Article Text

Abstract
Objective Patients with IBD display substantial heterogeneity in clinical characteristics. We hypothesise that individual differences in the complex interaction of the host genome and the gut microbiota can explain the onset and the heterogeneous presentation of IBD. Therefore, we performed a case–control analysis of the gut microbiota, the host genome and the clinical phenotypes of IBD.
Design Stool samples, peripheral blood and extensive phenotype data were collected from 313 patients with IBD and 582 truly healthy controls, selected from a population cohort. The gut microbiota composition was assessed by tag-sequencing the 16S rRNA gene. All participants were genotyped. We composed genetic risk scores from 11 functional genetic variants proven to be associated with IBD in genes that are directly involved in the bacterial handling in the gut: NOD2, CARD9, ATG16L1, IRGM and FUT2.
Results Strikingly, we observed significant alterations of the gut microbiota of healthy individuals with a high genetic risk for IBD: the IBD genetic risk score was significantly associated with a decrease in the genus Roseburia in healthy controls (false discovery rate 0.017). Moreover, disease location was a major determinant of the gut microbiota: the gut microbiota of patients with colonic Crohn's disease (CD) is different from that of patients with ileal CD, with a decrease in alpha diversity associated to ileal disease (p=3.28×10−13).
Conclusions We show for the first time that genetic risk variants associated with IBD influence the gut microbiota in healthy individuals. Roseburia spp are acetate-to-butyrate converters, and a decrease has already been observed in patients with IBD.
- INFLAMMATORY BOWEL DISEASE
- GENETICS
- BACTERIAL INTERACTIONS
- INTESTINAL BACTERIA
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The gut microbiota plays a key role in the pathogenesis of IBD.
Known and presumed epidemiological risk factors for developing IBD such as mode of birth, breast feeding, smoking, hygiene, infections, antibiotics, diet and stress are all known to cause gut microbial perturbations.
The large heterogeneity between patients with IBD is likely to result from individual differences in the complex interaction between the host genome and the gut microbiota.
Discovering gene–microbiota interactions is difficult due to the large number of genomic markers as well as microbial taxa, requiring stringent multiple testing corrections.
What are the new findings?
Gut microbial changes could precede the onset of IBD. A high IBD genetic risk score is associated with a decrease in the genus Roseburia in the gut microbiota of healthy controls without gut complaints.
Disease localisation is a major determinant of the IBD-associated gut microbiota composition.
The use of a large well-phenotyped healthy control cohort next to an IBD cohort leads to an improved list of IBD-associated gut microbial differences.
How might it impact on clinical practice in the foreseeable future?
Better understanding of gene–microbiota interactions and proinflammatory gut microbial changes that precede the onset of IBD can lead to new IBD therapeutics, and perhaps even microbial prevention strategies.
Background and aims
IBD, comprising Crohn's disease (CD) and ulcerative colitis (UC), is a chronic inflammatory disorder of the GI tract. In CD, inflammation can occur throughout the GI tract, whereas in UC, inflammation is confined to the mucosal layer of the colon. The clinical characteristics of IBD vary greatly between individuals with respect to disease location, disease activity and disease behaviour. The origin of this heterogeneous clinical presentation remains poorly understood.1 ,2
The pathogenesis of IBD consists of an exaggerated immune response in a genetically susceptible host to the luminal microbial content of the gut. Driven by rapidly evolving genotyping and next-generation sequencing technologies, tremendous progress has been made in deciphering the host genomic landscape of IBD.3 ,4 Systems biology approaches to genomic and biological data clearly show the importance of the interaction between the host genome and the microbial exposure in the gut.5 Moreover, known and presumed epidemiological risk factors for developing IBD such as mode of birth (vaginal vs caesarean section), breast feeding, smoking, hygiene, infections, antibiotics, diet, stress and sleep pattern are all known to cause microbial perturbations, suggesting a key role for the gut microbiota in the pathogenesis of IBD.6–9
Previous studies have shown a reduced biodiversity in the gut microbial composition of patients with IBD, characterised by a reduction of known beneficial bacteria, such as Faecalibacterium prausnitzii, Roseburia intestinalis and other butyrate producers, and an increase of pathogens or pathobionts, for example, adherent-invasive Escherichia coli and Shigella species of the Enterobacteriaceae family. However, these studies used a relatively small number of controls, who were usually selected from the patient population of the gastroenterology department after excluding those with IBD.10 Because recent gut microbiome research has shown significant effects of stool consistency and functional complaints on the gut microbiota,11–13 previous results could have been influenced by their method of selection of controls.
While the main composition of the gut microbiota in CD has been studied extensively, the composition of the gut microbiota in patients with UC has received less attention.10 ,14 ,15 Furthermore, the relationship between the gut microbiota and the clinical characteristics of IBD, including disease activity, disease duration and disease behaviour has only been studied in an exploratory manner.
Recent studies have begun to unravel the complex interaction of host genetics and the gut microbiota. These links between specific genetic variants and the abundance of specific bacteria are called microbiota quantitative trait loci (microbiotaQTLs). Twin studies show that the abundances of bacterial families Ruminococcaceae and Lachnospiraceae containing butyrate producers and acetate-to-butyrate converters are, to a certain degree, heritable.16–18 Animal studies in mice specifically designed to discover microbiotaQTLs show the influence of genomic loci on several microbial genera.19 Moreover, gut microbiota similarities in twins both concordant and discordant for IBD have been shown in several studies, further suggesting that host genetics can influence the gut microbiota.20–22 Furthermore, preliminary data show that specific variants of the NOD2 gene are associated with changes in the abundance of the Enterobacteriaceae family in patients with IBD.23
We hypothesise that the large heterogeneity between patients with IBD is likely to result from individual differences in the complex interaction between the host genome and the gut microbiota. Therefore, improving our knowledge of this interaction is crucial for our understanding of the pathogenesis of IBD.14 So far, very few studies have been able to elucidate this interaction in an integrated manner. Here, we present a large single-centre case–control analysis of the luminal gut microbiota, the host genetics and clinical phenotypes of both CD and UC. To ensure optimal data quality, we adopted a rigorously standardised approach to collect and process fresh frozen faecal samples of 313 patients with IBD from a single hospital in the north of the Netherlands and 582 truly healthy controls from the same geographical area. For all individuals, extensive clinical data, laboratory and endoscopic findings were collected. In addition, host genomic risk variants and risk scores were obtained in both the patients with IBD and the healthy controls to analyse host genomic influences on the gut microbial composition.
Methods
Cohorts
In total, 357 patients with IBD were recruited from the specialised IBD outpatient clinic at the Department of Gastroenterology and Hepatology of the University Medical Center Groningen (UMCG) in Groningen, the Netherlands. All patients with IBD were diagnosed based on accepted radiological, endoscopic and histopathological evaluation. We excluded 44 patients with IBD who had a stoma, pouch or short bowel syndrome from further analyses. Healthy controls were selected from the 1174 participants of LifeLines DEEP, a cross-sectional general population cohort in the northern provinces of the Netherlands.24 Data about medical history, medication use and gut complaints were meticulously reviewed by a medical doctor to ensure controls did not have any severe gut complaints or diseases, and did not use any medication that could confound our analysis of the gut microbiota. The selection process is described in detail in the online supplementary appendix. Pseudonymised data from patients with IBD and healthy controls were provided to the researchers. This study was approved by the Institutional Review Board of the UMCG (IRB number 2008.338). All participants signed an informed consent form.
Supplementary appendix
Clinical characteristics and medication use of patients with IBD
Extensive data on clinical characteristics and medication use were available for all patients with IBD at the time of stool sampling. Pseudonymised data were retrieved from the IBD-specific electronic patient records of the IBD Center at the Department of Gastroenterology and Hepatology of the UMCG. Disease activity at the time of sampling was determined by standardised and accepted clinical activity scores: the Harvey–Bradshaw index (HBI) for patients with CD and the Simple Clinical Colitis Activity Index (SCCAI) score for patients with UC. C reactive protein (CRP) and faecal calprotectin measurements were also available as indicators of disease activity. Disease localisation and behaviour were described according to the Montreal classification. Disease duration was determined as date of stool sampling in the study minus the date of diagnosis. IBD treatment at the time of sampling was scored (mesalazine, steroids, thiopurines, methotrexate, tumour necrosis factor α (TNF-α) inhibitors and other biologicals) as well as the use of other medications: proton pump inhibitors (PPIs), antidiarrhoeal medication (loperamide), bile salts, iron, minerals and vitamins at the time of sampling, and antibiotics use within the previous 3 months. Extraintestinal manifestations and complications of IBD were scored in several categories: (1) eye; (2) mouth; (3) skin; (4) joints; (5) Other (details in online supplementary appendix).
Serological measurements for antineutrophil cytoplasmic antibodies and anti-Saccharomyces cerevisiae antibodies were determined by immunofluorescence. Information on mode of birth, breast feeding during infancy and self-reported diets (see online supplementary appendix) was collected through questionnaires.
The association between a phenotype and the gut microbiota was only analysed if there were five or more patients with IBD with that phenotype. A list of all phenotypes can be found in the online supplementary appendix.
Stool sample collection and faecal DNA extraction
Stool samples were collected for 313 cases with IBD and 582 controls. Identical protocols were used to collect and process all stool samples. All participants were asked to produce a stool sample at home. These were frozen by the participant within 15 min after stool production in the participant's home freezer. A research nurse visited each participant shortly after stool production to collect the sample on dry ice for transport to the UMCG at −80°C. Samples were subsequently stored at −80°C in the laboratory. All samples remained frozen until DNA isolation for which aliquots were made, and microbial DNA was isolated using the Qiagen AllPrep DNA/RNA Mini Kit cat # 80204 as previously described.10
Host genotyping, variant selection and genetic risk modelling
Host DNA was available for all patients with IBD and healthy controls. Host DNA was isolated from peripheral blood as previously described.25 Genotyping was performed using the Immunochip, an Illumina Infinium microarray comprising 196 524 single nucleotide variants (SNPs) and a small number of insertion/deletion markers, selected based on results from genome-wide association studies of 12 different immune-mediated diseases including IBD. Normalised intensities for all samples were called using the OptiCall clustering program.26 The genotype prediction was improved via stringent calling with BeagleCall using recommended settings.27 Marker and sample quality control were performed as previously described.3 Human leucocyte antigen (HLA) imputation was performed using SNP2HLA. The Type 1 Diabetes Genetics Consortium genotype data were used as a reference panel for imputation. The SNP2HLA imputes the classical HLA alleles and amino acid sequences within the major histocompatibility complex (MHC) region on chromosome 6.28
To overcome statistical problems inherent to multiple testing when combining both genome-wide and 16S rRNA microbiota data, we adopted an approach of analysing a set of selected SNPs based on (i) their involvement in IBD, (ii) their predicted functional consequences and (iii) their role in bacterial sensing and signalling in the gut.23
Eleven known IBD genetic risk variants were selected for our genome–microbiota interaction analyses. We selected these risk variants ensuring that the selected IBD risk SNPs (as identified in the International IBD Genetics Consortium Immunochip analysis or targeted resequencing studies) are functional variants or are in strong linkage disequilibrium with functional variants that are implicated in the interaction of the host with the gut microbiota.3 ,29 We included the following seven genetic variants in NOD2: rs104895431 (S431L), rs2066844 (R702W), rs5743277 (R703C), rs104895467 (N852S), rs2066845 (G908R), rs5743293 (fs1007insC) and rs104895444 (V793M). The variant rs10781499 in CARD9 was selected because Card9 has been shown to mediate intestinal epithelial cell restitution, T helper 17 responses and control of intestinal bacterial infection in mice.30 Two variants in FUT2, rs516246 and rs1047781, were selected because these variants have been shown to influence colonic mucosa-associated microbiota in CD.31 SNPs rs11741861 in IRGM and rs12994997 in ATG16L1 were included because of their role in decreased selective autophagy that results in altered cytokine signalling and decreased antibacterial defence.32 ,33
In addition to these 11 genetic variants, we also created risk scores for all 200 known IBD risk variants.3 ,5 We also analysed the influence of the HLA-DRB1*01:03 haplotype on the gut microbial composition in colonic disease, because this recently identified haplotype is associated with both UC and colonic CD and is suggested to be involved in appropriately controlling the immune response to colonic microbiota.34
Determining the gut microbial composition
Illumina MiSeq paired-end sequencing was used to determine the bacterial composition of the stool samples. Forward primer 515F [GTGCCAGCMGCCGCGGTAA] and reverse primer 806R [GGACTACHVGGGTWTCTAAT] of hypervariable region V4 of the 16S rRNA gene were used. Custom scripts were used to remove the primer sequences and align the paired-end reads.10
Operational taxonomic units: operational taxonomic unit-picking and filtering
The operational taxonomic unit (OTU) selection was performed using the QIIME reference optimal picking, using Usearch (V.7.0.1090) to perform the clustering at 97% of similarity. Greengenes V.13.8 was used as a reference database. In all, 12 556 OTUs were identified. Samples with less than 10 000 counts were removed. OTUs that were not present in at least 1% of our samples or with a low abundance (<0.01% of the total counts) were filtered out.
Function prediction
The functional imputation tools PICRUSt and HUMAnN were used to investigate the functional implications of the gut microbiota of patients with IBD. More information about the function prediction and the software can be found in the online supplementary appendix.
Statistical analysis
The richness and the β-diversity of the microbiota dataset were analysed using QIIME.35 The Shannon diversity index and the number of observed species per sample were used as α-diversity metrics. β-diversity was calculated using unweighted Unifrac distances and represented in Principal Coordinate Analyses (PCoA). The Wilcoxon test and Spearman correlations were used to identify differences in the Shannon index and the relations between the principal coordinates. χ2 tests, Fisher’s exact tests, Spearman correlations and Wilcoxon–Mann-Whitney tests (WMW tests) were used to determine the differences in the clinical characteristics of patients with IBD. QIIMETOMAASLIN was used to convert the OTU counts into relative taxonomical abundance. OTUs representing identical taxonomies were aggregated, and higher taxon levels were added when multiple OTUs represented that taxon. Due to the limitations of the resolution on taxonomical classification using 16S gene sequencing, we restricted our analysis to the genus level and above. The initial 12 556 OTUs were classified into 250 taxonomical levels.
We used MaAsLin to identify differentially abundant taxa and pathways: (1) between patients with IBD and healthy controls, (2) between different IBD phenotypes and (3) between individuals with diverse amounts of IBD genetic risk variants.15 MaAsLin performs boosted additive general linear models between metadata and microbial abundance data. The default settings of MaAsLin were used in all analyses. We used the Q-value package implemented in MaAsLin to correct for multiple testing. A false discovery rate (FDR) of 0.05 was used as the cut-off value for significance. The effect of the IBD diagnosis (CD or UC) on the gut microbiota composition was analysed by adding the IBD diagnosis versus healthy as a discrete predictor in the MaAsLin general linear mixed model analysis. Unweighted genetic risk scores were calculated for every participant by summing up the risk alleles of the above-mentioned SNPs (risk allele=1; IBD protective allele=0).25 Weighted genetic risk scores were calculated for every participant by summing up the log-normalised odds of the genetic variants of the same above-mentioned SNPs. Both risk scores were added as a predictor to the additive general linear model in MaAsLin. The analyses of the host genome and the microbiota composition were performed separately in patients with IBD and healthy controls.
Correction for factors influencing the gut microbiota
Parameters that potentially influence the gut microbiota were identified by statistical analysis of cohort phenotypes, univariate MaAsLin analyses and literature search, and subsequently added as cofactors to the additive linear model. In every analysis, the parameters age, gender, body mass index, read-depth, PPI use, antibiotics use and IBD medication (mesalazine, steroids, thiopurines, methotrexate and TNF-α inhibitors) were added as covariates. Stool consistency also affects the gut microbiota. However, since stool consistency, mainly the occurrence of diarrhoea, is a key characteristic of increased IBD disease activity, stool consistency was not used as a covariate in all models. However, stool consistency was incorporated in the analyses, since the clinical disease activity scores used––the HBI for CD and the SCCAI––take the number of liquid stools per day (in the HBI) and the number of bowel movements during the day and during the night (in the SCCAI) into account.
Results
The clinical characteristics of patients with IBD and the selection of healthy controls
The cohort consists of 313 patients with IBD (188 patients with CD, 107 patients with UC and 18 patients with IBD intermediate/IBD undetermined (IBDI/IBDU)) and 582 healthy controls selected from the population cohort LifeLines DEEP (selection criteria can be found in the online supplementary appendix).24 Patients with CD were younger than healthy controls (41.3 vs 45.9 years; p=1×10−4, WMW test), while patients with UC were not older than healthy controls (p=0.32, WMW test). At the time of sampling, 81 patients with IBD (25.8%) had active disease, defined as an HBI of higher than 4 in patients with CD or an SCCAI score higher than 2.5 in patients with UC. Of the patients with IBD, 23.7% had used antibiotics within the last 3 months. PPI use was more frequent in patients with IBD (24.5%) than in healthy controls (4.7%) (p<0.001, χ2 test). Extensive information on all clinical characteristics and medication use is presented in table 1.
Clinical characteristics of patients with IBD and healthy controls
Overall composition of the gut microbiota in patients with IBD and healthy controls
The predominant phyla in both patients with IBD and healthy controls were Firmicutes (73% in patients with IBD, 75% in healthy controls), Actinobacteria (9% in patients with IBD, 13% in healthy controls) and Bacterioidetes (14% in patients with IBD, 8% in healthy controls). Clostridia was the most abundant class (64% in patients with IBD, 68% in healthy controls). An overview of the abundances at all taxonomic levels can be found in online supplementary table S1.
Supplementary table S1
Alpha diversity
A statistically significant decrease in the Shannon index was observed in patients with IBD compared with healthy controls as depicted in online supplementary figure S1 (p=5.61×10−14, Wilcoxon test) and figure 1.

α-diversity (Shannon index) of the gut microbiota of healthy controls, patients with UC, patients with colonic Crohn's disease (CD), patients with ileocolonic CD and patients with ileal CD. α-diversity is not decreased in colonic disease (UC and colonic CD) compared with healthy controls. In contrast, in patients with ileal and ileocolonic CD, the α-diversity is statistically significantly decreased (patients with ileal CD vs healthy controls p=3.28×10−13 and patients with ileocolonic CD vs healthy controls p=3.11×10−11, Wilcoxon test).
Supplementary figure S1
Principal coordinate analysis
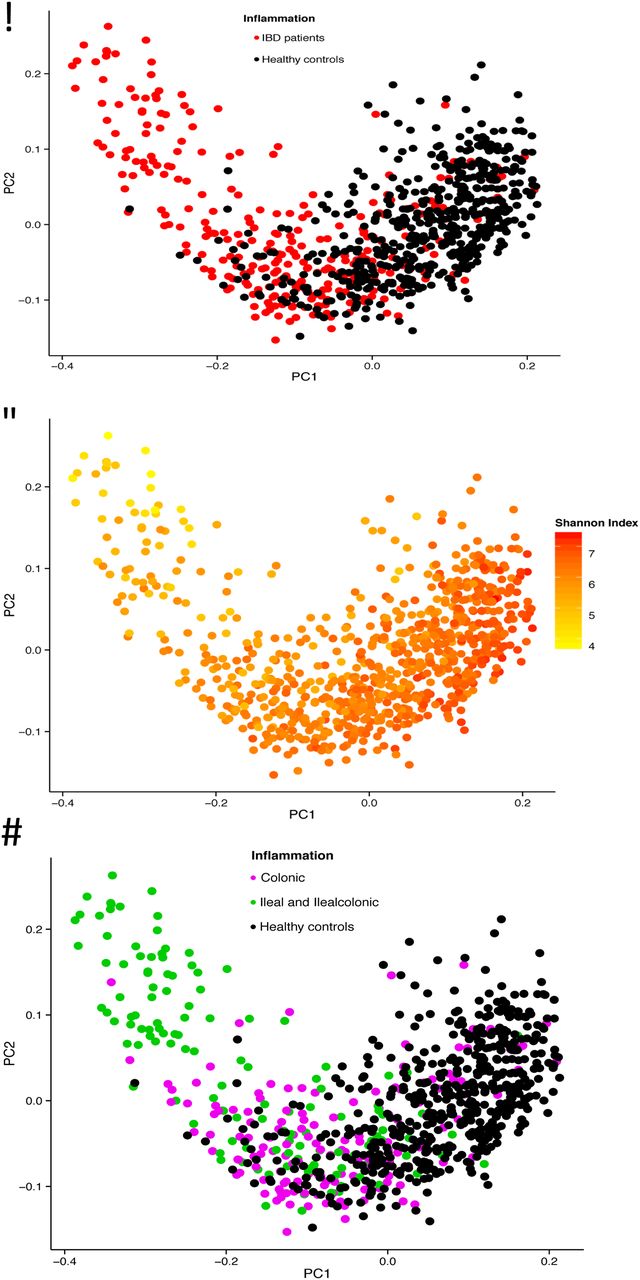
The differences in gut microbial composition between patients with IBD and healthy controls were also observed in the PCoA analysis. Statistically significant differences were found in the first three components (PCoA1 p=2.62×10−68, PCoA2 p=0.033, PCoA3 p=1.50×10−10, Wilcoxon test). The gut microbiota of healthy controls clustered together, while the gut microbiota of patients with IBD were more heterogeneous, partially overlapping the healthy controls. The shape of the PCoA plot is mainly explained by the disease location and the Shannon index (see results below), as depicted in figure 2A–D.

Principal coordinate analysis (PCoA) of stool samples of 313 patients with IBD and 582 healthy controls. (A) The gut microbiota of patients with IBD is different from the gut microbiota of healthy controls, with only partial overlap. (B) The first component is related to the Shannon index. (C and D) There is more overlap between colonic disease (UC and colonic Crohn's disease (CD) combined) and healthy controls than between ileal disease (ileal CD and ileocolonic CD combined) and healthy controls. The first component is related to disease location (PCoA1 r=0.63, p=7.39×10−91, Spearman correlation) and patients with colonic CD differ from patients with ileal CD (p=5.42×10−9).
IBD genetic risk variants are associated to unfavourable gut microbiota changes in healthy controls
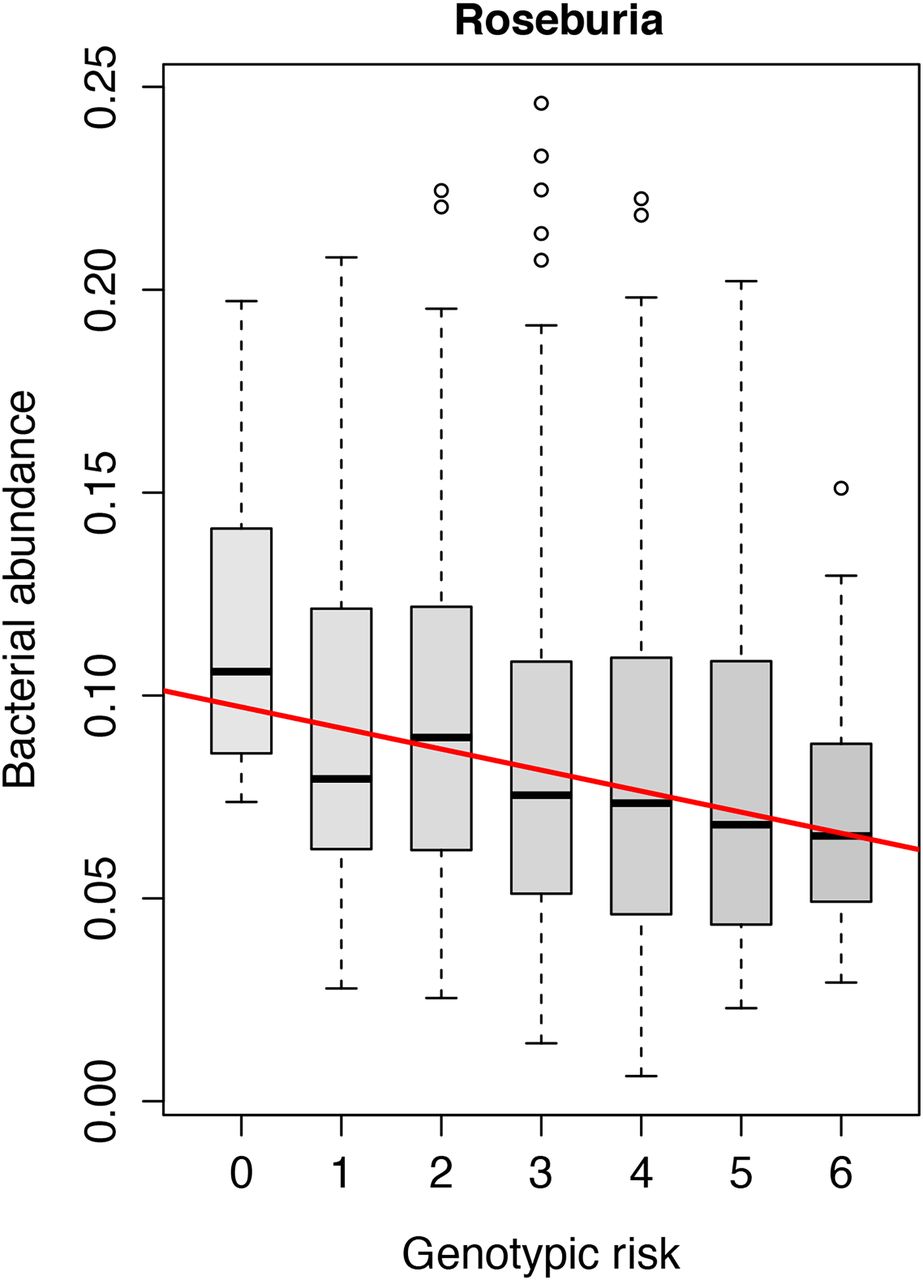
The role of 11 functional genomic variants associated to IBD in the genes NOD2, CARD9, ATG16L1, IRGM and FUT2 was investigated. In the unweighted analysis in healthy controls, a higher number of IBD risk alleles was associated with a decrease in the abundance of the genus Roseburia of the phylum Firmicutes (FDR=0.017) as depicted in figure 3. In patients with IBD as well as subsets of patients with IBD (patients with CD, patients with UC, patients with ileal CD, patients with ileocolonic CD and patients with colonic CD), neither the single genetic risk variants, the HLA-DRB1*01:03 haplotype, nor the weighted or unweighted composite scores of genetic risk alleles showed any statistically significant effect on the gut microbiota composition. All results of the analyses with the risk scores of 11 SNPs can be found in online supplementary table S2. Risk scores including all 200 IBD risk SNPs did not show any significant relations with the gut microbiota composition.
Supplementary table S2

Increased risk score of 11 IBD-related genetic variants in gut bacterial handling genes (NOD2, CARD9, IRGM, ATG16L1 and FUT2) is statistically significantly associated to decreased abundance of Roseburia spp. in healthy controls (false discovery rate=0.017).
Dysbiosis in patients with CD and UC: new associations
Crohn's disease
Compared with healthy controls, 69 taxa were statistically significantly altered in patients with CD (genus and above; 28%; FDR<0.05). These alterations are presented in table 2 and depicted in the cladogram in online supplementary figure S2A. The phyla Bacteroidetes (FDR=1.12×10−14) and Proteobacteria (FDR=2.71×10−22) were increased, while the phyla Actinobacteria (FDR=7.15×10−10) and Tenericutes (FDR=1.90×10−12) were decreased. Within the phylum Bacteroidetes, the order Bacteroidales was increased (FDR=1.12×10−14) as well as the genus Parabacteroides within the family Porphyromonadaceae (FDR=0.0016). Within the order Clostridiales of the phylum Firmicutes, seven families were decreased: Mogibacteriaceae, Christensenellaceae, Clostridiaceae, Dehalobacteriaceae, Peptococcaceae, Peptostreptococcaceae and Ruminococcaceae (FDR<0.05). The family Enterobacteriaceae of the phylum Proteobacteria, containing many known gut pathogens, was increased (FDR=0.0020). The genera Bifidobacterium, Ruminococcus and Faecalibacterium were also decreased in patients with CD (FDR=2.16×10−6, 4.70×10−5 and 7.82×10−23, respectively).
Supplementary figure S2
Comparison of altered taxa in patients with Crohn's disease (CD) compared with healthy controls: family level and above
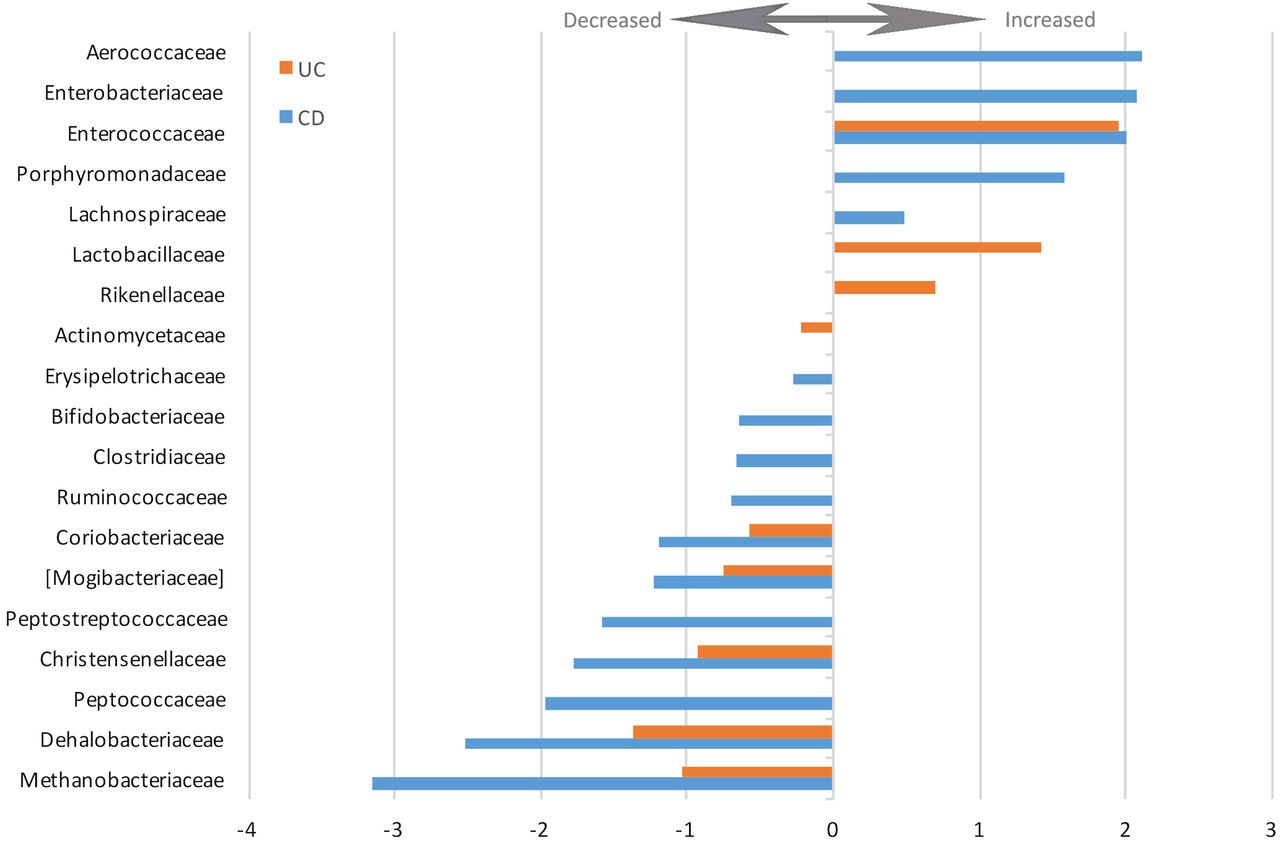
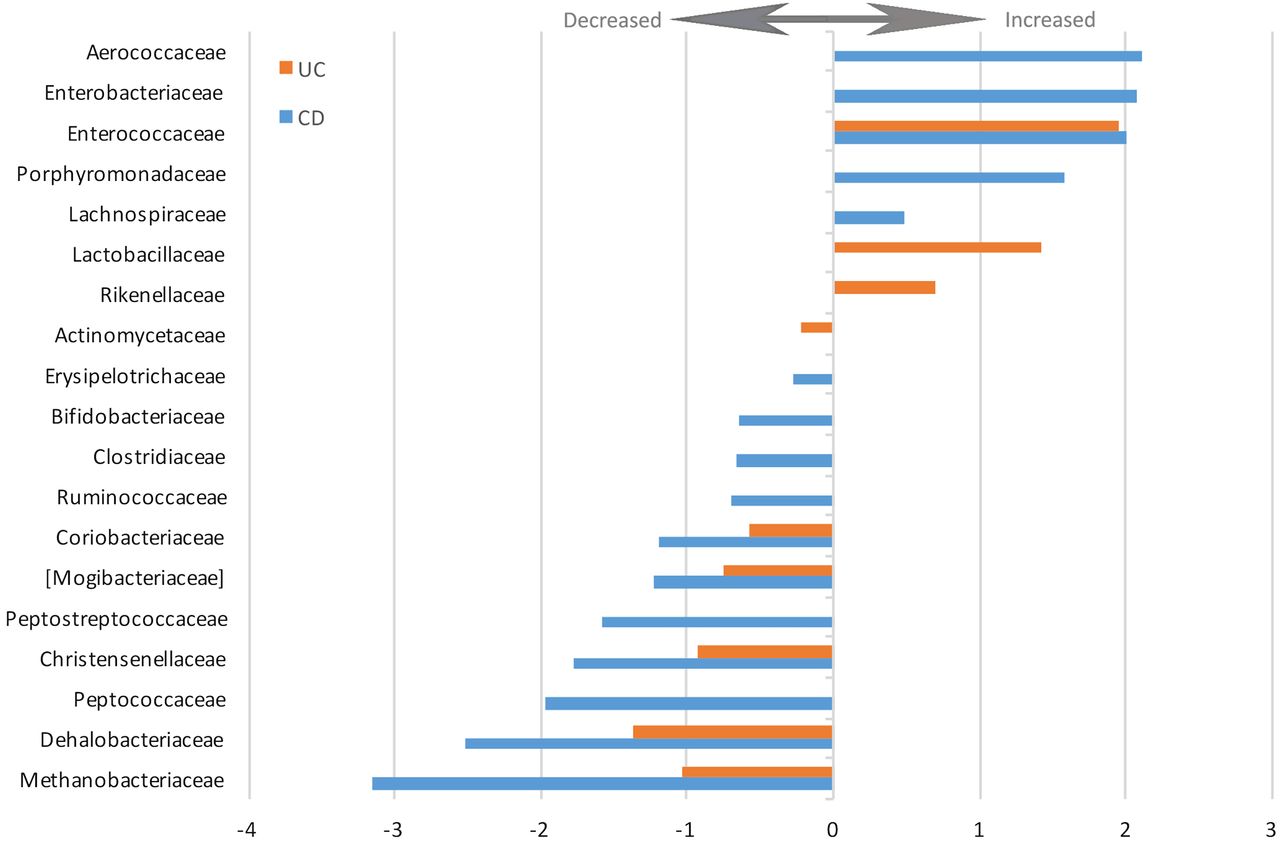
The changes in relative abundance of the statistically significantly altered families are depicted in figure 4. The complete list of increased and decreased taxa including direction, coefficient and FDR values is presented in online supplementary table S3.
Supplementary table S3
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Log2-fold change of increased and decreased bacterial families in patients with UC and Crohn's disease versus healthy controls (false discovery rate<0.05).
Ulcerative colitis
In patients with UC, 38 of the taxa were statistically significantly altered compared with healthy controls (genus and above; 12%; FDR<0.05). These alterations are presented in table 3 and depicted in a cladogram in online supplementary figure S2B. Similar to the patients with CD, the abundances of the phyla Bacteroidetes (FDR=8.87×10−13) and Proteobacteria (FDR=4.06×10−5) were increased, while the phylum Firmicutes (FDR=0.0079) was decreased in patients with UC. Within the phylum Bacteroidetes, the order Bacteroidales (FDR=8.87×10−13), the family Rikenellaceae (FDR=0.025) and the genus Bacteroides (FDR=1.72×10−18) were all increased compared with healthy controls. Lachnobacterium and Roseburia, genera in the order Clostridiales of the phylum Firmicutes, were also increased in patients with UC (FDR=0.023 and FDR=0.00056, respectively). The changes in relative abundance of the altered families are depicted in figure 4 (FDR<0.05). The complete list of increased and decreased taxa, including direction, coefficient and FDR values, is presented in online supplementary table S3.
Comparison of significant taxa associations in patients with UC: family level and above
Disease location is a major determinant of the gut microbiota in patients with IBD
The PCoA depicted in figure 2C shows the difference between the gut microbiota of patients with colonic disease (colonic CD and UC combined) and patients with ileal disease (ileal CD and ileocolonic CD combined). There is overlap between healthy controls and patients with colonic disease, while in concordance with the α-diversity analysis in figure 1, the gut microbiota of patients with ileal disease deviates more from healthy controls. The statistical analysis of the PCoA supports this result: the first component is related to disease location (PCoA1 r=0.63, p=7.39×10−91, Spearman correlation), and patients with colonic CD differ from patients with ileal CD (p=5.42×10−9). The α-diversity analysis shows similar results: the gut microbiota of patients with IBD with colonic disease is not statistically significantly decreased compared with healthy controls (Shannon index in patients with UC=6.41 vs Shannon index in healthy controls=6.50, p=0.06; Shannon index in patients with colonic CD=6.38 vs Shannon index in healthy controls=6.50, p=0.08, Wilcoxon test). On the contrary, patients with IBD with ileal disease show a statistically significant decrease in α-diversity (patients with ileal CD vs healthy controls p=3.28×10−13 and patients with ileocolonic CD vs healthy controls p=3.11×10−11, Wilcoxon test), as depicted in figure 1.
Whether the IBD genetic risk was associated with disease location was also tested. The genetic risk could not explain the disease location (colonic IBD vs ileal involved IBD; unweighted genetic risk score using 200 SNPs; Spearman correlation; r=0.045; p=0.47). The taxonomy analysis of disease location is presented in the online supplementary appendix.
Effects of IBD disease activity on the gut microbiota
We analysed several read-outs for disease activity at the time of sample collection: the clinical HBI scores for patients with CD and SCCAI scores for patients with UC, as well as CRP and faecal calprotectin level measurements for all patients with IBD. A higher HBI was associated with an increase of the family Enterobacteriaceae in patients with CD (FDR=0.036). No significant associations were found between the gut microbiota and the SSCAI in patients with UC. Neither CRP nor faecal calprotectin was statistically significantly associated with altered bacterial abundances in the gut. Details of the disease activity analyses can be found in online supplementary tables S4 and S5.
Supplementary table S4
Supplementary table S5
Effects of IBD disease duration on the gut microbiota
The disease duration in patients with IBD was measured from the date of diagnosis up to the date of sample collection. A longer duration of the disease, corrected for age, was associated with a higher abundance of the phylum Proteobacteria (FDR=0.045) (see online supplementary table S6).
Supplementary table S6
Analysis of other IBD subphenotypes
Other gut microbial associations with other IBD subphenotypes including medication, smoking behaviour and extraintestinal manifestations can be found in the Results section of the online supplementary appendix.
Pathway prediction and gut microbiota function changes in patients with IBD
Multiple metabolic pathways including butyrate metabolism, endotoxin metabolism and antibiotic resistance pathways were differentially expressed between patients with IBD, UC, CD, ileal CD, ileocolonic CD and colonic CD as compared with healthy controls. These altered Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways are presented in online supplementary figure S3 and table S7. The metabolism of short chain fatty acids was decreased in patients with IBD, as indicated by the decrease of the propanoate (also known as propionate) metabolism in patients with CD and UC (ko00640; CD: FDR=2.74×10−11 and UC: FDR=3.59×10−5), the decrease of the butanoate (also known as butyrate) metabolism in patients with CD (ko00650; FDR=5.31×10−9) and the decreased fatty acid metabolism in patients with CD (ko00071; FDR=4.28×10−18). Lipopolysaccharide or endotoxin biosynthesis was increased in both patients with CD and UC (ko00540; CD: FDR=4.69×10−7 and UC: FDR=0.027). β-lactam resistance metabolism was increased in patients with CD (ko00312; FDR=4.69×10−7). There were no significant pathway increases or decreases related to the clinical disease activity score, the HBI, for patients with CD (see online supplementary table S8). More detailed information on the predicted pathways can be found in the Results section of the online supplementary appendix.
Supplementary figure S3A
Supplementary figure S3B
Supplementary table S7
Supplementary table S8
Supplementary table S9
Supplementary table S10
Supplementary table S11
Supplementary table S12
Supplementary table S13
Supplementary table S14
Supplementary table S15
Supplementary table S16
Supplementary table S17
Conclusions
By performing this extensive integrated case–control analysis of the gut microbiota, the host genome and the clinical characteristics of IBD, we have identified new gut microbial associations with IBD and are now able to refine our understanding of the findings of previous studies. We found a relation between host genetic IBD susceptibility variants and the gut microbiota composition in healthy individuals and observed the effect of disease location on the gut microbiota. Moreover, we report microbial associations with multiple IBD subphenotypes.
Onset of IBD: genetic risk factors for IBD associated with proinflammatory gut microbiota alterations in healthy individuals
Discovering gene–microbiota interactions is difficult due to the large number of genomic markers as well as microbial taxa, requiring stringent multiple testing correction, thus limiting the possibility of finding statistically significant results. To resolve this issue, we created risk scores of known functional IBD risk variants proven to be involved in the bacterial handling in the gut. This hypothesis-based gene–microbiota approach limits the number of tests that need to be done and has proven to be successful.
The gut microbiota interacts with the intestinal epithelium and the host immune system.18 ,36–39 Recently, it was hypothesised that the interaction of the immune system with the gut microbiota goes two ways: ‘good’ gut microbiota can ameliorate immune responses, but the gut immune system can also ‘farm’ good bacteria in order to maintain immune–microbe homeostasis.36 ,37 We can show support for this hypothesis: in healthy individuals, an increased genetic burden in functional variants in genes involved in bacterial handling (NOD2, IRGM, ATG16L1, CARD9 and FUT2) is associated with a decrease of the acetate-to-butyrate converter Roseburia spp.
The species Roseburia intestinales is one of the 20 most abundant species in the gut microbiota.40 Importantly, a decrease in Roseburia spp. is already associated to the gut microbiota of patients with IBD.10 ,15 In an in vitro model, Roseburia spp. specifically colonised the mucins, which govern mucosal butyrate production.41 Butyrate derived from Clostridium clusters IV, VIII and XIVa to which Roseburia spp. belong has been shown to induce Treg cells, preventing or ameliorating intestinal inflammation.38 ,39 The abundances within the family Lachnospiraceae, to which Roseburia spp. belong, are significantly more similar in monozygotic twins than in dizygotic twins.17 Moreover, unaffected siblings of patients with CD share a decrease in Roseburia spp.22
This finding in healthy individuals carrying IBD genetic risk variants has implications for our understanding of the onset of IBD. We hypothesise that genetic risk factors of the gut immune system lead to ‘farming’ of a more proinflammatory gut microbiota and increased susceptibility to IBD. Subsequent unfavourable microbial perturbations due to environmental risk factors could further disturb the immune–microbe homeostasis in the gut, eventually leading to IBD.
In addition to our genetic risk score based on specific functions, analyses using genetic risk scores of all 200 known IBD susceptibility variants, many of whose function is unknown, did not yield any statistically significant results in either patients with IBD or in healthy controls. We could not detect any gene–microbiota interactions in patients with IBD, probably due to the already well-established dysbiosis as a consequence of the inflammation in the gut. Another complication is the interrelatedness of the genotypes and phenotypes in IBD. For example, NOD2 risk variants are known to be associated with ileal CD, and we show that ileal CD has a specific microbial signature. After correction for treatment, disease activity and disease location, we could not find any statistically significant genome–microbiota relations in patients with IBD.
Dysbiosis in patients with CD and UC: new associations identified, previous associations corrected
The dysbiosis of the gut microbiota in patients with IBD is profound: the abundances of 69 taxa in patients with CD and 38 taxa in patients with UC were altered compared with healthy individuals (FDR<0.05). We compared our results on the phylum, class, order and family levels with two previous studies looking into the gut microbiota of patients with IBD.10 ,15 ,20 This comparison is presented in tables 2 (patients with CD) and 3 (patients with UC). An important new finding of our study is the increase in the phylum Bacteroidetes in both patients with CD and UC. Increased levels of Bacteroidetes have recently been discovered in patients with IBS.13 Since the control groups used in the previous IBD studies also had functional GI complaints (ie, IBS), this would have confounded any comparisons between Bacteroidetes levels in patients with IBD and controls, masking any meaningful enrichment in IBD.
The genus Bacteroides within the phylum Bacteroidetes is increased in our patients with UC. The involvement of Bacteroides spp in the pathogenesis of IBD has been implied in animal studies. In NOD2 knockout mice, the exaggerated inflammatory response in the small intestine was dependent on Bacteroides vulgatus.42 Bacteroides thetaiotaomicron induced colitis in HLA-B27 transgenic rats.43 Another study looking into the effects of the vitamin D receptor in mice found increased levels of Bacteroides spp in colitis and increased levels of Bacteroides fragilis in colon biopsies of patients with UC.44
Increased abundance of the families Streptococcaceae, Micrococcaceae and Veillonellaceae, previously associated with IBD, is now associated to PPI use in our study. PPI use is overrepresented in patients with IBD.45 Since previous studies did not correct for PPI use, we assume that alterations in the abundances of these taxa were wrongly assigned to the effect of IBD.
Our study is the largest gut microbiota study in patients with UC to date, and within it we can now begin to resolve the landscape of the UC gut microbiota. We were able to find many new associations, including the association with a decreased abundance of phylum Tenericutes, which we also find to be associated with more extensive UC.
Disease location is a major determinant of the gut microbial composition in IBD
We showed the importance of disease location for the composition of the gut microbiota in patients with IBD. In our PCoA, the gut microbiota of patients with colonic CD is more similar to the microbiota of patients with UC than to that of patients with ileal CD. While different clusters of gut microbiota samples are also observed in recent IBD metagenomics research, we have been able to relate these clusters to the disease location phenotype.46 The importance of disease location also matches recent insights into host genetics, in which, based on genetic risk scores, colonic CD lies between UC and ileal CD.4 We found that the gut microbiota composition in stool could explain the differences in IBD disease location, while the genetic risk variants in our cohort could not. Moreover, there is important overlap in the clinical presentation of colonic CD and UC, for example, the risk of developing colorectal carcinoma in colonic CD is similar to UC, but different from ileal CD.47 Based on both the previous genetic findings and our current microbiota findings, it is becoming more apparent that colonic CD and ileal CD are different diseases within the IBD spectrum.
Through careful selection of healthy controls, meticulous standardisation of stool collection, extensive phenotyping and host genotyping, we were able to successfully perform analyses and gain insight into the gut microbiota as the key mediator of the IBD pathogenesis. For the first time, we find evidence for the role of the gut microbiota in the onset of IBD: healthy individuals with a high genetic risk load for IBD also have unfavourable changes in their gut microbiota. This relationship warrants further investigation as it might be both a potential target for treatment and a possibility for prevention of IBD in genetically susceptible hosts or their families.
Acknowledgments
The authors thank all the participants of the UR-IBD and Lifelines DEEP cohorts for contributing stool samples; Dianne Jansen, Jacqueline Mooibroek, Anneke Diekstra, Brecht Wedman, Rina Doorn, Astrid Maatman, Tiffany Poon, Wilma Westerhuis, Daan Wiersum, Debbie van Dussen, Martine Hesselink, Ettje Tigchelaar, Soesma A. Jankipersadsing, Maria Carmen Cenit and Jackie Dekens for logistics support, laboratory support, data collection and data management; the research group of Morris Swertz for providing the high-performance computing infrastructure including the Calculon cluster computer; the Parelsnoer Institute for supporting the IBD biobank infrastructure; Timothy Tickle, Curtis Huttenhower, Alexandra Sirota, Chengwei Luo and Aleksander Kostic for their help in training the first and second authors; Marten Hofker and Eelke Brandsma for contributing to the scientific discussion. This article was edited for language and formatting by Kate McIntyre, Associate Scientific Editor in the Department of Genetics, University Medical Center Groningen.
References
Footnotes
FI and AVV are shared first authors.
AZ and RKW are shared last authors.
Contributors RKW, DG, AZ, GD, CH and RJX designed the study. FI, MCV, LMS, HMvD, RWFTS, GD and RKW collected the data. FI, AVV, MJB, RA and JF analysed the data. FI, AVV, EAMF and RKW drafted the manuscript. CW, JF, EAMF, LF, DG, AZ, GD, CH, RJX and RKW critically reviewed the manuscript.
Funding RKW, JF and LF are supported by VIDI grants (016.136.308, 864.13.013 and 917.14.374) from the Netherlands Organization for Scientific Research (NWO). EAMF is funded by a career development grant from the Dutch Digestive Foundation (MLDS) (No. CDG-014). Sequencing of the LifeLines deep cohort was funded by a Top Institute Food and Nutrition grant GH001 to CW. CW is further supported by an ERC advanced grant (ERC-671274). AZ holds a Rosalind Franklin fellowship (University of Groningen) and a CardioVasculair Onderzoek Nederland grant (CVON 2012-03).
Competing interests None declared.
Ethics approval Institutional Review Board of the University Medical Center Groningen.
Provenance and peer review Not commissioned; externally peer reviewed.