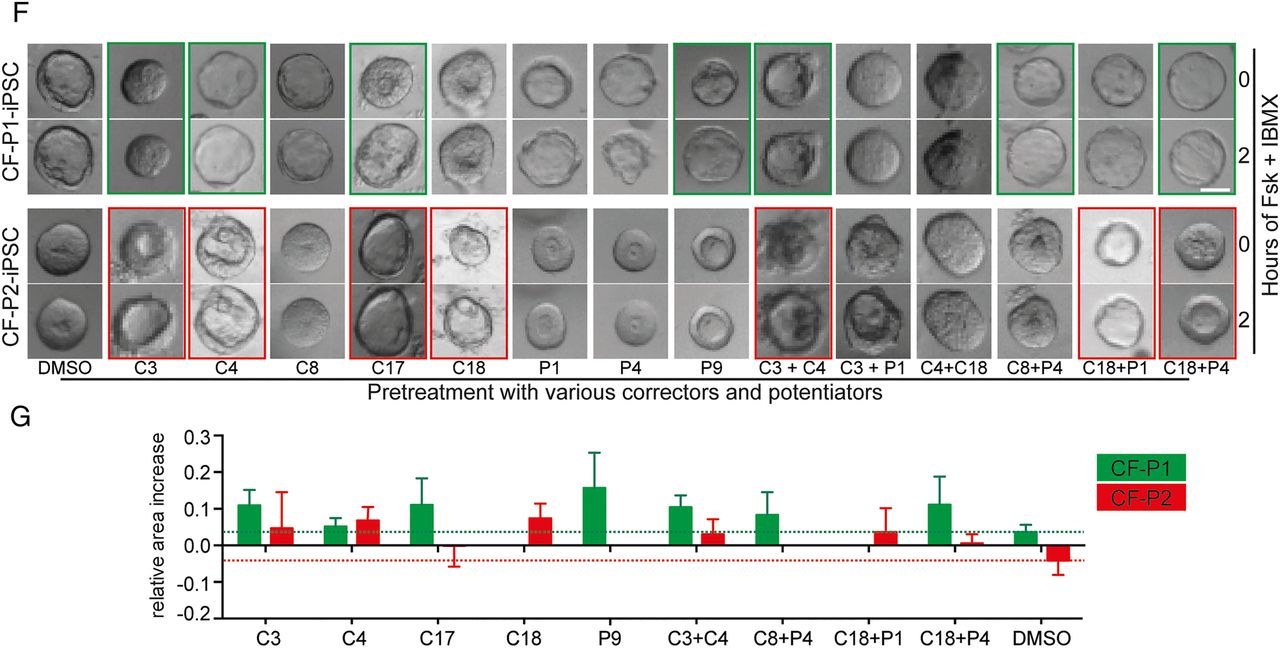
Continued.
Next, we searched for transcriptional evidence of a CF phenotype in patient-derived organoids. Previous transcriptome studies from F508del mutated native nasal epithelial cells identified characteristic gene signatures in human CF airway.35 We applied the latter gene sets for gene set enrichment analysis to the transcriptomes of CF and control PO samples. Indeed, we noted similar enrichment patterns in our PO samples for cell proliferation and defence response as well as for ESR1-target genes (figure 5E). Thus, molecular signatures established in CF nasal epithelium could be reidentified in POs with mutant CFTR, indicating common disease mechanisms across the affected organs in CF.
A compound screen in POs to rescue the CF phenotype
The pronounced swelling difference upon CFTR activation between control and CF organoids makes our platform an interesting model to identify novel drugs ameliorating pancreatic CFTR function. Accordingly, we compiled a set of small-molecule compounds that should improve cellular processing (termed CFTR correctors) and/or gating function of the CFTR protein (termed CFTR potentiators)40 to simulate such a proof-of-concept screen (for detailed compound description, see online supplementary table S1). Applying various drug combinations to our cultures, we faithfully rescued the CF phenotype (figure 5F). Notably, CF-P1 organoids harbouring a premature stop codon in the CFTR gene seemed to be less amenable for corrector compound-mediated rescue (figure 5F, G; green and red boxes). Therefore, our newly developed culture system is not only suitable to address developmental questions but also represents a novel drug-screening platform to develop and test future therapeutic options.
Modified mRNAs allow CFTR gene supplementation in POs
The most desirable treatment approach for an inherited disease is curative gene therapy. However, current regimens for gene repair such as DNA-based gene therapy are either hampered by safety concerns41 and/or low gene transfer efficiency.42 The application of nucleotide chemically modified mRNA (cmRNA), circumvents these caveats and further ensures high stability, thus representing a promising therapeutic tool. Previous work by our group and others has shown that delivery of cmRNA leads to therapeutic levels of protein expression as a result of high gene transfer efficiency, higher stability and/or low immunogenicity, and hence, can even be used for life-saving genome editing in vivo.18 Unfortunately, labour and cost-intensive testing is required to identify organ-optimised and personalised cmRNAs for gene supplementation in vivo. Organoids, however, would be a desirable system to screen and validate cmRNAs in vitro for subsequent in vivo applications, although the utility of such a system remains to be validated. To address the latter, we aimed to establish cmRNAs optimised for POs in an attempt to rescue CFTR function in CF POs. We tested a set of different modifications (unpublished data) using mRNAs encoding for dsRed. With one modification, we obtained robust dsRed protein expression even 7 days after transfection in subsequently developed organoids independent of the culture regimen (figure 6A). This modification was used for CFTR gene supplementation in CF organoids and indeed upon activation of CFTR using forskolin and IBMX in the organoid-swelling assay described above a significant rescue could be observed in CFTR mutants (figure 6B, C).
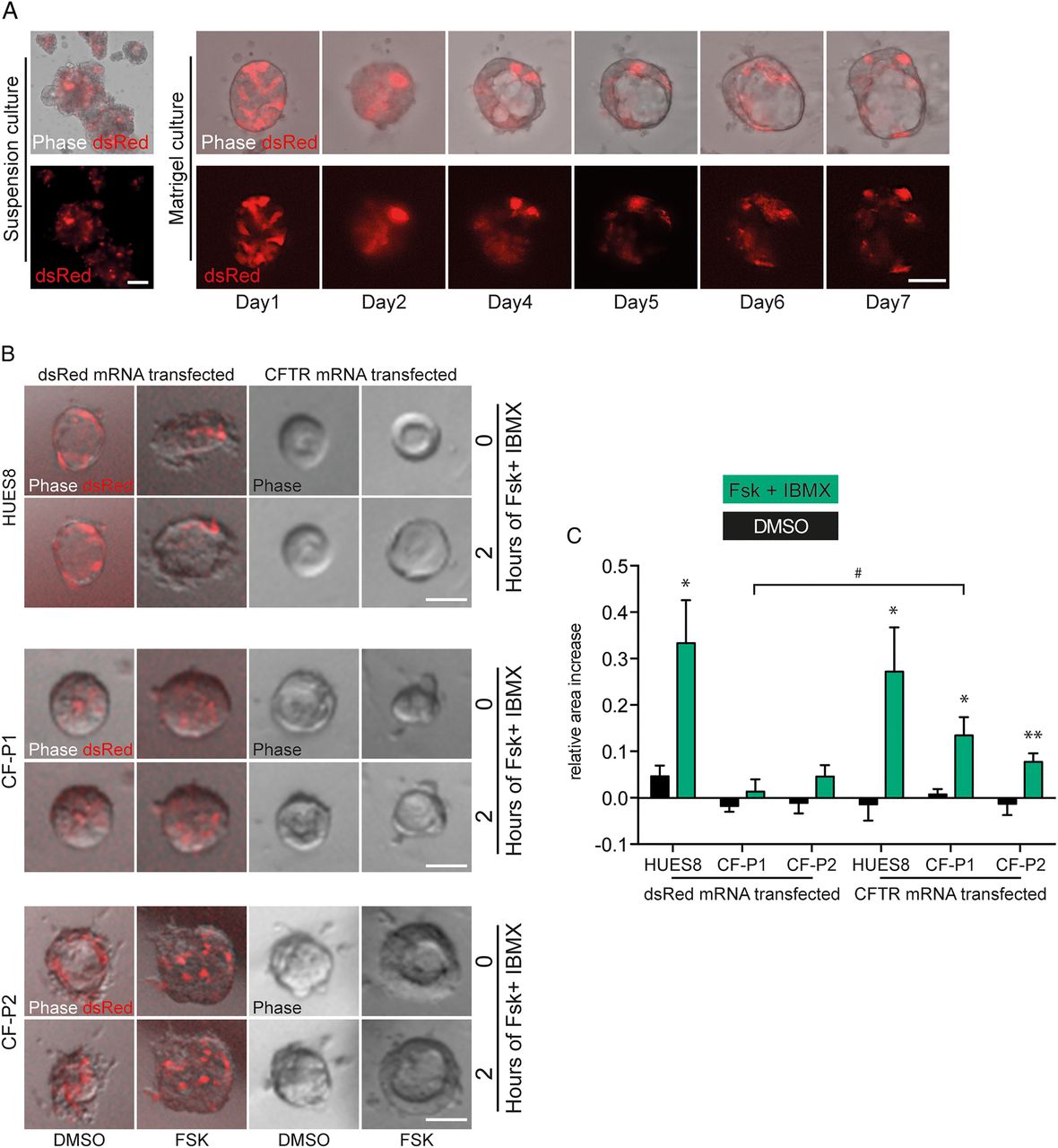
mRNA-mediated gene transfer in pancreatic organoids. (A) Representative image of pancreatic organoids (POs) in suspension culture 7 days after transfection with a specifically modified dsRed mRNA. Right, indicates subsequent development of a similarly transfected organoid in matrigel culture over time. (B and C) Rescue of defective cystic fibrosis transmembrane conductance regulator (CFTR) function in CF-human-induced pluripotent stem cell (iPSC)-derived pancreatic organoids as assessed by the forskolin-induced swelling assay 4 days after transfection with a CFTR mRNA with similar modifications as tested in (A). (B) Images of POs before and after 2 hours treatment with forskolin and IBMX and (C) corresponding quantification in indicated genotypes. Values represent mean±SEM of 12 organoids out of 3 independent wells analysed per condition. Asterisks denote significant difference between FSK/IBMX and DMSO treatment. Hash indicates effect of CFTR compared with dsRed-mRNA treatment. Statistical significance was determined by two-tailed t-test, */#p<0.05, **p<0.01. Scale bars 50 µm.
POs resemble human fetal pancreas upon orthotopic transplantation in mice
Finally, we aimed for a xenotransplantation approach in an attempt to generate a humanised animal model for pancreatic disease. Upon orthotopic PO transplantation into the murine pancreas (schematic outline in figure 7A), large grafts of human tissue readily visible with the naked eye could be observed. Human origin of the grafts was visualised with human-specific CK8 staining (figure 7B). Trilineage differentiation potential of PP cells, which gave rise to POs, is shown by immunohistochemistry for AMY, CK19 and insulin (figure 7B). Overall, morphology and graft size in H&E staining between WT and CF-derived organoids did not differ (figure 7B). Consequently, detailed immunophenotyping of the grafts was performed. In line with morphological appearance, acinar-like cells stained positive for several acinar marker genes such as AMY and chymotrypsin C, while ductal structures expressed CK19 (figure 7C, D and online supplementary figure S10A). To assess the maturity of the grafts, co-staining for acinar and ductal lineage markers with various progenitor cell markers such as SOX9,43 PDX144 and NKX6.111 was performed. Most acinar and ductal cells still stained positive for SOX9,43 while only acinar cells showed remaining PDX1 positivity.44 Vice versa, we noted a mutual exclusive expression pattern for NKX6.1 and acinar markers such as AMY, indicating that these cells lost their acinar potency as described previously.11 This observation fits well with the data obtained in lineage tracing studies done in mice11 ,43 ,44 and would indicate a developmental stage of a human Carnegie Stage CS20 to CS23 (figure 7D and online supplementary figure S10B). Staining for α-smooth muscle actin (α-SMA) also revealed signs of neovascularisation in the grafts (figure 7C). Similar to in vitro cultured organoids, apoptotic cells were hardly detectable, while Ki67-positive cells could be frequently found in both genotypes (figure 7B and online supplementary figure S10C). As expected, CFTR staining was more diffuse in CF grafts compared with the luminal, membranous pattern in WT grafts (figure 7E, F). Overall, there were no observable differences in the expression of the investigated developmental markers between WT and CF-derived grafts at least based on the performed semi-quantitative analyses in the not fully maturated grafts. In summary, our novel PO platform exceeds previous findings obtained in zebrafish45 that the perinatal CF phenotype results from functional CFTR defects due to defective fluid secretion and not from defective pancreatic development. Further maturation time of the grafts in vivo is required to provide additional data.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Orthotopic organoid transplantation in mice. (A) Schematic presentation of experimental set-up and pictures of pancreas tissue explanted 5 weeks after injection of pancreatic organoids (POs) into the pancreas of mice (left: overview, right: zoom-in). Dotted line represents putative graft. (B) Immunohistochemical analysis of orthotopic pancreas grafts. Indicated markers show trilineage potential and proliferative capacity of the transplanted POs (amylase-acinar, cytokeratin 19 (CK19)-ductal, insulin-endocrine, Ki67-proliferation). Genotype and markers as indicated. Human-specific cytokeratin 8 (CK8) confirms human origin. Red arrowheads mark few endocrine cells being insulin positive. H&E, scale bars 400 µm, immunohistochemical images (human CK8, amylase, CK19, insulin, Ki67, scale bars 100 µm). (C–F) Immunofluorescence analysis of orthotopic pancreas grafts. Genotype and markers as indicated. (C) Human-specific cytokeratin 8 (CK8) confirms human origin. α-smooth muscle actin (α-SMA) marks myofibroblasts indicating putative vessel formation around exocrine structures. SOX9/CK19 co-labelling marks ductal structures (D) PDX1 signal in CPA1-positive acinar structures indicates remaining progenitor marker activity in this lineage as known in mice.44 The majority of present NKX6.1-positive cells (arrowheads) stain negative for amylase (AMY), indicating that the acinar lineage has lost this marker as expected from developmental studies in mice.11 (E and F) Apical cystic fibrosis transmembrane conductance regulator (CFTR) expression (arrowhead) in ductal structures derived from grafted (E) wild-type organoids is lost in (F) cystic fibrosis (CF) (asterisk marks the lumen). Dashed lines indicate graft/host tissue border. Transplantation experiments were performed with pancreatic organoids (d21) generated from HUES8 (wild-type) or CF-P1 iPS cells (CF) in FN-based suspension culture. Scale bars: 40 µm.
Discussion
Progress to generate acinar/ductal cells from multipotent PPs remains slow in light of a recently published study investigating cancer organoids derived from PSC.14 Our study represents progress to the field in terms of providing a new disease-modelling platform. We present a serum-free and a largely small-molecule-based approach to derive pancreatic acinar-like and duct-like cells using a cost-effective 3D organoid culture system. These organoids faithfully represent human pancreas on a global gene expression level and upon orthotopic transplantation into immune-deficient mice. The stepwise commitment process in generating organoids complemented with subsequent maturation in vivo can pinpoint developmental defects to a distinct time frame and therefore may facilitate the study of pancreatic diseases in humanised mice as shown here for CF. The impressive success of graft implantation could be further improved by repetitive transplantations at various sites of the pancreas, although it remains unclear whether human pancreatic progeny will also functionally integrate within the murine host. In that sense, it is necessary to further define maturity and purity of PO-derived grafts in the future. This is indispensable for the modelling of a less-well-characterised disease.
PO cultures derived from primary cancer specimens have been shown to be a superior model for pancreatic cancer.46 These organoids recapitulate the pancreatic niche extremely well, but exhibit a limited take rate in culture after endoscopic fine-needle biopsies and are strongly dependent on a set of highly expensive growth factors whose omission leads to culture deterioration.46 ,47 Still, techniques to derive and culture organoids obtained from human adult epithelium are far more developed and better characterised than PSC-derived organoids and have unclosed novel avenues for translational research as shown recently in a rectal biopsy-derived organoid screen for CF drugs.48 However, organoids generated from PSCs require less growth factor supplementation most likely due to the fact that the multipotent nature of PPs together with incomplete purity allows for the generation of a self-organising niche producing the required factors. Indeed, similar observations have been made when intestinal organoids were derived from either PSCs or tissue biopsies.49 ,50 Heterogeneous pancreatic commitment of individual organoids also provides room for further improvement. While we noted virtually pure POs, structures containing non-pancreatic progeny were also observed in our cultures. During further culture, obvious signs of extra-pancreatic lineage differentiation such as outgrowing neurons (data not shown) became apparent. This is most likely due to incomplete purity at the PP stage underlining the necessity to further tailor this crucial step.
To provide proof of concept of the utility of this model for studying pancreatic diseases, we decided to study pancreatic facets of CF in a physiologically relevant environment. In vivo, CFTR expression can be detected early on in the human blastocyst stage51 while CF phenotypes become evident perinatally. Current animal models lack the possibility to investigate whether mutated CFTR may affect stepwise commitment to a mature pancreatic gland. Here, we challenge this hypothesis as pancreatic development in CF seems to occur uninterrupted in vitro and in vivo at least when there is no functional activity of the pancreas triggered by nutrients. This observation is supported by a recent study in zebrafish.45 Of note, our marker-based characterisation allots a Carnegie Stage 20-23 to our grafts, a differentiation stage still far away from fully matured pancreas.
Various CFTR correctors and activators are about to enter the clinic (https://clinicaltrials.gov/ct2/); however, randomised controlled clinical trials are hindered by the imbalance between the high number of compounds and genetically matched patients, creating a demand for a personalised ex vivo drug testing system. To our knowledge, all current drugs have been primarily designed to rescue the lung phenotype of CF while pancreatic phenotypes of CF have not been explored due to the lack of access to bona fide pancreatic cells and premature patient death caused by the lung disease. Our reprogramming system using plucked human hair as a cell source allows the fast and efficient generation of CF-patient-specific iPSCs.38 ,39 ,52 In turn, subsequent differentiation towards POs would eventually allow preclinical and individualised drug testing to evaluate whether a patient qualifies for a particular therapy or not. Proof of concept has been recently given.48 Focusing on a curative approach, gene therapy is a relevant topic in CF and other inherited pancreatic diseases, but this approach is limited by possible off-target effects and a residual integration risk. The application of nucleotide cmRNA, however, circumvents these threats and further ensures high stability, thus representing a promising therapeutic tool. Previous work by our group and others has shown that delivery of cmRNA leads to therapeutic levels of protein expression as a result of high gene transfer efficiency, higher stability and/or low immunogenicity, and hence, can even be used for life-saving genome editing in vivo. Here, we complement POs with cmRNA-based CFTR gene supplementation as an ex vivo test tool for various mRNA modifications in an organ-specific context.
In summary, we provide a patient-specific platform to study pancreatic development in a human context. Moreover, it provides the ability to mimic, investigate and rescue pancreatic phenotypes of inherited pancreatic disease in a dish but also in a xenotransplantation assay generating human pancreas in mice.
Acknowledgments
The authors thank Ralf Köhntop, Stefanie Fischer, Sabine Conrad and Claudia Ruhland for excellent technical assistance and Ronan Russell for helpful discussions. They also thank Paul Walther and the Electron Microscopy Group of Materials Science at the University of Ulm. The authors acknowledge Madsen, O.D. and the Developmental Studies Hybridoma Bank (DSHB) for providing the NKX6.1 antibody.
References
Footnotes
↵SL and AK jointly supervised this work and contributed equally.
Correction notice This article has been corrected since it published Online First. The funding section has been updated.
Contributors SL and AK contributed equally to this article. Study concept and design: AK, AI and SL. Acquisition of data: JSA, PCH, BS, MW, AL, JM, MS, C-CK, IGC, LP, MB, MH, TM, MS, SR, MZenker, ML, MM and JR. Analysis and interpretation of data: MH, MW, MK, SL, AK, AI and LP. Drafting of the manuscript: AK, AI, MH and SL. Critical revision of the manuscript for important intellectual content: MZenke, MH, AK, AI, TS, SL and MW. Study supervision: AK, AI and SL.
Funding This study was funded by the Deutsche Forschungsgemeinschaft (DFG, K.L. 2544/1-1 and 1-2), the Forschungskern SyStaR to AK, BIU (Böhringer Ingelheim Ulm to AK), the Fritz-Thyssen Foundation (Az. 10.15.2.040), the German Cancer Aid (111879) and the Else-Kröner-Fresenius-Stiftung (2011_A200). AK is indebted to the Baden-Württemberg Stiftung for the financial support of this research project by the Eliteprogramme for Postdocs. AK is also an Else-Kröner-Fresenius Memorial Fellow. LP is supported by a research fellowship of the Else-Kröner-Fresenius-Stiftung. MH was supported by the International Graduate School in Molecular Medicine and the Bausteinprogramme (L.SBN. 110), Ulm University. MM is supported by a grant of Ulm University (Baustein for Senior Clinician Scientists). IGC is funded by the Interdisciplinary Center for Clinical Research (IZKF Aachen) and Start Program, RWTH Aachen University Medical School, Aachen, Germany. AK was supported by an UEG Top Abstract Prize awarded at the United European Gastroenterology Week 2015.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Local ethics committee at Ulm University.
Provenance and peer review Not commissioned; externally peer reviewed.