Article Text

Abstract
Objective Both non-alcoholic fatty liver disease (NAFLD) and the multitarget complexity of microRNA (miR) suppression have recently raised much interest, but the in vivo impact and context-dependence of hepatic miR-target interactions are incompletely understood. Assessing the relative in vivo contributions of specific targets to miR-mediated phenotypes is pivotal for investigating metabolic processes.
Design We quantified fatty liver parameters and the levels of miR-132 and its targets in novel transgenic mice overexpressing miR-132, in liver tissues from patients with NAFLD, and in diverse mouse models of hepatic steatosis. We tested the causal nature of miR-132 excess in these phenotypes by injecting diet-induced obese mice with antisense oligonucleotide suppressors of miR-132 or its target genes, and measured changes in metabolic parameters and transcripts.
Results Transgenic mice overexpressing miR-132 showed a severe fatty liver phenotype and increased body weight, serum low-density lipoprotein/very low-density lipoprotein (LDL/VLDL) and liver triglycerides, accompanied by decreases in validated miR-132 targets and increases in lipogenesis and lipid accumulation-related transcripts. Likewise, liver samples from both patients with NAFLD and mouse models of hepatic steatosis or non-alcoholic steatohepatitis (NASH) displayed dramatic increases in miR-132 and varying decreases in miR-132 targets compared with controls. Furthermore, injecting diet-induced obese mice with anti-miR-132 oligonucleotides, but not suppressing its individual targets, reversed the hepatic miR-132 excess and hyperlipidemic phenotype.
Conclusions Our findings identify miR-132 as a key regulator of hepatic lipid homeostasis, functioning in a context-dependent fashion via suppression of multiple targets and with cumulative synergistic effects. This indicates reduction of miR-132 levels as a possible treatment of hepatic steatosis.
- FATTY LIVER
- NONALCOHOLIC STEATOHEPATITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic fatty liver disease (NAFLD) is one of the most important liver diseases of this decade, affecting approximately 25% of adults globally, but its pathogenesis is incompletely understood.
The small non-coding microRNAs (miRs) repress the expression of target mRNA transcripts, including metabolic ones. Each miR can target many genes and each target can be silenced by numerous miRs, but the impact of this complexity is still an enigma.
miR-132 is causally involved in cell proliferation, epigenetic regulation and intestinal inflammation, but has not been studied in the metabolic context.
What are the new findings?
Conditional miR-132 overexpressing transgenic mice, NAFLD and non-alcoholic steatohepatitis (NASH) mouse models, and human liver tissues from patients with NAFLD, all show increased hepatic miR-132, and decreased metabolism-related miR-132 targets, thus validating miR-132 as an important regulator of hepatic lipid homeostasis.
In vivo suppression of miR-132 in diet-induced obese mice reversed hepatic steatosis and hyperlipidaemia, unlike the partial impact of suppressing individual miR-132 targets.
miR-132 operates high in the hierarchy of hepatic lipid homeostasis by simultaneously suppressing multiple regulatory transcripts.
How might it impact on clinical practice in the foreseeable future?
miR-132 levels can serve as a biomarker for assessment of NAFLD/NASH.
miR-132 may become a target for developing and assessing novel NAFLD/NASH treatment strategies.
Introduction
MicroRNAs (miRs) are small non-coding RNAs that interact with short motifs in the 3′-untranslated region of target mRNAs to repress their expression. miRs play key roles in development and metabolism1 of eukaryotes.2 They regulate entire pathways by simultaneously repressing multiple target mRNAs.3 Assessing the contribution of each target to miR-mediated phenotypes is challenging and has mainly been addressed in cell culture,4 leaving the in vivo impact of this complexity and the hierarchy of miRs5 largely unknown. Nevertheless, addressing this matter is a prerequisite for understanding miR-related molecular mechanisms and enabling miR-targeted therapeutics.
Non-alcoholic fatty liver disease (NAFLD) affects about 25% of adults globally,6 with sharp increases over the last two decades, in part attributed to a sedentary lifestyle and high calorie diet. NAFLD begins with hepatic steatosis, a key predisposing factor for non-alcoholic steatohepatitis (NASH), cirrhosis and subsequent hepatocellular carcinoma (HCC),7 ,8 and is a leading indication for liver transplantation in the Western world.7 NAFLD is characterised by the accumulation of triglyceride lipid droplets in the hepatocyte cytoplasm.7 The underlying mechanisms leading to NAFLD are still debated.9 ,10 NAFLD associates with insulin resistance, hyperlipidaemia, obesity and type 2 diabetes, symptoms of metabolic syndrome (MetS).9 ,11 ,12 Treatments are well established for diabetes and hyperlipidaemia, but not for NAFLD and NASH, where the main therapeutic intervention is weight loss,7 highlighting the unmet need for novel prophylactic and therapeutic agents for chronic NAFLD and NASH.
We studied the miR-132 gene, transcribed from an intron of a non-coding transcript located on chromosome 11 in mice and chromosome 17 in humans.13 ,14 miR-132 targets mRNAs which regulate key biological processes including metabolism,15–18 cell proliferation,13 epigenetic regulation19 and inflammation (via suppression of acetylcholinesterase (AChE)),20 as well as nervous system-related cholinergic functions.21 Brain miR-132 levels increase under psychological stress, which impairs cognition.22 Interestingly, miR-132 changes were observed in metabolic disorder, depression and anxiety,23 and its levels increase in inflamed intestinal tissue from patients with IBD.24 Hepatic miR-132 is elevated in a mouse model of alcoholic liver disease25 and in human HCC.26 Notably, miR-132 targets such as Pten and Sirt1 associated with hepatic steatosis, hyperlipidaemia and glucose regulation.15 ,16 ,27 ,28 Nevertheless, the direct in vivo role of miR-132 in hepatic metabolism, and the context dependency of its function in different disease stages and states have not been investigated.
Materials and methods
For additional information, see the online supplementary materials and methods.
supplementary data
Human samples: frozen liver samples from NAFLD or apparently healthy patients were obtained from donors with proper ethical approvals from institutional review boards (ProteoGenex, Culver City, California, USA).
Animal studies were approved by the ethics committees of The Hebrew University of Jerusalem and Munchen GmbH. NASH model C57Bl/6J mice were fed a methionine-deficient and choline-deficient diet (MCD), a choline-deficient and ethionine-supplemented diet (CDE), a high fat and high sucrose diet (HFHS) or a choline-deficient high fat diet (CDHFD).
Inducible miR-132 transgenic mice were generated by cloning the mouse miR-132 precursor17 into the pTRE-tight plasmid (Clontech, Mountain View, California, USA). Six transgenic founders were crossed with mice expressing the reverse tetracycline controlled transactivator (rtTA) under the Gt(ROSA)26Sor promoter (The Jackson Laboratory, Bar Harbor, Maine, USA). Inducible miR-132 transgenic mice were fed a regular chow diet (RCD). C57bl/6J control mice were fed an RCD or a high fat diet (Harlan Teklad, Madison, Wisconsin, USA) for 9–11 weeks to reach diet-induced obesity.
Oligonucleotides were locked nucleic acid (LNA, Exiqon, Vedbaek, Denmark) or 2-O-methyl phosphorothioated (Sigma-Aldrich, Israel), complementary to mature miR-132 (AM132, 16-mer) or to the primate-specific miR-608 (AM608, 15-mer). DIO mice were injected intravenously with one dose of 10 mg/kg or three successive daily doses of 3.3 mg/kg of AM132 or AM608.
Glucose metabolism, lipid and liver enzyme assays are detailed in online supplementary materials and methods. Quantitative RT-PCR (qRT-PCR) mRNA levels were determined as previously described.20
Statistical analyses included Student’s t-test or one-way and two-way analysis of variance (ANOVA) with least significant difference (LSD) post hoc. Average±SEM is shown. Partial least square regression (PLS-R) analysis was based on MATLAB Pearson’s correlation and one-way ANOVA with Bonferroni post hoc test.
Results
Peripheral miR-132 excess lowers its target levels and induces hepatic steatosis
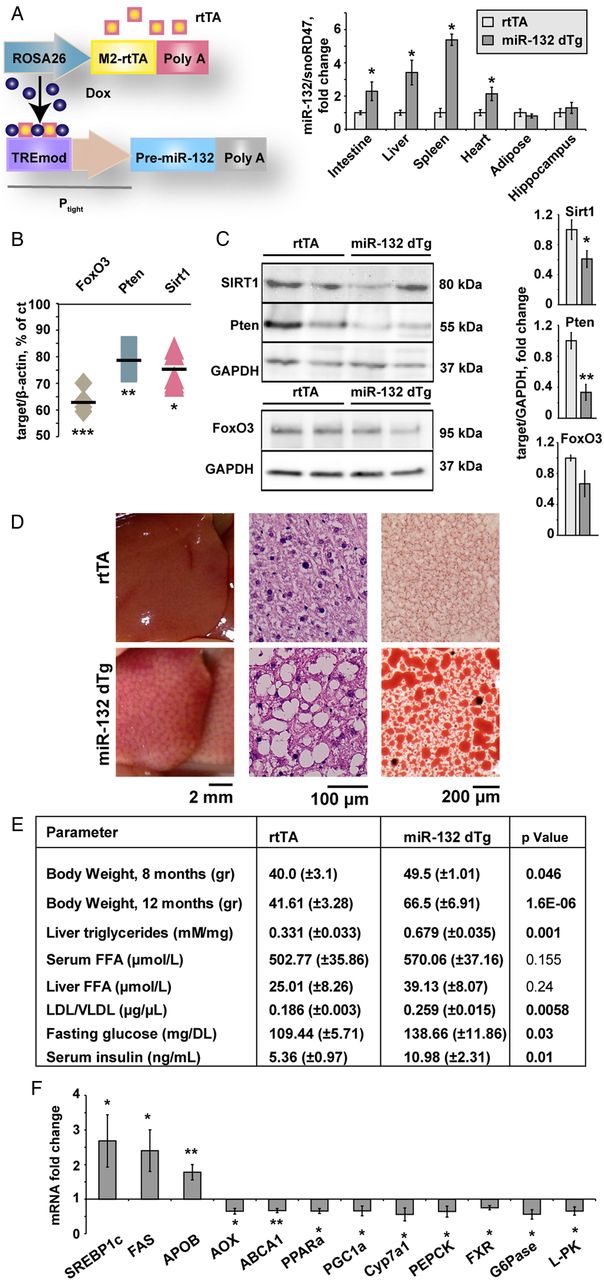
We induced conditional miR-132 overexpression in peripheral tissues exclusively by engineering transgenic mice carrying the pre-miR-132 gene under control of the Tet-responsive promoter (figure 1A). Intercrossing with mice constitutively expressing the doxycycline (Dox)-inducible rtTA protein under the Gt(ROSA)26Sor promoter, enabled expressing rtTA in peripheral tissues but not the brain.29 Only 4.8% of the offspring mice were double transgenic, significantly below the expected Mendelian ratio of 25% (see online supplementary figure S1A). This may reflect leakiness of the Tet-On system, and the detrimental effect of excess miR-132 on embryonic development. Surviving double transgenic progeny (miR-132 dTg mice) showed miR-132 overexpression after doxycycline administration to all animals, in numerous peripheral tissues including intestine, liver, spleen and heart, but not in adipose tissue and hippocampus (see figure 1A and online supplementary figure S1B).

Peripheral miR-132 excess leads to multiple target reductions in diverse tissues, and associates with hepatic steatosis and impaired lipid homeostasis. (A) Schematic representation and expression levels of the engineered doxycycline-inducible double transgenic miR-132 system: the reverse tetracycline controlled transactivator (rtTA) followed by a β-globin Poly A sequence is located downstream of the Gt(ROSA)26Sor promoter and the ColA1 locus. Pre-miR-132 under the Tet-responsive Ptight promoter followed by an SV40 Poly A was expressed in progeny of rtTA-miR-132 double transgenic mice (miR-132 dTg). The rtTA protein (orange squares) binds in trans to the TREmod element in the presence of doxycycline, allowing pre-miR-132 transcription. Excess miR-132 levels in rtTA and miR-132 dTg mice were observed in the intestine, liver and spleen and heart but not adipose tissue and hippocampus (n=12 for rtTA tg, n=5 for miR-132 dTg, n=3 for rtTA and miR-132 dTg for adipose tissue). (B) Reduced mRNA levels of the miR-132 targets Sirt1, Pten and FoxO3 in liver from miR-132 dTg mice (n=3). Levels are normalised to the housekeeping β-actin and expressed as percentage of control non-transgenic littermates. (C) Western blot and quantification (normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) of reductions in Sirt1, Pten and FoxO3 levels in livers of miR-132 dTg mice (n=3–6). (D) Lipid accumulation in miR-132 dTg mice compared with rtTA littermates: from left to right, representative image of fatty liver, H&E stained liver sections and Oil Red O stained liver sections. (E) Body weight and lipid profiles of miR-132 dTg mice aged 12 months showing greater weight, elevated liver triglycerides and low-density lipoprotein (LDL)/very low-density lipoprotein (VLDL) elevated serum glucose and insulin, and insignificantly changed liver and serum free fatty acid (FFA) levels (n=3). (F) Elevated and reduced transcripts in miR-132 dTg mice compared with rtTA, normalised to multiple housekeeping genes and then to rtTA controls. Data were obtained by Fluidigm analysis and is representative of three experiments (n=3 per group, *p<0.05, **p<0.01, ***p<0.001 determined by two-tailed Student's t-test). Values are expressed as mean±SEM.
miR-132 dTg mice showed reduced hepatic levels of the miR-132 target gene transcripts FoxO3, Pten and Sirt1, all validated miR-132 targets shown to be associated with steatosis, by 37%, 21% and 24%, respectively (see figure 1B and online supplementary figure S1C,D).These mice also showed pronounced decreases in hepatic protein levels of Sirt1 and Pten, a slight decrease in hepatic FoxO3 protein compared with littermates (figure 1C), and decreased hydrolytic activity of AChE, another target of miR-132, in intestine, spleen and serum but not liver, heart or hippocampus (see online supplementary figure S1E,F shows these and additional targets that are not significantly changed).
miR-132 dTg mice of three independent transgenic lines fed an RCD had enlarged pale livers with macrovesicular liver steatosis (see figure 1D and online supplementary figure S2A, table S1 for NAFLD activity score (NAS)30), excluding the transgene insertion site as a cause of the phenotype. Both male and female miR-132 dTg mice showed increased body weight (figure 1E), with some weight increase also seen in the absence of doxycycline (see online supplementary figure S2B), suggesting some leakiness.
miR-132 dTg mice showed elevated liver triglycerides, serum low-density lipoprotein/very low-density lipoprotein (LDL/VLDL) cholesterol, fasting glucose and insulin and unchanged levels of serum HDL and liver or serum free fatty acids (see figure 1E and online supplementary figure S2C). A microfluidic high-throughput qRT-PCR analysis (Fluidigm) revealed elevation of SREBP1c, fatty acid synthase (FAS) and apolipoprotein B (APOB), and reduced acyl-coenzyme A oxidase (AOX), ABCA1, peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) and peroxisome proliferator activated receptor α in the miR-132 dTg mice (see figure 1F and see online supplementary figure S2D for additional transcripts).
miR-132 dTg mice also presented a significantly lower respiratory quotient and higher fat oxidation (see online supplementary figure S3A,B), indicating higher use of fat for energy production compared with rtTA controls. Although their activity profile did not differ from controls, with both groups showing similar ambulatory activity such as wheel running, meal size and sip volume (see online supplementary figure S3D–F), miR-132 dTg mice displayed a significantly lower total energy expenditure (see online supplementary figure S3C). Together, our data suggest that peripheral overexpression of miR-132 initiates lower metabolic rate as well as enhanced lipolysis, which in turn increases the transport of free fatty acids from adipose tissue to the liver, resulting in ectopic fat accumulation in the liver.
Hepatic miR-132 is elevated and its targets reduced in human NAFLD samples
We quantified the levels of miR-132 in four human liver samples of patients with NAFLD and five normal tissue or normal tissue adjacent to a tumour samples (mean age 65 and 64.8 years, respectively, all of Caucasian ethnic origin, online supplementary table S2). Hepatic miR-132 levels were increased 13-fold in NAFLD tissues compared with controls (figure 2A). Quantifying those steatosis-associated target transcripts of miR-132 which were decreased in the transgenic mice revealed reduced levels of FoxO3, Pten and Sirt1 (by 37%, 22% and 29%, respectively), whereas P300 levels were elevated by 30% (figure 2B–E). Principal component analysis showed clear segregation of NAFLD tissues from controls based on expression data of miR-132 and all target transcripts (see online supplementary figure S4A). These data, and our transgenic mouse results, suggested that both miR-132 excess and reduced miR-132 target transcript levels play a role in NAFLD pathogenesis.

Hepatic miR-132 is elevated and its targets are reduced in human non-alcoholic fatty liver disease (NAFLD). (A) Hepatic miR-132 levels in postmortem tissue from human patients diagnosed with NAFLD, compared with apparently healthy tissues. (B–E) Hepatic miR-132 target transcript levels in the same human tissue samples: FoxO3 (B), Pten (C), Sirt1 (D) and P300 (E). n=4–5, *p<0.05, **p<0.01 (two-tailed Student's t-test). Ctrl, control.
Knockdown of individual miR-132 targets partially mimics the hepatic impact of miR-132 excess
Next, we quantified hepatic miR-132 and validated miR-132 targets in several NAFLD, short term and chronic NASH mouse models.7 ,8 We used diet-induced obese mice (DIO),31 mice fed an HFHS diet,32 an MCD diet,33 a CDE diet34 or a CDHFD diet.8 These five models differ in their levels of pathology, steatosis and inflammation, as well as extent of concomitant metabolic syndrome; but all showed elevated hepatic miR-132 and reduced FoxO3, Pten and Sirt1 levels compared with age-matched regular chow-fed controls (see online supplementary figure 4B–D and table S1). This suggests miR-132 involvement in diverse NAFLD-NASH states, although not necessarily through the same targets.
To explore the relative contribution of specific validated targets to the development of steatosis, we used LNA antisense (AS) GapmeRs to knock down each of several targets separately. Five different GapmeRs were screened for each target mRNA in C2C12 myoblast mouse cells to identify the most potent GapmeR (see online supplementary figure S5A); 10 mg/kg of each selected GapmeR targeting either FoxO3, Pten, P300 or Sirt1, was intravenously injected into DIO C57bl/6J mice (figure 3A and online supplementary figure S5B). At 7 days post-treatment, mice injected with FoxO3-AS and Pten-AS showed increased liver/body weight (figure 3B), whereas P300-AS injected mice showed reduced liver/body weight and Sirt1-AS injected mice showed no change. The treated mice showed no significant change in body weight (see online supplementary figure S5D). Treatment with FoxO3-AS and Pten-AS led to macrovesicular and microvesicular steatosis, respectively, whereas treatment with P300-AS and Sirt1-AS led to reduced vacuole numbers or no visible change, respectively (figure 3C). FoxO3-AS and Pten-AS treatment also increased serum LDL/VLDL but not HDL levels, and liver triglycerides showed a similar but statistically insignificant trend (see figure 3D and online supplementary figure S5C) with levels similar to those seen in miR-132 dTg mice that were fed with regular chow (figure 1E). Thus, suppressing several individual miR-132 targets failed to mimic the phenotype caused by miR-132 elevation.
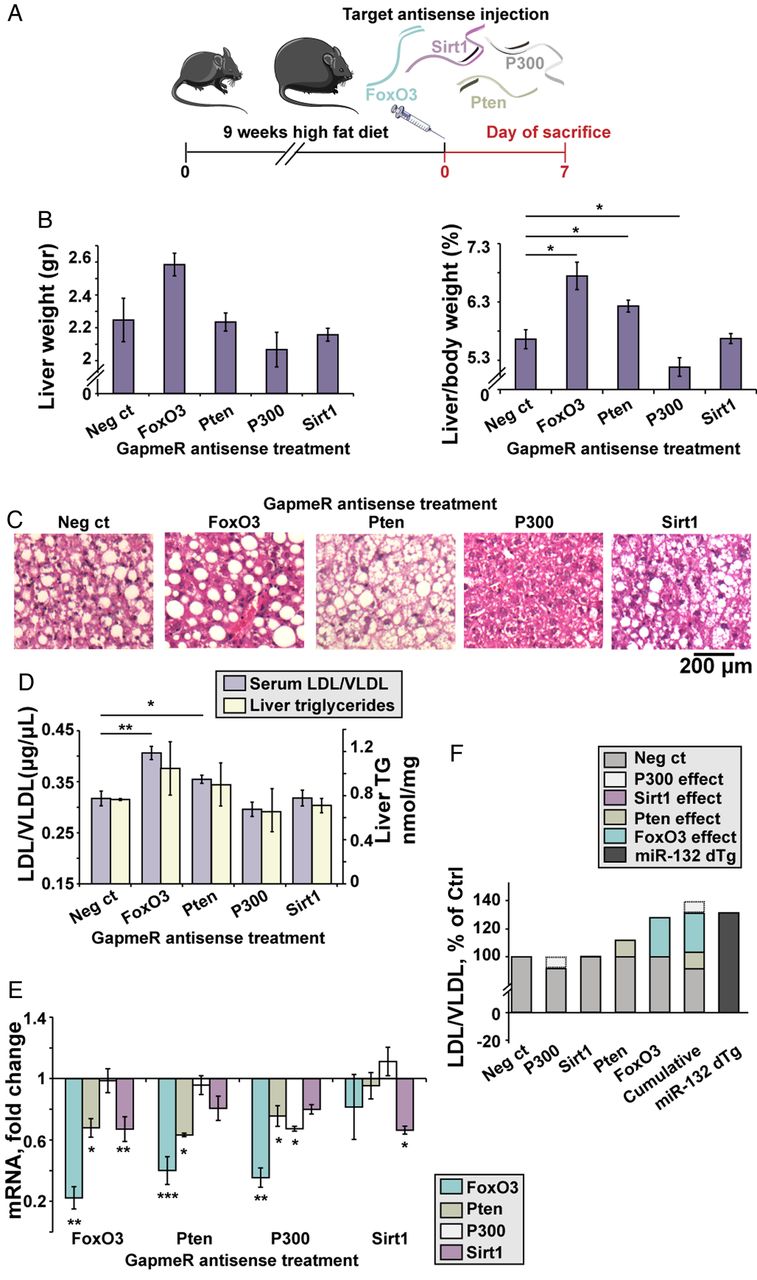
Individual miR-132 target knockdowns in diet-induced obese mice (DIO) mice fail to fully mimic the impact of transgenic miR-132 excess. (A) Experimental design: C57Bl/6J mice were fed with a high fat diet for 9 weeks, followed by a single intravenous injection of 10 mg/kg antisense (AS) GapmeRs for FoxO3, Pten, P300, Sirt1 or a negative control oligonucleotide. Mice were sacrificed 7 days post-treatment. (B) Liver weight and normalised liver/body weight in GapmeR-treated mice, showing elevation in FoxO3-AS-treated and Pten-AS-treated mice, reduction in P300-AS and no change in Sirt1-AS-treated mice (n=4). (C) H&E staining of liver sections showing variable size of fat vacuoles, with a decrease seen in Pten-AS-treated and P300-AS-treated mice. (D) Serum low-density lipoprotein/very low-density lipoprotein (LDL/VLDL) and liver triglycerides (TG) in mice treated with target AS GapmeRs (n=4). (E) Target mRNA levels, obtained from Fluidigm analysis, normalised to multiple housekeeping genes and then to negative control-treated mice (n=4), showing cross-target effects of the GapmeRs. (F) Individual and cumulative effects of each AS GapmeR on the levels of LDL/VLDL (as shown in D) compared with miR-132 dTg mice.*p<0.05, **p<0.01, ***p<0.001 (one-way analysis of variance with LSD post hoc). Bars show mean±SEM.
Next, we measured the hepatic mRNA levels of all four targets in the GapmeR-injected mice. FoxO3-AS knockdown reduced FoxO3 levels by 78% and also reduced Pten and Sirt1 levels; Pten-AS knockdown reduced Pten by 37% while reducing FoxO3 levels by 60%, a larger reduction than that of Pten; and P300-AS knockdown reduced P300 by 33% while reducing FoxO3 and Pten (figure 3E). Sirt1 knockdown did not markedly reduce the other targets. These targets do not show sequence similarity, indicating the existence of complex interactions both between miR-132 and its target genes and between the genes themselves. These findings indicated that the development of hepatic steatosis may reflect the cumulative complex impact of various miR-132 targets, with P300 acting inversely, and Sirt1 having no effect in this mouse model.
To explore the cumulative effect on serum lipoproteins, we compared the outcome of suppressing each of these miR-132 targets following each single GapmeR treatment of the DIO C57BL/6J mice with the combined LDL/VLDL levels of miR-132 dTg mice (figure 3F). This ‘virtual cumulative effect’ of multiple target knockdowns in DIO mice yielded similar LDL/VLDL levels to those observed in miR-132 dTg mice, although those mice were fed an RCD (figure 3F), tentatively indicating an additional impact of miR-132 upregulation over the cumulative effect of downregulating specific hepatic targets.
AS oligonucleotide suppression of miR-132 reverses liver steatosis and hyperlipidaemia
To examine the impact of targeting miR-132 in hepatic steatosis conditions, we tested if suppressing miR-132 elevation could reverse steatosis. We intravenously injected C57BL/6J male DIO mice, which were fed a high fat diet for 11 weeks starting at the age of 6–7 weeks, with a 16-mer LNA AS oligonucleotide complementary to miR-132 (AM132) or with a 16-mer LNA-based oligonucleotide complementary to miR-608 (a stress-regulating primate-specific miRNA35 with no predicted metabolism-related mouse targets), which served as a control (Ctrl). Both agents were injected at 3.3 mg/kg for three successive days and mice were sacrificed at different time points after the last injection, to allow a kinetic study of the effect (figure 4A). DIO mice presented significant steatosis, elevated triglycerides and higher LDL/VLDL levels compared with littermate controls fed with RCD, with no change in total body weight or serum HDL (see online supplementary figure S6B). By 7 days after AM132 treatment DIO mice showed a striking reduction in liver miR-132 levels (figure 4B), indicating efficient miR-132 knockdown in vivo. Moreover, DIO mice treated with AM132 showed progressive decreases in liver weight at 7 and 14 days post-treatment, bringing these parameters close to those of lean RCD-fed mice (see figure 4C–E and online supplementary table S1 for NAS). They also showed higher levels of miR-132 target transcripts (see online supplementary figure S6A). Comparison of Ctrl-treated mice with double distilled water (DDW)-treated ones showed a similar phenotype, with no change in liver and body weight, serum LDL/VLDL or HDL levels, excluding a non-specific oligonucleotide effect (see figure 4C–4E and online supplementary figure S6CF). Histological analysis and Oil Red O staining of liver sections from AM132-treated mice showed reduction in diffuse microvesicular and macrovesicular steatosis at 5–14 days post-treatment, although the mice were fed a high fat diet until sacrificed. Steatosis occurred mainly in zone 3, the pericentral zone, where lipogenesis and glycolysis occur and where steatosis usually begins before it progresses with increasing severity of the disease towards the portal tract36 (figure 4C–F and online supplementary figure S8A). AM132 but not Ctrl treatment also reduced liver triglyceride, free fatty acid and serum LDL/VLDL levels (although not serum HDL) levels, with kinetics similar to those of liver weight changes, reaching levels of RCD-fed mice at days 7–14 (see figure 5A–C and online supplementary figure S6E,F). A chronic regime of AM132 treatment, with two injections of 10 mg/kg every 14 days was able to extend the impact of AM132 on liver steatosis for up to 28 days post-treatment (see online supplementary figure S7A,B). Taken together, these results indicate that in DIO mice miR-132 is high in the hierarchy of causal factors leading to liver steatosis and hyperlipidaemia.

miR-132 suppression reduces liver weight and fat vacuoles. (A) Experimental design: C57Bl/6J mice fed a high fat diet (diet-induced obese mice (DIO) mice) for 11 weeks were injected with AM132 or control (Ctrl) oligonucleotide for three consecutive days and sacrificed at various days post-treatment. (B) Liver miR-132 levels in DIO mice 7 days post-AM132 are reduced compared with the Ctrl oligonucleotide treatment (n=4 each). (C) Representative liver pictures, H&E and Oil Red O stained liver sections from DIO mice at increasing days post-treatment. (D) Reduced liver weight of AM132-treated DIO mice (n=8 for regular chow diet (RCD), 4 for days 0 and 2, 5 for day 5, 9 for day 7 and 6 for days 14, 21 and 28). (E) Normalised liver/body weight of these mice. (F) Oil Red O quantification results (n=3). All data represent three experiments, *p<0.05, **p<0.01, ***p<0.001 AM132-treated mice compared with controls (two-tailed Student's t-test). Bars show mean±SEM. Staining quantifications were based on 10 fields each.
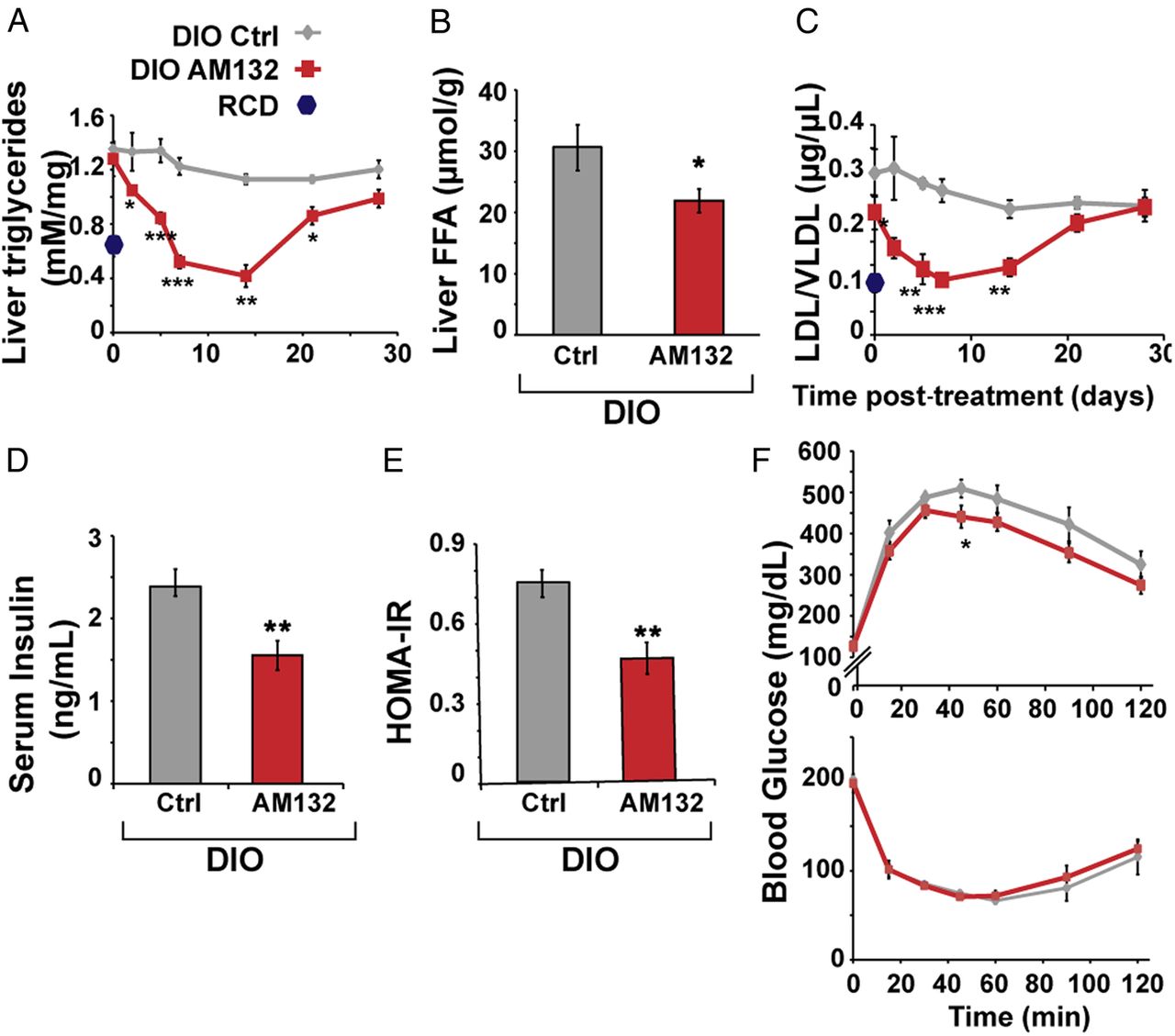
miR-132 suppression reduces triglycerides, serum low-density lipoprotein/very low-density lipoprotein (LDL/VLDL) and insulin. (A) AM132 treatment leads to gradual and transient reduction of liver triglycerides (n=8 for regular chow diet (RCD), 4 for days 0 and 2, 5 for day 5, 9 for day 7 and 6 for days 14, 21 and 28). (B) Hepatic free fatty acids are reduced in AM132-treated mice at 7 days post-treatment (n=9). (C) Reduced LDL/VLDL in sera of AM132-treated diet-induced obese mice (DIO) mice (n=4–9). (D) Reduced fasting insulin levels in AM132-treated DIO mice (n=8). (E) Reduced sera values of calculated homeostatic model assessment (HOMA)-insulin resistance (IR) in these mice (n=8). (F) Top: Slightly improved glucose tolerance in AM132-treated DIO mice versus Control (Ctrl)-treated mice 7 days post-treatment (n=8). Bottom: Unchanged insulin tolerance in these mice (n=8.). All data represent three experiments, *p<0.05, **p<0.01, ***p<0.001 AM132-treated mice compared with controls (two-tailed Student's t-test). Bars show mean±SEM. Staining quantifications were based on 10 fields each.
Both AM132 and Ctrl-treated mice maintained aspartate aminotransferase (AST) activity similar to that of untreated RCD mice, excluding overt liver toxicity in the LNA-treated mice (see online supplementary figure S8B). A few small macrophage aggregates were seen in livers of some AM132-treated mice, suggesting mild inflammation (see online supplementary figure S6D), although mRNA levels of the pro-inflammatory cytokine interleukin-1β were unchanged at 7 days post-treatment (see online supplementary figure S8C).
RCD mice injected with AM132, Ctrl or DDW showed no change in liver histology, liver and liver/body weight, AST, liver triglycerides or serum LDL/VLDL and HDL (see online supplementary figure S9A–E). Also, miR-132 levels in the visceral adipose tissue of the treated DIO mice remained unchanged (see online supplementary figure S8D).
DIO mice exhibit MetS-like characteristics including insulin resistance and glucose intolerance.37 Therefore, we measured insulin and glucose levels in AM132-treated and control mice. At 7 days post-treatment, AM132-treated DIO mice presented lower fasting serum insulin levels and a lower homeostatic model assessment index compared with Ctrl-treated mice (1.55±0.17 vs 2.43±0.16 ng/mL, figure 5D and 0.45±0.05 vs 0.74±0.05, figure 5E). An intravenous glucose tolerance test showed slightly reduced levels in AM132-treated mice, but intravenous insulin tolerance test (ITT) and fasting glucose levels were similar to those of Ctrl-treated mice (see figure 5F and online supplementary figure S8E). Thus, at its peak effective time point, AM132 affected glucose homeostasis only minimally, possibly through its target Sirt1 which is known to improve insulin sensitivity.38
AM132 elevates miR-132 target levels
We quantified the hepatic levels of miR-132 targets and of metabolic transcripts by high-throughput qRT-PCR. At 7 days post-AM132 treatment, FoxO3, Pten, Sirt1 and Cyp2e1 were all upregulated (figure 6A). Cyp2e1 is notably involved in metabolic function,39 and our current findings highlight its relationship to hepatic steatosis. Protein levels of FoxO3, Sirt1, Pten and P300 were also elevated at 7–14 days post-treatment (see figure 6B,C and online supplementary figure S7C for 21 days post-treatment). ABCA1, AOX, PGC1a and Ehhadh transcripts were elevated at 7 days post-AM132 treatment, whereas glycerol-3-phosphate acyltransferase (GPAT) and SREBP2 transcripts were lowered (figure 6D, and additional transcripts in online supplementary figure S8F),40 ,41 indicating generally enhanced lipolysis, possibly via elevated PGC1a which activates farnesoid X receptor (FXR) and leads to triglyceride metabolism,42 elevated AOX and Ehhadh which lead to fatty acid oxidation,43 some reduction in lipogenesis due to reduced levels of GPAT which catalyses the first step of glycerolipid biosynthesis44 and SREBP2 which regulates both cholesterogenesis and lipogenesis.45 Interestingly, some transcripts modified in AM132-treated DIO mice showed inverse changes to those observed in the transgenic miR-132 dTg mice, reiterating the causal involvement of liver miR-132 in hepatic steatosis.

miR-132 suppression upregulates its liver targets. (A) Elevated liver miR-132 target transcripts in AM132-treated diet-induced obese mice (DIO) mice, 7 days post-treatment, determined by Fluidigm quantitative PCR and normalised to multiple housekeeping genes and then to control (Ctrl) treatment (n=4). (B) Western blot and quantification of elevated liver FoxO3, Sirt1 and Pten in AM132-treated DIO mice, 7 and 14 days post-treatment. (C) Immunofluorescence and quantification of elevated FoxO3 and P300 in liver sections. Arginase 1 (Arg1) served as a hepatocyte marker (n=3–4) and quantification was performed in areas that did not contain fat vacuoles. (D) Increased and reduced metabolic transcripts in AM132-treated DIO mice 7 days post-treatment (n=4). (E) The ratio of measured versus predicted serum low-density lipoprotein/very low-density lipoprotein (LDL/VLDL) values was calculated with a partial least squares regression (PLS-R) linear model in the five different in vivo experiments (GapmeRs, purple; DIO non-treated (NT), dark grey; DIO treated with AM132, red; miR-132 dTg, light grey). Elevating (dTg) or lowering (AM132 treated) miR-132 leads to serum LDL/VLDL levels that are higher or lower, respectively, than when reducing levels of a single miR-132 targets. Mean square error for training groups (T-MSE) was calculated according to GapmeR knockdown in vivo experiments. p<0.01 (Bonferroni post hoc). Data represent three experiments, *p<0.05, **p<0.01, ***p<0.001 (two-tailed Student's t-test). Values shown are mean±SEM.
AM132 affects serum LDL/VLDL levels through a multitarget mechanism
To assess the individual and cumulative effect of each miR-132 target in the various models we studied, we constructed a PLS-R linear model based on the expression profiles of the miR-132 mRNA targets in the four in vivo GapmeR experiments (figure 6E). This model allowed us to reduce the complexity of multiple target interactions while taking into account the simultaneous changes in expression of Pten, Sirt1, FoxO3 and P300 in each GapmeR experiment. Based on the combined expression patterns of these targets, we calculated the predicted serum LDL/VLDL levels for each experiment. These predicted levels were correlated with the measured levels of serum LDL/VLDL (R=0.71, Pearson’s correlation p<0.05, online supplementary figure S8F). Thus, individual mRNA target changes reliably estimated LDL/VLDL variations between GapmeR experiments. However, liver triglycerides failed to show such correlation (R=0.37, online supplementary figure S8F), indicating the causal effect of different elements on this parameter.
To test whether the serum LDL/VLDL modifications in response to miR-132 changes could be explained only by the combined effect of these four targets, we applied the model to our other experimental groups. In DIO non-treated mice, the measured LDL/VLDL did not differ from predicted levels, but strikingly, in AM132-treated DIO mice the measured serum LDL/VLDL levels were lower than predicted by our model, whereas in miR-132 dTg mice the measured serum LDL/VLDL levels were higher than predicted. That the effect of miR-132 perturbation was larger than the sum contribution of these four targets indicates a synergistic multitarget effect of miR-132, suggesting its high rank in the hierarchy of lipid metabolism regulation.
AM132 treatment acts in a dose-dependent manner
Mice injected with AM132 for three successive days with escalating doses of 0.8, 1.6 and 3.3 mg/kg/day, or with a single 10 mg/kg dose, presented a dose-dependent reduction of liver and liver/body weights, with the single 10 mg/kg treatment being as effective as that of the 3×3.3 mg/kg dose (see online supplementary figure S10A,B). Also, both the 10 mg/kg dose and the 3×3.3 mg/kg dose completely abolished microvesicular steatosis and reduced liver triglycerides and serum LDL/VLDL levels, but did not change serum HDL levels (see online supplementary figure S10C–F). DIO mice treated with fully 2-O-methylated and phosphorothioated oligonucleotides of identical sequence and length to AM132 and the control oligonucleotide showed similar reductions in liver weight, liver/body weight and serum LDL/VLDL levels (see online supplementary figure S11A–C). Thus, AM132 operates in a sequence and dose-dependent manner and regardless of its specific chemical structure.
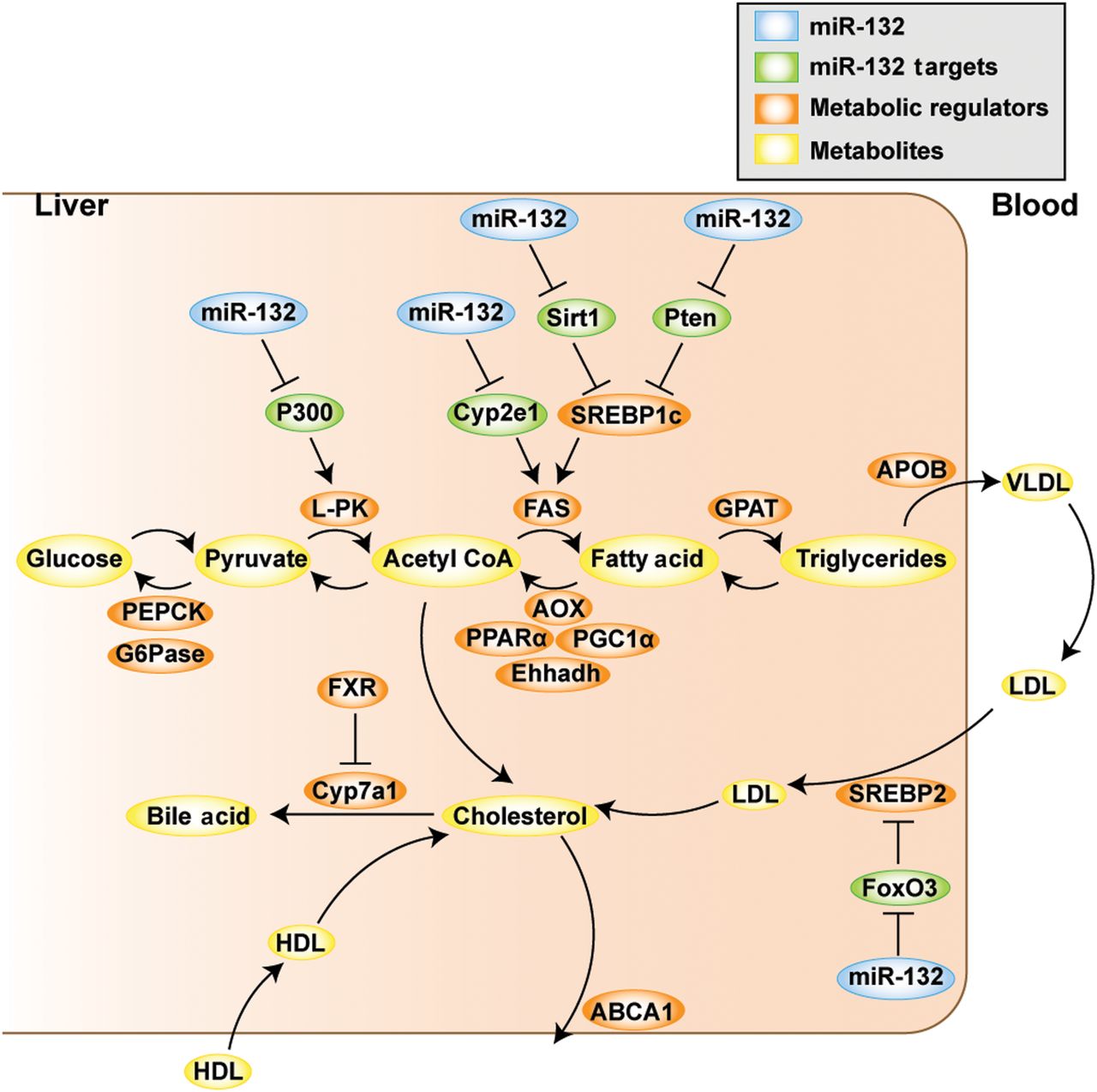
Last, we combined our metabolic transcript quantifications in the different experiments with published studies1 ,7 ,9 ,46 ,47 to construct a scheme depicting miR-132 regulation of lipid metabolism. This analysis (figure 7) suggests that the direct targets of miR-132 are upstream regulators of metabolically active genes, proposing that this miRNA plays an important role in initiating diverse processes that contribute to NAFLD pathogenesis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the role of miR-132 in regulating lipid metabolism in liver and blood. By post-transcriptional regulation of its direct targets (shown in green), miR-132 (blue) initiates downstream processes that lead to changes in transcripts regulating metabolism (orange), which results in altered serum and hepatic lipid profiles (components thereof in yellow).
Discussion
We have identified a novel role for miR-132 in hepatic lipid metabolism and NAFLD/NASH pathogenesis. Compatible with the overlap between anxiety and metabolism-controlling miRs,23 we have shown hepatic elevation of miR-132 and reduction of several validated metabolism-related miR-132 targets in conditional miR-132 overexpressing transgenic mice, in several NAFLD and NASH mouse models and in human liver tissue from patients with NAFLD. In vivo suppression of miR-132 reversed these changes in mouse models, but suppression of individual miR-132 targets only partially recapitulated the effect of peripheral miR-132 overexpression. These findings demonstrate inverse multitarget effects on liver steatosis by upregulation and downregulation of miR-132, in a number of models and diet conditions, indicating that the stress-inducible miR-132 operates high in the hierarchy of hepatic lipid homeostasis by suppressing multiple regulatory transcripts.
The reversal of fatty liver phenotype in AM132-treated DIO mice reflects the cumulative effect of modest increases in a number of mRNA targets of miR-132 and the corresponding decrease in protein levels. This cumulative or synergistic effect could be instigated by context-dependent changes in the level of multiple, but not necessarily identical target genes. The elevated weight of transgenic mice overexpressing miR-132 on and off doxycycline suggests leakiness in the Tet-On system and reflects the multitissue expression pattern of the ROSA26 promoter,29 as was supported by the similar food consumption of miR-132 dTg and control rtTA mice. Control experiments excluded toxicity and indicated sequence specificity of AM132, and in vivo treatment did not decrease body weight, perhaps reflecting selective liver targeting of the injected oligonucleotide,48 as the liver is the major site for oligonucleotide deposition after intravenous administration.49 Although it is possible that a minor fraction of the oligonucleotide also affects additional organs, penetration into the brain is apparently minimal.50
Silencing of single miR-132 targets did not always phenocopy the impact of miR-132 overexpression. In FoxO3 and Pten knockdown, we saw increased liver/body weight and LDL/VLDL, whereas P300 knockdown led to reduced liver/body weight and Sirt1 knockdown did not show a significant effect. Some of the targets studied in this work have previously been linked to liver pathology: knockouts of Pten and Sirt1 were reported to cause hepatic steatosis in mice15 ,16, although only when challenged with high fat diet, in contrast to our miR-132 dTg mice which developed steatosis when fed with regular chow. FoxO3 is known to have an important role in glucose metabolism, and its knockout yielded hyperlipidaemia.27 In comparison, the role of P300 in hepatic steatosis is widely debated, with some claiming that its inhibition could be beneficial in treating hepatic steatosis,46 while others report its altered function in metabolic disease states.51 In our experiments, knockdown of P300 in DIO mice led to a certain improvement in steatosis. Others reported that liver-specific deletion of Sirt1 leads to hepatic steatosis.16 That in our system Sirt1 knockdown did not show any significant effect may indicate differences in experimental layout.
The miR-132 targets that were modulated in our study overlap only partially with those in other reports, possible reflecting differences in administered diets, genetic background and various unknown compensation mechanisms in the miR-132 dTg mice. This suggests that distinct targets of miR-132 may contribute to its dyslipidemic impact in different states, due to the influence of distinct downstream effectors and to their interactions with partner proteins. Future studies will be required to identify and determine the role of additional miR-132 targets in hepatic steatosis. Replication of the results seen in human liver samples in an additional cohort will be of importance in determining the therapeutic potential of miR-132 modification in treatment of NAFLD.
Other miRNAs have been associated with liver steatosis: miR-122 has been well studied and is known to target SLC7A1 and ADAM17; miR-33 targets numerous genes in the fatty acid β-oxidation and cholesterol efflux pathways;1 ,3 miR-34a affects lipid metabolism through its target Sirt1,3 and miR-148a targets ABCA1, thus regulating circulating lipoprotein levels.47 miRNAs can either downregulate mRNA levels or directly repress translation,52 which may explain why after AM132 treatment in the DIO model, changes in mRNA levels of some targets do not correspond to the changes in protein levels. Therapies being developed for NASH include obeticholic acid, an FXR agonist, but this treatment causes concurrent elevation in LDL cholesterol.53 Interestingly, our AM132-treated DIO mice showed elevated FXR along with the reduced liver steatosis and LDL cholesterol.
In conclusion, we have discovered a unique function of miR-132 as a novel context-dependent rheostat that regulates hepatic lipid metabolism by finely tuning multiple targets in diverse conditions, hopefully establishing the basis for a deeper understanding and an innovative treatment of hepatic lipid metabolism disorders of various aetiologies.
References
Footnotes
Contributors GH planned and performed all of the experiments, analysed and interpreted data and wrote the manuscript; NY constructed the model and performed the partial least square and principal component analysis (PCA) analysis; YT performed in vivo experiments and contributed to the biochemical analysis; RH performed quantitative PCR, planned and performed the Fluidigm analyses and contributed to the in vivo experiments and biochemical analysis; ERB performed western blots, planned some of the experiments and edited the manuscript; SU and JT planned, performed and analysed metabolic cage data; RZ measured miR-132 levels; RZ and BE contributed to some of the in vivo experiments; YRK contributed methionine-deficient and choline-deficient diet, high fat and high sucrose diet and choline-deficient and ethionine-supplemented diet mouse tissues and histology; EK contributed choline-deficient high fat diet (CDHFD) mouse tissues and histology; OP performed pathological analysis; ES contributed to the clinical aspects; EP advised and contributed mouse non-alcoholic steatohepatitis (NASH) samples; MH contributed murine CDHFD liver NASH samples; DSG planned the experiments, interpreted data and edited the manuscript; HS guided the project, planned the experiments, interpreted data and edited the manuscript; all coauthors read and commented on the manuscript contents.
Funding This study was supported by the European Research Council Advanced Award CholinomiRs 321501, the European Commission FP-7 Health-2013-Innovation Grant #602133, the European Union's Horizon 2020 research and innovation programme 639314, the Israel Science Foundation grant 817/13 and The Hebrew University's Translational Research programme (to HS). MH received an European Research Council (ERC) consolidator grant HepatoMetaboPath.
Competing interests None declared.
Patient consent Obtained.
Ethics approval ProteoGenex, Culver City, California, USA.
Provenance and peer review Not commissioned; externally peer reviewed.