Article Text

Abstract
Objective Tofacitinib is an oral, small-molecule Janus kinase inhibitor that is being investigated for IBD. We evaluated the efficacy and safety of tofacitinib for induction and maintenance treatment in patients with moderate-to-severe Crohn's disease (CD).
Design We conducted two randomised, double-blind, placebo-controlled, multicentre phase IIb studies. Adult patients with moderate-to-severe CD were randomised to receive induction treatment with placebo, tofacitinib 5 or 10 mg twice daily for 8 weeks. Those achieving clinical response-100 or remission were re-randomised to maintenance treatment with placebo, tofacitinib 5 or 10 mg twice daily for 26 weeks. Primary endpoints were clinical remission at the end of the induction study, and clinical response-100 or remission at the end of the maintenance study.
Results 180/280 patients randomised in the induction study were enrolled in the maintenance study. At week 8 of induction, the proportion of patients with clinical remission was 43.5% and 43.0% with 5 and 10 mg twice daily, respectively, compared with 36.7% in the placebo group (p=0.325 and 0.392 for 5 and 10 mg twice daily vs placebo). At week 26 of maintenance, the proportion of patients with clinical response-100 or remission was 55.8% with tofacitinib 10 mg twice daily compared with 39.5% with tofacitinib 5 mg twice daily and 38.1% with placebo (p=0.130 for 10 mg twice daily vs placebo). Compared with placebo, the change in C-reactive protein from baseline was statistically significant (p<0.0001) with 10 mg twice daily after both induction and maintenance treatments.
Conclusions Primary efficacy endpoints were not significantly different from placebo, although there was evidence of a minor treatment effect. No new safety signals were observed for tofacitinib.
Trial registration numbers NCT01393626 and NCT01393899.
- CLINICAL TRIALS
- IMMUNOLOGY
- CROHN'S DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Crohn's disease (CD) is a relapsing IBD, which may lead to progressive bowel damage and patient disability.
The current treatment goal is to induce and maintain remission. A substantial proportion of patients with moderate-to-severe CD do not respond or lose response to currently available therapies over time, and novel therapeutic alternatives are required.
What are the new findings?
After 8 weeks of induction therapy, small treatment effects versus placebo in proportions of patients in remission were observed with tofacitinib 5 and 10 mg twice daily.
After 26 weeks of maintenance therapy, clinical response-100 or remission was observed in a higher proportion of patients receiving tofacitinib 10 mg twice daily than placebo, although the difference was not statistically significant.
A minor treatment effect with tofacitinib was demonstrated by secondary clinical endpoints and supported by changes in biomarkers.
No new safety signals were observed for tofacitinib compared with those seen previously in studies of other indications.
How might it impact on clinical practice in the foreseeable future?
Despite the non-significant results, the improvement in measures of disease activity observed in the current study supports the development of further studies investigating the efficacy and safety of Janus kinase inhibition for CD.
Introduction
Crohn's disease (CD) is an IBD that is often progressive, with a remitting/relapsing course leading to complications such as strictures or fistulae.1–3 The treatment goal is to induce and maintain remission, limit steroid exposure, induce mucosal healing and prevent complications that may lead to surgery or disability.4 ,5
Although biological therapies have been a major advance in the treatment of CD, there is a high incidence of non-response and/or loss of response over time.6 ,7 Currently available treatment options have not shown a convincing reduction in surgical rates.8 ,9 Additionally, safety issues are associated with both traditional and biologic therapies,10–12 and novel treatment options are clearly needed.
Tofacitinib is an oral, small-molecule Janus kinase (JAK) inhibitor that is being investigated for IBD. JAKs are involved in cytokine signal transduction via phosphorylation of transcription factors of the signal transducer and activator of transcription family.13 JAK inhibitors modulate signalling of several cytokine receptors at the same time, leading to systemic immunosuppression. Tofacitinib is a potent inhibitor of JAK1 and JAK3.14 A phase II study,15 and two identical phase III studies,16 demonstrated that patients with moderately to severely active UC receiving tofacitinib were more likely to achieve remission at 8 weeks than those receiving placebo. A previous 4-week phase II study, carried out in patients with moderate-to-severe CD, did not show efficacy for tofacitinib 1, 5 or 15 mg twice daily in inducing clinical response; however, a surprisingly high placebo response was observed.17
We now report phase IIb induction and maintenance randomised placebo-controlled trials to investigate the efficacy and safety of tofacitinib 5 and 10 mg twice daily for inducing and maintaining clinical remission in patients with moderate-to-severe CD.
Methods
Study design
Patients were enrolled into two sequential and integrated phase IIb, randomised, double-blind, placebo-controlled, parallel-group, dose-ranging, multicentre trials to evaluate the efficacy and safety of tofacitinib for induction (induction study) and maintenance (maintenance study) treatment in adults with moderately to severely active CD. The studies were conducted at 80 sites in 18 countries (see online supplementary file). In the induction study, eligible patients were initially randomised (3:2:2:4) to receive placebo, tofacitinib 5, 10 or 15 mg twice daily for 8 weeks. The unbalanced allocation ratios were determined based on trial simulations for the fitting of an Emax model for dose–response curve. After 16 patients were enrolled in the tofacitinib 15 mg twice daily group, the protocol was amended and enrolment in the tofacitinib 15 mg twice daily dose group was stopped to focus the tofacitinib CD development programme on the 5 and 10 mg twice daily dose levels. Subsequently, eligible patients were randomised (1:1:1) to receive placebo, tofacitinib 5 or 10 mg twice daily for 8 weeks (figure 1).
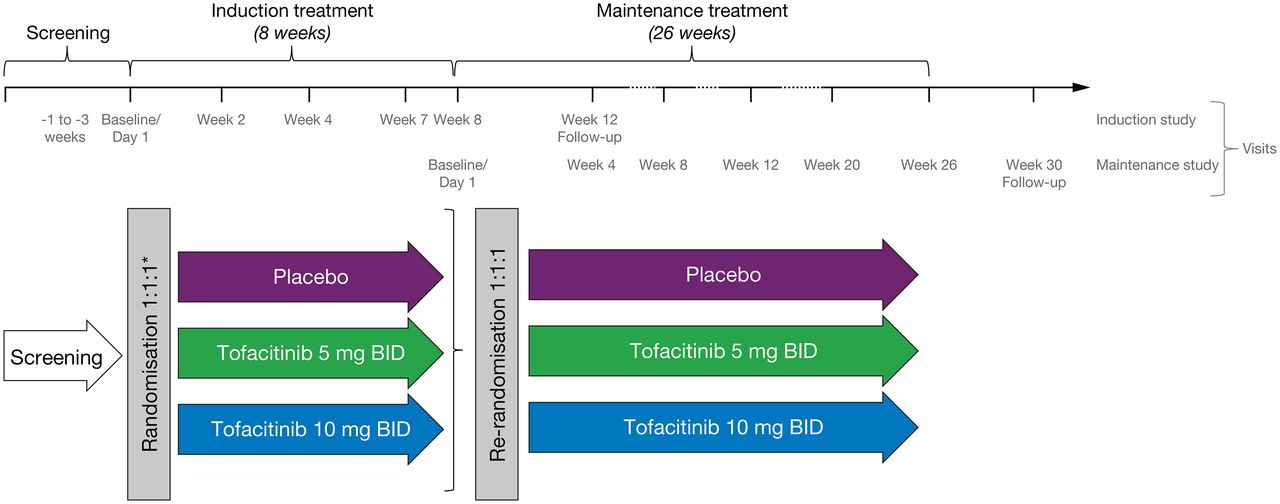
Study design. Patients who completed the 8-week induction study and achieved clinical response-100 (decrease in Crohn's disease activity index (CDAI) score at week 8 of at least 100 points from baseline) and/or clinical remission (CDAI <150) and met eligibility criteria, were potentially eligible to enter the 26-week maintenance study. Patients were followed up for 4 weeks after completion or early withdrawal of the induction or maintenance study. *Patients were initially randomised (3:2:2:4) to receive placebo, tofacitinib 5, 10 or 15 mg twice daily. A protocol amendment was implemented to stop enrolment in the tofacitinib 15 mg twice daily dose group after 16 patients were enrolled. BID, twice daily.
supplementary file
Patients receiving azathioprine, 6-mercaptopurine or methotrexate were required to stop these agents at least two weeks before randomisation. Concomitant tumour necrosis factor inhibitor (TNFi) were not allowed, and a wash-out period of at least eight 8 weeks prior to randomisation was required. Concomitant corticosteroids were allowed up to oral prednisone-equivalent 30 mg/day or budesonide 9 mg/day, provided a stable dose had been taken for at least two weeks prior to baseline. Patients were required to have completed the induction study and achieved a clinical response-100 (Crohn's disease activity index (CDAI) decrease from baseline ≥100 points) or clinical remission (CDAI <150 points) at week 8, regardless of whether they received active treatment or placebo, to be eligible to enter the maintenance study (subject to meeting other selection criteria; see online supplementary file).
In the maintenance study, patients were re-randomised (1:1:1) to receive treatment with placebo, tofacitinib 5 or 10 mg twice daily for 26 weeks. A mandatory steroid taper algorithm was applied for patients receiving oral corticosteroids, starting at baseline of the maintenance period with the dose decreased by 5 mg prednisolone-equivalent per week until the dose reached 20 mg/day and then by 2.5 mg/week to 10 mg/day. Further taper was at the discretion of the investigator. Patients who completed the double-blind treatment period or fulfilled criteria for treatment failure were potentially eligible to transfer to an open-label extension study. Patients who withdrew from the maintenance study early and did not fulfil treatment failure criteria, or who declined to participate in the extension study, were followed-up for 4 weeks after the end of their treatment and assessed for safety.
Study patients
In the induction study, eligible patients were adult patients with moderate-to-severe CD (CDAI ≥220 to ≤450, and intestinal ulceration documented by colonoscopy within six weeks prior to screening by the local practitioner). Patients had to have history of inadequate response or intolerance to at least one of the following: corticosteroids, azathioprine/6-mercaptopurine, methotrexate or TNFi. Patients with active (draining) fistulae or intra-abdominal or pelvic abscesses were excluded. Full lists of inclusion/exclusion criteria and permitted/prohibited concomitant therapies are provided in the online supplementary file.
Efficacy outcomes
Calculation of CDAI score was based on eDiary entries recorded by the subject over seven consecutive days prior to a particular visit. Efficacy endpoints were measured at baseline, weeks 2, 4 and 8 of the induction study, and baseline, weeks 4, 8, 12, 20 and 26 of the maintenance study (figure 1).
Induction study
The primary efficacy endpoint was clinical remission (CDAI <150) at week 8. Secondary endpoints included clinical remission at weeks 2 and 4; clinical response-70 (decrease in CDAI ≥70 from baseline), clinical response-100 (decrease in CDAI ≥100 from baseline) and clinical response-100 or clinical remission at weeks 2, 4 and 8. In addition, post hoc analyses were carried out to investigate patient-reported outcomes (PRO)2-75 (clinical remission defined as the sum of stool frequency score and abdominal pain score <75) and PRO3-80 (clinical remission defined as the sum of stool frequency score, abdominal pain score and general well-being score <80) outcomes at week 8.
Maintenance study
The primary endpoint was clinical response-100 (decrease in CDAI ≥100 from induction study baseline) or clinical remission at week 26. Secondary endpoints included clinical response-100 or clinical remission at weeks 4, 8, 12 and 20; clinical response-100 or clinical remission both at weeks 4, 8, 12, 20 and 26; sustained clinical remission and sustained clinical response at both weeks 20 and 26. Other secondary endpoints common to both studies included CDAI scores over time, serum C-reactive protein (CRP) and faecal calprotectin (FCP) levels over time.
Safety outcomes
Safety endpoints included incidence and severity of adverse events (AEs). Clinical laboratory parameters were monitored and events, confirmed by adjudication related to cardiovascular safety, opportunistic infections, malignancy, GI perforation and hepatic injury, were recorded. Malignancies were confirmed by central laboratory pathologist review of biopsies, when relevant biopsy specimens were available.
Sample size
The induction study aimed to randomise approximately 275 patients. Following removal of the tofacitinib 15 mg twice daily group from the study, it was estimated that at least 80 patients would be assigned to each of the three remaining groups, with the total planned number of patients remaining unchanged at 275. Under the assumption that the placebo remission rate was 19%, 80 patients per group would provide 80% power at a level of 0.05 to detect a 20% difference.
Based on the expected response proportions to tofacitinib and placebo at the end of the induction study, it was expected that approximately 90 patients from the tofacitinib groups would be re-randomised in the exploratory maintenance study. With 30 patients per group, and the assumption of 55% patients in the tofacitinib groups achieving remission or clinical response-100 at week 26, the half-width of the two-sided 80% CI of the primary endpoint in each tofacitinib group was <12%. With an additional assumption that the primary endpoint was 35% in the placebo group at week 26, the half-width of the two-sided 80% CI for the difference between each of the tofacitinib groups and the placebo group was <16.5%.
Randomisation and blinding
Assignment of subject identification number and study drug were managed by a tele-randomisation system, by which the subject was enrolled online or via a telephone call. At the baseline visit, provided all inclusion and exclusion criteria were met, the subject was randomised to trial medication. In the induction study, patients were stratified by whether or not they had previous exposure to TNFi. In the maintenance study, patients were stratified and randomised according to their treatment assignments in the induction study, and their clinical remission status at week 8 of the induction study. Study treatment was blinded to patients, investigators and the sponsor. Treatment randomisation information remained confidential and was not released to the investigator or study staff until the conclusion of the studies.
Statistical methods
In the induction study, the stratified Cochran-Mantel-Haenszel χ2 test was used for efficacy data to test the superiority of each dose of tofacitinib to placebo. Stratification was based on prior use of TNFi treatments at baseline. Two-sided 95% CIs for the difference between each dose of tofacitinib and placebo were calculated at weeks 2, 4 and 8, based on normal approximation to the binomial distribution. A total of 16 patients had been randomised to the tofacitinib 15 mg twice daily group, in the induction study, when this group was removed. Due to the small number of patients treated with tofacitinib 15 mg twice daily, the data for this group were not included for comparison with placebo in the efficacy analyses but were included in the safety analyses.
In the maintenance study, binary efficacy endpoints were descriptively summarised by dose group and study visit. Normal approximations were used to form the two-sided 80% CIs for the treatment difference and p values from two-sided Fisher's exact test for the comparison of each dose of tofacitinib versus placebo.
The continuous endpoints such as CDAI scores and biomarkers that were measured overtime were analysed using linear mixed-effects models that require no imputation of missing data. For binary efficacy endpoints derived from the CDAI score, patients with missing values were treated as non-responders or non-remitters in both studies. The changes from baseline for CDAI scores measured repeatedly over time were analysed using a linear mixed-effects model assuming an unstructured covariance matrix. Biomarker data were log-transformed (natural logarithm) and changes analysed using a linear mixed-effects model. The adjusted estimate for the difference between each tofacitinib dose and placebo, as well as the corresponding two-sided 95% CIs and p values, were reported. Descriptive summaries of CRP and FCP in the original scale were also presented by dose group for observed values and for change from baseline at each time point.
Analyses were performed for the full analysis set (FAS, all randomised patients who received at least one dose of study medication) of the induction study, and for the modified FAS (mFAS, excluding placebo responders from the induction study) population of the maintenance study. Safety endpoints were summarised for the safety analysis set, which included all patients who received at least one dose of study medication.
Results
Study patients
Induction study
Between October 2011 and March 2015, 567 patients were screened and 280 randomised in the induction study (figure 2). Most common reasons for exclusion included CDAI out of range, presence of fistulae/abscesses and laboratory parameters out of range.

{kind=link}
{kind=link}
Flow diagram.
Of the 92 patients allocated to the placebo group, 1 did not meet the entry criteria and did not receive the placebo study treatment. A total of 188 patients were allocated to the tofacitinib groups (5 mg twice daily, n=86; 10 mg twice daily, n=86; 15 mg twice daily, n=16), and all received their allocated treatment. A total of 236 patients completed the study; main reasons for discontinuation included insufficient clinical response (n=17) and AEs (n=8) (see online supplementary file for full details on discontinuations).
Maintenance study
Among patients who completed the induction study, 180 were re-randomised in the maintenance study conducted between March 2012 and July 2015, including 128 patients who had received tofacitinib in the induction study. All patients randomised in the maintenance study received their assigned treatments (placebo, n=59; tofacitinib 5 mg twice daily, n=60; tofacitinib 10 mg twice daily, n=61); 97 patients completed the study (placebo, n=27; tofacitinib 5 mg twice daily, n=32 and 10 mg twice daily, n=38). Main reasons for discontinuation from the maintenance study included insufficient clinical response (n=63), AEs (n=9) and patients’ request to withdraw from the study (n=5) (see online supplementary file for full details on discontinuations).
Patients' baseline demographics and disease characteristics were comparable between groups in both studies, with high proportions of previously TNFi-exposed patients across all treatment groups (table 1; see online supplementary table S1). The placebo group of the induction study had a numerically higher number of females (65.9%) compared with the tofacitinib groups (5 mg twice daily, 37.2%; 10 mg twice daily, 54.7%; 15 mg twice daily, 43.8%). In addition, although patients entering the maintenance study had a clinical response-100 or clinical remission, relatively high levels of CRP and FCP were observed in all treatment groups at maintenance study baseline (table 1; see online supplementary table S1).
Baseline demographics and disease characteristics
Efficacy outcomes
Clinical outcomes
Induction study
The observed proportions of patients with clinical remission at week 8 were 36.7% (95% CI 26.8% to 47.5%), 43.5% (95% CI 32.8% to 54.7%) and 43.0% (95% CI 32.4% to 54.2%) in the placebo, tofacitinib 5 and 10 mg twice daily groups, respectively) (table 2; see online supplementary table S3). Among TNFi-experienced patients, the observed proportions of those with clinical remission were 42.4% and 38.2% with tofacitinib 10 mg twice daily and 5 mg twice daily, respectively, and were not significantly different from placebo (36.2%). At the end of the induction phase (week 8) and compared with placebo, the observed proportions of clinical response-100 and -70 were 12–16% higher with tofacitinib 5 mg twice daily (p<0.05 for both endpoints) and 10 mg twice daily (not significant for either endpoint). Also at week 8, the proportions of patients receiving placebo, tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily achieving clinical remission or clinical response-100 were 55.6%, 71.8% and 69.8%, respectively; the difference between placebo and tofacitinib 5 mg twice daily was significant (p<0.05). Mean decreases from baseline in CDAI score at week 8 were significantly larger with both tofacitinib doses (both p<0.05) compared with placebo. Data from the small number of patients who received tofacitinib 15 mg twice daily are described in the online supplementary table S4.
Efficacy outcomes and biomarkers analyses
Post hoc analyses using alternative endpoints to measure remission showed significant differences in PRO2-75 with both tofacitinib 5 and 10 mg twice daily versus placebo, and in PRO3-80 for tofacitinib 5 mg twice daily versus placebo (table 2).
Maintenance study
The proportions of patients with clinical response-100 or remission at week 26 was 55.8% (80% CI 44.9% to 66.3%) with 10 mg twice daily vs 38.1% (80% CI 27.9% to 49.2% with placebo (p=0.130)) (table 2). The corresponding value for tofacitinib 5 mg twice daily was 39.5% (80% CI 29.4% to 50.5%; not significant vs placebo). In the TNFi-experienced subgroup, the observed proportion of patients with clinical response-100 or remission at week 26 was 48.6% with tofacitinib 10 mg twice daily versus 40.7% with placebo (not significant); 37.1% of patients receiving tofacitinib 5 mg twice daily achieved clinical response-100 or remission. Compared with placebo, the observed proportion of patients with clinical response-100 was 20.1% higher with tofacitinib 10 mg twice daily at week 26 (not significant). Adjusted estimates of change from baseline at week 26 in CDAI score were 69.5, 63.5 and 19.1 for placebo, tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily, respectively (not significant).
Biomarkers
Induction study
By week 8, mean decreases from baseline CRP concentration were significantly larger with tofacitinib 5 and 10 mg twice daily (p<0.001 and <0.0001, respectively) compared with placebo. Mean FCP decreases from baseline were not significantly higher with either dose of tofacitinib compared with placebo (table 2).
Maintenance study
At week 26 of the maintenance study, there was a significant difference in the change from baseline in CRP levels in patients treated with tofacitinib 10 mg twice daily (p<0.0001) compared with the placebo group; this resulted from an increase in level in the placebo group while levels in the tofacitinib 10 mg twice daily group remained relatively stable. In addition, mean changes in FCP from baseline with tofacitinib 5 and 10 mg twice daily were significantly different from placebo (p<0.05 and <0.0001, respectively) (table 2).
Safety outcomes
No deaths were reported in either study. The proportions of patients with treatment-emergent AEs were 60.4%, 58.1% and 60.5% in the induction placebo, tofacitinib 5 and 10 mg twice daily groups, respectively; and 74.6%, 83.3% and 78.7% in the maintenance placebo, tofacitinib 5 and 10 mg twice daily groups, respectively (table 3). Data from the small number of patients who received tofacitinib 15 mg twice daily are described in the online supplementary table S5. In both studies, the most common AEs were GI AEs and infections. Of the GI AEs, the most frequent events were nausea and flare of CD. The most common infections were nasopharyngitis and urinary tract infections.
Summary of treatment-emergent adverse events (all causalities)
The proportions of patients with serious AEs (SAEs) were numerically higher in the maintenance study (10.0–13.1%) compared with the induction study (3.3–11.6%). In both studies, these proportions were similar between the tofacitinib 5 mg twice daily and placebo groups (3.5% and 3.3%, respectively in the induction study; 10.0% and 11.9%, respectively, in the maintenance study), and numerically higher with tofacitinib 10 mg twice daily (11.6% and 13.1% in the induction and maintenance study, respectively) (table 3). The most common SAEs in the tofacitinib 10 mg twice daily groups were GI AEs and infections.
Two patients in each of the induction study treatment groups had serious infections (placebo: one cytomegalovirus infection and one perianal abscess; tofacitinib 5 mg twice daily: one abdominal abscess and one gastroenteritis; tofacitinib 10 mg twice daily: one perianal abscess and one pneumonia influenza) (table 3). Of these, one case (cytomegalovirus infection in the placebo group) was confirmed by adjudication as an opportunistic infection. One case of malignancy confirmed by adjudication (breast cancer, in the tofacitinib 10 mg twice daily treatment group) was reported in the induction study.
In the maintenance study, there were no reports of serious infections or special events of interest in the placebo group. Three and two patients in the tofacitinib 5 and 10 mg twice daily groups, respectively, had serious infections (tofacitinib 5 mg twice daily: one Clostridium difficile colitis, one C. difficile infection and one septic shock all in the same patient, and two perianal abscesses; tofacitinib 10 mg twice daily: one perianal abscess and one pneumonia influenza infection). No cases of opportunistic infections were confirmed by adjudication in the maintenance study. There was one case of large intestinal perforation confirmed by adjudication in the tofacitinib 5 mg twice daily group of the maintenance study and two cases of non-serious herpes zoster (tofacitinib 10 mg twice daily) were reported in the maintenance study. Finally, no cases of cardiovascular events were confirmed by adjudication in either study.
At week 8 of the induction study, larger increases in total cholesterol, high-density lipoprotein cholesterol and low-density lipoprotein cholesterol levels were observed with tofacitinib 5 and 10 mg twice daily (8.4% and 11.5% increase from baseline in total cholesterol, respectively) compared with placebo (3.4%). A 10.5% increase from baseline in triglyceride levels was reported in the placebo group at the end of the induction study compared with 1.1% and 0.1% in the tofacitinib 5 and 10 mg twice daily groups, respectively. At week 26 of the maintenance study, mean per cent changes from baseline in the placebo, tofacitinib 5 and 10 mg twice daily groups were −12.3%, 3.3% and 0.2%, respectively, for total cholesterol levels and −1.5%, 4.2% and 8.0%, respectively, for triglyceride levels.
Discussion
The proportion of patients in clinical remission (CDAI <150) after 8 weeks of induction therapy, with tofacitinib 5 and 10 mg twice daily, was not statistically significant compared with placebo. The proportion of patients achieving clinical remission or clinical response-100 at week 8 was only significant for tofacitinib 5 mg twice daily versus placebo. A modest but consistent treatment effect of tofacitinib was demonstrated for the secondary clinical endpoints of clinical response-70 and clinical response-100 at week 8. This treatment effect was supported by significantly greater reductions from baseline in CRP concentration with both doses of tofacitinib versus placebo although no significant reductions from baseline in FCP concentration were observed. In the subsequent 26-week maintenance study, the primary endpoint of clinical response-100 or clinical remission at week 26 was observed in a higher proportion of patients receiving tofacitinib 10 mg twice daily than placebo, although again the difference was not statistically significant. It should be noted, however, that this study was not powered for comparisons between treatment groups. Together with the results observed in patients with moderately to severely active UC in which the proportion of patients achieving remission with tofacitinib 10 mg twice daily was significantly higher compared with placebo,16 changes from baseline in the biomarkers CRP and FCP concentrations (secondary endpoints) supported a treatment effect with tofacitinib 10 mg twice daily in CD.
The proportion of patients in the placebo group achieving or maintaining clinical remission or clinical response-100 was substantially higher than that reported in other recent phase II or III studies assessing other agents in CD and using CDAI as the efficacy endpoint.18–25 This unexpectedly high placebo response prevented a thorough evaluation of the dose-response relationship of tofacitinib in inducing and maintaining response in clinical endpoints and biomarkers.
A number of factors could have contributed to the high placebo response observed in these studies. First, although there was a requirement for visible ulceration for inclusion, there was no requirement for a centrally read endoscopy at study entry, and no protocol-defined minimal requirement for the extent or severity of ulceration at baseline. There was also no protocol-defined threshold for FCP or CRP levels at baseline, as an objective marker of disease activity. A previous study has shown a greater treatment effect in patients with endoscopically confirmed ulcers and elevated CRP levels at baseline.11 A high proportion of patients (>30%) in the tofacitinib 5 and 10 mg twice daily and placebo groups were receiving corticosteroids at baseline. Although there was a requirement for a stable dose to have been received for at least two weeks prior to baseline and to be maintained during the induction trial, it is possible that the response seen in some patients receiving placebo was due to steroids, which are known to be effective in inducing remission.4 ,26 ,27 A slow tapering with prolonged corticosteroid therapy may have led to a higher proportion of responders in the placebo group. Lastly, the lack of correlation between CDAI and disease activity may have contributed to an elevated number of remitters in the placebo group. This possibility is supported by the demonstration of a statistically significant treatment effect for both 5 and 10 mg twice daily using PRO2-75 remission endpoint and for 5 mg twice daily using the PRO3-80 remission endpoint.
No new safety signals were detected for tofacitinib in either study compared with those previously reported in other indications,16 ,28 ,29 although three AEs of special interest (one case of large intestinal perforation confirmed by adjudication and two cases of non-serious herpes zoster) were seen in patients receiving tofacitinib as maintenance therapy. Most AEs were either GI AEs or infections. There were no cases of opportunistic infection confirmed by adjudication in patients treated with tofacitinib in either study, although one such infection (cytomegalovirus infection) was reported in the placebo group of the induction study. The proportions of patients with SAEs were numerically higher with tofacitinib 10 mg twice daily (11.6% and 13.1% in the induction and maintenance study, respectively) compared with the tofacitinib 5 mg twice daily (3.5% and 10.0%) and placebo (3.3% and 11.9%) groups.
In summary, some evidence of minor clinical efficacy for tofacitinib in inducing and maintaining remission in moderate-to-severe CD was observed in these two phase IIb clinical studies. There were no unexpected safety findings. The minor improvement in measures of disease activity observed in these studies supports further investigation of the efficacy and safety of JAK inhibition for CD.
Acknowledgments
The authors would like to thank the patients, investigators and study teams who were involved in both clinical trials.
References
Footnotes
Contributors PH, BES, WJS, EM, GC, WW and AM substantially contributed to the conception or design of the work. All authors substantially contributed in the acquisition, analysis or interpretation of data. All authors drafted the work and/or revised it critically for important intellectual content; approved the final published version of the manuscript; and are accountable for all aspects of this work.
Funding Medical writing support, under the direction of the authors, was provided by Sandrine M Dupré, PhD, of Complete Medical Communications and funded by Pfizer.
Competing interests JP: received consulting fees from AbbVie, Boehringer Ingelheim, Galapagos, GlaxoSmithKline, Janssen, MSD, Pfizer, Second Genome, Takeda, TiGenics and Topivert Pharma; received lectures and/or speaker bureau fees from AbbVie, Janssen, MSD, Pfizer and Takeda. WJS: received grant support from Receptos, Exact Sciences, Amgen, the American College of Gastroenterology and the Broad Foundation; received grant support and personal fees from Prometheus Laboratories, AbbVie, Boehringer Ingelheim, Takeda, Atlantic Pharmaceuticals, Janssen, Bristol-Myers Squibb, Genentech, Pfizer and Nutrition Science Partners; and personal fees from Kyowa Hakko Kirin, Millennium Pharmaceuticals, Celgene Cellular Therapeutics, Santarus, Salix Pharmaceuticals, Catabasis Pharmaceuticals, Vertex Pharmaceuticals, Warner Chilcott, Gilead Sciences, Cosmo Pharmaceuticals, Ferring Pharmaceuticals, Sigmoid Biotechnologies, Tillotts Pharma, Am Pharma BV, Dr. August Wolff, Avaxia Biologics, Zyngenia, Ironwood Pharmaceuticals, Index Pharmaceuticals, Nestle, Lexicon Pharmaceuticals, UCB Pharma, Orexigen, Luitpold Pharmaceuticals, Baxter Healthcare, Ferring Research Institute, Amgen, Novo Nordisk, Mesoblast, Shire, Ardelyx, Actavis, Seattle Genetics, MedImmune (AstraZeneca), Actogenix NV, Lipid Therapeutics GmbH, Eisai, Qu Biologics, Toray Industries, Teva Pharmaceuticals, Eli Lilly, Chiasma, TiGenix, Adherion Therapeutics, Immune Pharmaceuticals, Celgene, Arena Pharmaceuticals, Ambrx, Akros Pharma, Vascular Biogenics, Theradiag, Forward Pharma, Regeneron, Galapagos, Seres Health, Ritter Pharmaceuticals, Theravance, Palatin, Biogen and the University of Western Ontario (owner of Robarts Clinical Trials). SS: received consulting fees from Ferring, AbbVie, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Janssen, Galapagos, MedImmune, MSD, Pfizer/Hospira, Shire, Takeda and UCB; received lectures and/or speaker bureau fees from Ferring, AbbVie, MSD, Takeda, UCB and Falk. BES: received consulting fees from AbbVie, Akros Pharma, Amgen, AstraZeneca, Boehringer Ingelheim, Celgene, Forest Research Institute, Lilly, MedImmune, Puretech Ventures, LLC, Receptos, Salix, Shire, Takeda, Topivert Pharma, Vedanta Biosciences, Bristol-Myers Squibb, Janssen R&D, Luitpold Pharmaceuticals, Pfizer, Prometheus Laboratories, Synergy Pharmaceuticals, Takeda, Theravance Biopharma and Tigenix; received research grants from AbbVie, Celgene, GlaxoSmithKline, Janssen R&D, Pfizer, Prometheus Laboratories and Takeda. SV: received consulting fees from Takeda, Roche/Genentech, Merck, Centocor, AbbVie, UCB, Pfizer, Ferring, Second Genome and Galapagos; received research grants from Centocor, AbbVie, Takeda and Merck; received lectures and/or speaker bureau fees from Merck, AbbVie, Takeda, Pfizer, Ferring, Falk and Centocor. GD: received consulting fees from AbbVie, Ablynx, Amakem, AM Pharma, Avaxia, Biogen, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Celltrion, Cosmo, Covidien/Medtronics, Ferring, DrFALK Pharma, Engene, Galapagos, Gilead, GlaxoSmithKline, Hospira, Immunic, Johnson and Johnson, Lycera, Medimetrics, Millennium/Takeda, Mitsubishi Pharma, MSD, Mundipharma, Novo Nordisk, Pfizer, Prometheus laboratories/Nestle, Receptos, Robarts Clinical Trials, Salix, Sandoz, Setpoint, Shire, Teva, Tigenix, Tillotts, Topivert, Versant and Vifor; received research grants from MSD, AbbVie, Takeda, Mundipharma, Ferring and Falk; received lectures and/or speaker bureau fees from AbbVie, Ferring, Johnson and Johnson, MSD, Mundipharma, Norgine, Pfizer, Shire, Millennium/Takeda, Tillotts and Vifor. RP: received consulting fees from AbbVie, Amgen, Aptalis, AstraZeneca, Baxter, Biogen, Bristol-Myers Squibb, Celgene, Cubist, Eisai, Ferring, Gilead, Janssen, Merck, Robarts Clinical Trials, Salix, Samsung Bioepis, Shire, Centocor, Elan, GlaxoSmithKline, UCB, Pfizer and Takeda; received research grants from AbbVie, Ferring, Janssen and Takeda; received lectures and/or speaker bureau fees from AbbVie, Aptalis, AstraZeneca, Ferring, Janssen, Merck, Prometheus, Shire and Takeda; received advisory board fees from AbbVie, Abbott, Amgen, Aptalis, AstraZeneca, Baxter, Eisai, Ferring, Genentech, Jansen, Merck, Schering-Plough, Shire, Centocor, Elan, GlaxoSmithKline, UCB, Pfizer, Bristol-Myers Squibb, Takeda, Cubist, Celgene and Salix. PDRH: received consulting fees from AbbVie, Amgen, Genentech, JBR Pharma and Lycera. J-FC: received consulting fees from Abbott Laboratories, ActoGeniX, Albireo Pharma, Amgen, AstraZeneca, Bayer AG, Biogen Idec, Boehringer Ingelheim, Bristol-Myers Squibb, Cellerix, Centocor, Chemocentryx, Cosmo Technologies, Danone Research, Elan Pharmaceuticals, Genentech, Giuliani SpA, Given Imaging, GlaxoSmithKline, Hutchison MediPharma, MSD, Millennium Pharmaceuticals (now Takeda), Neovacs, Ocera Therapeutics, Pfizer, Shire Pharmaceuticals, Schering-Plough, Prometheus Laboratories, Sanofi-Aventis, Synta Pharmaceuticals Corp, Teva, Therakos, UCB Pharma and Wyeth; received research grants from AstraZeneca, Ferring, Schering-Plough and UCB Pharma; received lectures and/or speaker bureau fees from Abbott Laboratories, Centocor, Elan Pharmaceuticals, Given Imaging, Otsuka America Pharmaceutical, MSD, Schering-Plough, Shire Pharmaceuticals, Tillotts Pharma and UCB Pharma; received advisory board fees from Abbott Laboratories, Centocor, Danone, Elan, MSD, Millennium Pharmaceuticals (now Takeda), Schering-Plough and UCB Pharma. BGF: received consulting fees from Abbott/AbbVie, Actogenix, Akros, Albireo Pharma, Amgen, AstraZeneca, Avaxia Biologics, Avir Pharma, Axcan, Baxter Healthcare Corp., Biogen Idec, Boehringer Ingelheim, Bristol-Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, EnGene, Ferring Pharma, Roche/Genentech, GiCare Pharma, Gilead, Given Imaging, GSK, Ironwood Pharma, Janssen Biotech (Centocor), JnJ/Janssen, Kyowa Kakko Kirin Co Ltd., Lexicon, Lilly, Lycera BioTech, Merck, Mesoblast Pharma, Millennium, Nektar, Nestle, Novo Nordisk, Pfizer, Prometheus Therapeutics and Diagnostics, Protagonist, Receptos, Salix Pharma, Serono, Shire, Sigmoid Pharma, Synergy Pharma, Takeda, Teva Pharma, TiGenix, Tillotts, UCB Pharma, Vertex Pharma, VHsquared Ltd., Warner-Chilcott, Wyeth, Zealand, Zyngenia; received research grants from Abbott/AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Janssen Biotech (Centocor), JnJ/Janssen, Roche/Genentech, Millennium, Pfizer, Receptos, Santarus, Sanofi, Tillotts and UCB Pharma; received lectures and/or speaker bureau fees from Abbott/AbbVie, JnJ/Janssen, Takeda, Warner-Chilcott and UCB Pharma; received advisory board fees from Abbott/AbbVie, Amgen, AstraZeneca, Avaxia Biologics, Bristol-Myers Squibb, Celgene, Centocor, Elan/Biogen, Ferring, JnJ/Janssen, Merck, Nestle, Novartis, Novo Nordisk, Pfizer, Prometheus Laboratories, Protagonist, Salix Pharma, Takeda, Teva, TiGenix, Tillotts Pharma AG, UCB Pharma; is a board of directors member of Robarts Clinical Trials. GC, MM, WW, WN, AM, PH and EM are all employees and stockholders of Pfizer Inc.
Ethics approval These studies were approved by the institutional review board or independent ethics committee for each centre and carried out according to the Declaration of Helsinki and in compliance with all International Conference on Harmonization (ICH) Good Clinical Practice (GCP) Guidelines.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All prespecified analyses of studies NCT01393626 and NCT01393899 are included in the main manuscript or online supplementary material.