Article Text

Abstract
Objective Accumulation of free fatty acids (FFAs) in hepatocytes induces lipotoxicity, leading to non-alcoholic fatty liver disease (NAFLD). This study aimed to investigate the underlying mechanisms by which FFA contributes to the pathogenesis of NAFLD via the regulation of 3-mercaptopyruvate sulfurtransferase (MPST), a key enzyme that regulates endogenous hydrogen sulfide (H2S) biosynthesis.
Design Hepatic MPST expression was evaluated in mice and patients with NAFLD. A variety of molecular approaches were used to study the effects of MPST regulation on hepatic steatosis in vivo and in vitro.
Results In vitro treatment of hepatocytes with FFAs upregulated MPST expression, which was partially dependent on NF-κB/p65. Hepatic MPST expression was markedly increased in high fat diet (HFD)-fed mice and patients with NAFLD. Partial knockdown of MPST via adenovirus delivery of MPST short hairpin RNA or heterozygous deletion of the Mpst gene significantly ameliorated hepatic steatosis in HFD-fed mice. Consistently, inhibition of MPST also reduced FFA-induced fat accumulation in L02 cells. Intriguingly, inhibition of MPST significantly enhanced rather than decreased H2S production, whereas MPST overexpression markedly inhibited H2S production. Co-immunoprecipitation experiments showed that MPST directly interacted with and negatively regulated cystathionine γ-lyase (CSE), a major source of H2S production in the liver. Mechanistically, MPST promoted steatosis via inhibition of CSE/H2S and subsequent upregulation of the sterol regulatory element-binding protein 1c pathway, C-Jun N-terminal kinase phosphorylation and hepatic oxidative stress.
Conclusions FFAs upregulate hepatic expression of MPST and subsequently inhibit the CSE/H2S pathway, leading to NAFLD. MPST may be a potential therapeutic target for NAFLD.
- fatty liver
- liver metabolism
- hepatocyte
- hydrogen sulfide
- oxidative stress
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Free fatty acids (FFAs) and their metabolites are important mediators of lipotoxicity, leading to the progression of non-alcoholic fatty liver disease (NAFLD).
The liver is an important organ in the regulation of hydrogen sulfide (H2S) metabolism, and H2S homeostasis plays an important role in hepatic lipotoxicity.
Reported evidence suggests that the cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) system, which contributes to endogenous H2S biosynthesis, may be regulated by fatty acids. The role of the CBS/CSE system in the pathogenesis of NAFLD has been actively investigated, and the system is proposed to serve as a potential therapeutic target for NAFLD.
3-Mercaptopyruvate sulfurtransferase (MPST) is also an important enzyme that contributes to the generation of H2S by using 3-mercaptopyruvate as substrate in a pyridoxal-5′-phosphate (PLP)-independent way, which is different from the CBS/CSE system by way of using cysteine as substrate that is dependent on PLP.
What are the new findings?
FFAs induced upregulation of hepatic MPST protein expression in high-fat diet (HFD)-fed mice and patients with NAFLD.
Hepatic H2S synthesis is impaired in NAFLD models in vivo and in vitro. Partial knockdown of MPST significantly elevated H2S levels by directly interacting with and negatively regulating CSE.
Partial knockdown of MPST via adenovirus delivery of MPST short hairpin RNA or heterozygous deletion of the Mpst gene significantly ameliorated hepatic steatosis in HFD-fed mice. Small interfering RNA-mediated partial knockdown of MPST reduced fat accumulation, while MPST overexpression exacerbated lipid accumulation in FFA-treated L02 cells.
MPST upregulates the sterol regulatory element-binding protein 1c pathway, C-Jun N-terminal kinase phosphorylation and hepatic oxidative stress, which is mediated by inhibition of CSE/H2S.
How might it impact on clinical practice in the foreseeable future?
MPST may be a potential therapeutic target for NAFLD.
Inhibition of MPST may be a novel therapeutic strategy for the treatment of NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disorder in Western countries, with a prevalence of 20%–30% in the general population.1 2 The disease encompasses a broad spectrum of clinicopathology, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH); the latter may further progress to liver cirrhosis and hepatocellular carcinoma.3 4 The number of patients with NAFLD is growing at alarming rates worldwide. However, its pathogenesis remains poorly understood but is of great interest, since current therapeutic options are limited.
Emerging data indicate that the flux of fatty acids through the liver is increased in patients with NASH.5 Free fatty acids (FFAs) and their metabolites are important mediators of lipotoxicity via the induction of lipid overaccumulation, which causes lipotoxic hepatocellular injury and the progression of NAFLD.6 However, the exact mechanisms by which FFAs cause lipotoxicity remain unclear.
Hydrogen sulfide (H2S), which has been known for centuries as a toxic gas with the smell of rotten eggs,7 has been established as the third gaseous signalling molecule following nitric oxide and carbon monoxide, participating in a wide range of physiological processes, including inflammation, apoptosis, vasorelaxation and neuromodulation.8–10 The role of H2S in NAFLD has been received great attention over the last two decades. The liver is an important organ in the regulation of H2S metabolism.11 Conversely, H2S plays critical roles in pathophysiology of liver diseases.12 H2S protected against liver injury induced by ischemia reperfusion and carbon tetrachloride in rodents.13–15 Recent studies revealed that the endogenous formation of H2S was impaired in high fat diet (HFD)-fed mice and rats with NASH.16 17 Treatment with sodium hydrosulfide (NaHS), a H2S donor, prevented NASH in rodents by abating oxidative stress and suppressing inflammation.17–19 These findings suggest that H2S homeostasis plays an important role in hepatic lipotoxicity. However, how H2S biosynthesis is regulated in NAFLD remains unclear.
In mammalian tissues, H2S can be produced from cysteine by pyridoxal-5′-phosphate (PLP)-dependent enzymes including cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE); the former is mainly expressed in the brain and the latter is expressed in the vasculature and liver.20 3-Mercaptopyruvate sulfurtransferase (MPST) is another enzyme PLP independently that uses 3-mercaptopyruvate as substrate to generate H2S.21 The CBS/CSE system, which may be regulated by several fatty acids, has been actively investigated in the pathogenesis of NAFLD and proposed to serve as a potential therapeutic target for NAFLD.22 However, the role of MPST in NAFLD has not been investigated. To date, whether MPST is involved in the regulation of hepatic lipid metabolism and in the pathogenesis of NAFLD remains unknown.
In this study, we investigated the underlying mechanisms by which fatty acids contribute to the pathogenesis of NAFLD, involving the role of MPST in the development of NAFLD. Our results provided novel evidence that FFAs stimulate the expression of hepatic MPST in NAFLD, and this upregulated MPST promotes NAFLD via inhibition of the CSE/H2S pathway.
Materials and methods
Human samples
Liver biopsies from liver transplant donors who were healthy adults (n=19) and from patients with NAFLD (n=37) who underwent liver biopsies for suspected NAFLD or NASH or during liver surgeries were randomly selected from the liver biopsy samples recruited at the First Affiliated Hospital, College of Medicine, Zhejiang University; The Affiliated Hospital of Hangzhou Normal University; Renji Hospital, School of Medicine, Shanghai Jiao Tong University; Hangzhou Sixth People’s Hospital; and Ningbo Medical Treatment Center Lihuili Hospital. The inclusion criteria and clinical characteristics of patients were provided in the online supplementary data. All liver tissue samples from patients used in this study were approved by the Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University, and all patients provided their written informed consent.
Supplementary file 1
Animal experiments
An intravenous hydrodynamic injection of adenovirus plasmids that contained green fluorescent protein (AD-GFP) coding sequence and MPST short hairpin RNA (AD-shMPST) was administered to male C57BL/6 mice to inhibit hepatic MPST expression. Heterozygous MPST-deficient (MPST+/-) mice on a C57BL/6 background were generated through frameshift mutation by TAL-effector nuclease system by Beijing ViewSolid Biotechnology (Beijing, China). Detailed explanations of generation strategy and all animal treatments are provided in the online supplementary data and online supplementary figure 1. All of the animal experiments were performed according to the guidelines approved by the Animal Care and Use Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University.
Other materials and methods are described in details in the online supplementary data.
Results
FFAs upregulate hepatic MPST expression in HFD-fed mice and in patients with NAFLD
A previous study suggests that the CBS/CSE system may be regulated by FFAs.22 Here we determined whether FFAs also regulate MPST. To test this hypothesis, we stimulated human hepatocyte cell lines with FFAs to establish in vitro models of hepatocyte steatosis (online supplementary figure 2A, B) and measured MPST expression. As illustrated in figure 1A and online supplementary figure 2C, FFA treatment markedly upregulated MPST protein expression in FFA-stimulated L02 cells and 7701 cells. The blockade of NF-κB, an important downstream signal of FFAs,23 reduced MPST expression (figure 1B), while the overexpression of NF-κB/p65 enhanced MPST expression in FFA-stimulated L02 cells (figure 1C). Next we examined whether FFAs also upregulate hepatic expression of MPST in vivo in HFD-fed mice, in which hepatic FFA levels were significantly elevated compared with controls (figure 1D). As illustrated in figure 1E, hepatic MPST protein expression was upregulated by approximately 1.5-fold in HFD-fed mice compared with control-diet-fed mice. Finally, we measured the levels of MPST protein in liver biopsies from patients with NAFLD, who exhibit increased FFA flux through the liver and elevated FFA concentrations in circulation (online supplementary table 1).5 24 As shown in figure 1F, immunohistochemistry experiments revealed markedly higher MPST protein expression within the cytoplasm of steatotic hepatocytes from patients with NAFLD compared with those from healthy controls, which was further confirmed by semiquantitative analysis of integral optical density of MPST positive areas. In addition, there was a positive correlation between the area of steatosis and MPST expression in the livers of patients with NAFLD (figure 1G). Collectively, these data suggest that hepatic steatosis is associated with the FFA-induced upregulation of MPST in NAFLD.
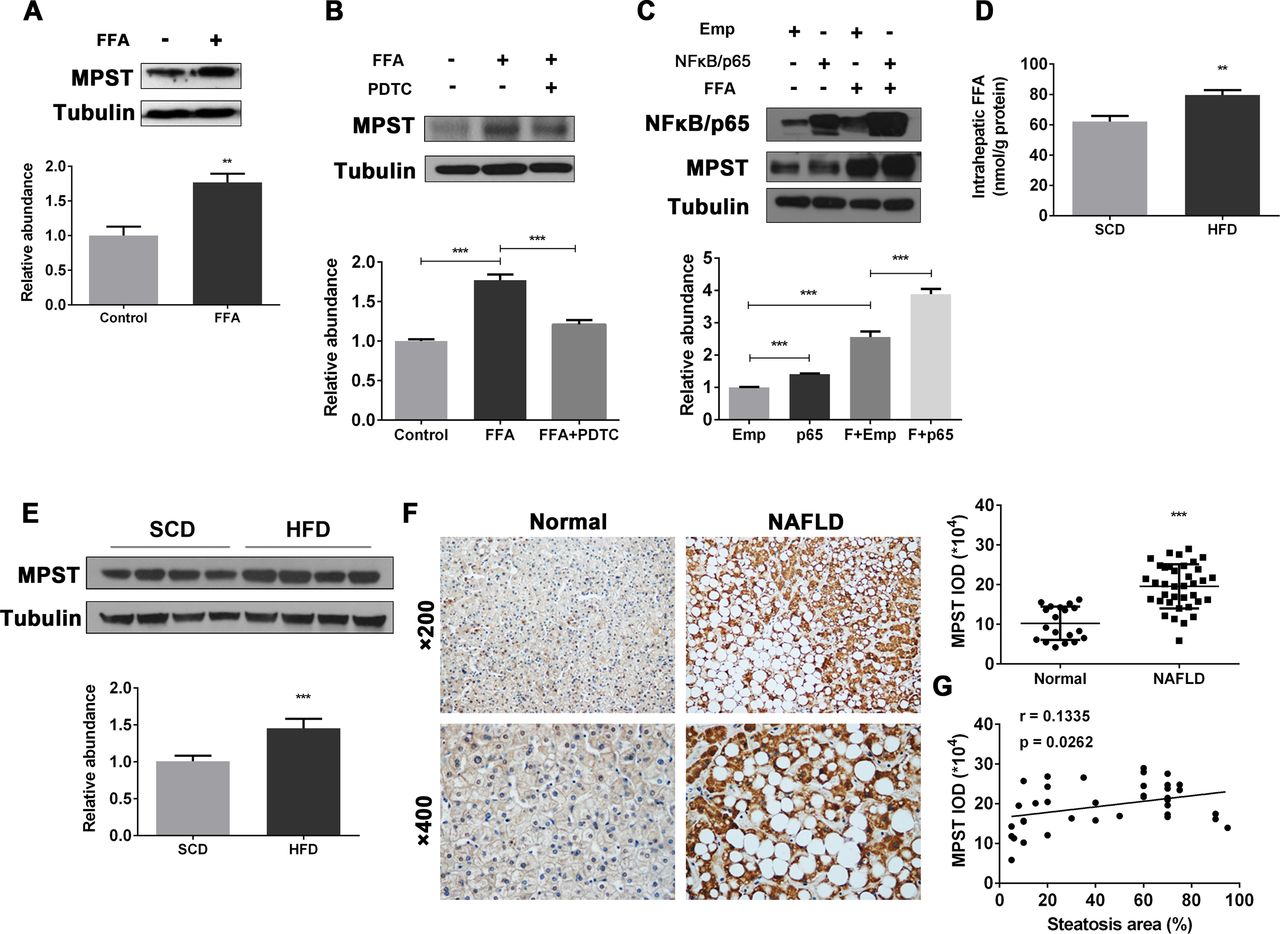
Upregulation of hepatic MPST expression in HFD-fed mice and patients with NAFLD: a role of FFAs.(A) L02 cells were treated with FFAs (1 mM) for 24 hours and then subjected to western blot analysis. (B) L02 cells were pretreated with pyrrolidine dithiocarbamate (NF-κB-specific inhibitor) (100 µM) for 1 hour and then treated with FFAs (1 mM) for 24 hours. Western blot analyses were performed. (C) pENTER-NF-κB/p65 plasmid or empty vector was transfected into L02 cells for 24 hours, followed by FFA treatment for 24 hours. Western blot analyses were performed. Liver tissues from 8-week HFD-fed and SCD-fed mice were obtained and subjected to intrahepatic FFA measurement (D) or western blot analysis (E). The bar graph indicates relative MPST abundance as quantified by densitometry from the immunoblots after normalisation to tubulin. (F) Representative images of immunohistochemistry staining of liver sections from controls (n=19) and patients with NAFLD (n=37). Original magnification, upper panel, ×200; lower panel, ×400. Semiquantitative analysis for the IOD of MPST positive areas is shown on the right of the images. (G) The area of steatosis positively correlated with MPST expression in the livers of patients with NAFLD. The results are expressed as the mean±SD of five mice per group or three independent experiments in A–E. *p<0.05, **p<0.01, ***p<0.001. FFA, free fatty acid; HFD, high fat diet; IOD, integral optical density; MPST, 3-mercaptopyruvate sulfurtransferase; NAFLD, non-alcoholic fatty liver disease; SCD, standard chow diet.
Partial knockdown of MPST markedly ameliorates hepatic steatosis in vivo
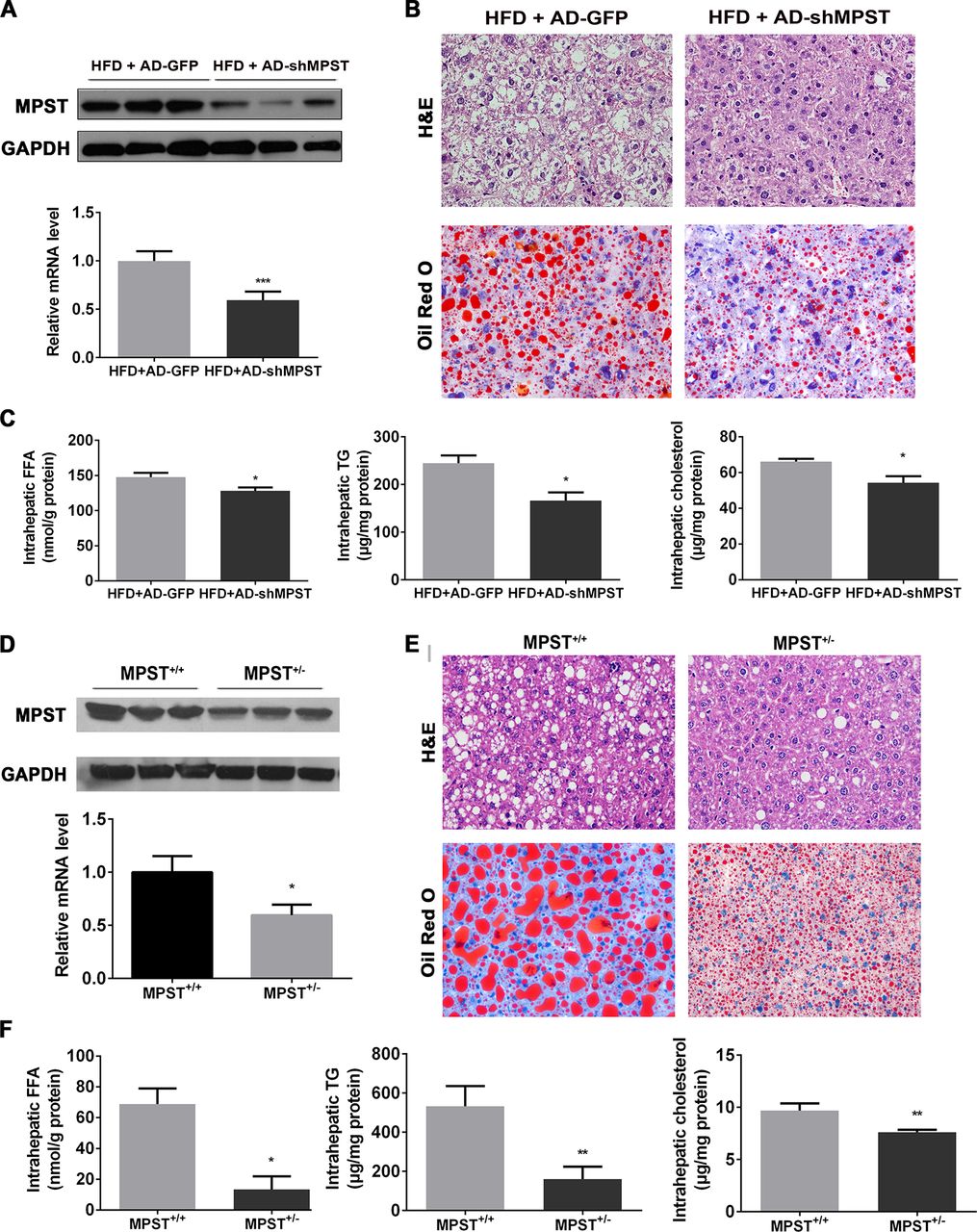
To investigate the role of MPST in NAFLD, we first applied a recombinant adenovirus-mediated RNA interference approach to inhibit hepatic MPST expression in HFD-fed mice. As illustrated in figure 2A, hepatic MPST expression was inhibited by approximately 40% at the messenger RNA (mRNA) level and 60% at the protein levels after the injection of the adenovirus. Notably, hepatic steatosis was improved after partial knockdown of hepatic MPST expression, as demonstrated by H&E and Oil Red O staining (figure 2B) and the measurement of triglyceride (TG) and total cholesterol (TC) levels (figure 2C). In addition, hepatic FFA levels and plasma TC levels were also decreased after MPST knockdown (figure 2C, online supplementary table 2). There was also a trend toward reduction of plasma transaminase levels after MPST knockdown, but the difference did not reach statistical significance (online supplementary table 2).

Partial knockdown of MPST markedly ameliorates hepatic steatosis in vivo. (A–C) HFD-fed C57/BL6 mice were given 1×109 plaque-forming units of AD-GFP or adenovirus-mediated knockdown of MPST (AD-shMPST) via the tail vein every 2 weeks from the beginning of the experiment and were sacrificed after 8 weeks of HFD feeding. (A) Liver tissues were subjected to western blot analyses of MPST protein level (upper panel) or real-time qPCR for MPST mRNA level determination (lower panel). (B) Representative images of H&E and Oil Red O staining of livers infected with AD-GFP and AD-shMPST in HFD-fed mice. Original magnification ×400. (C) Liver tissues were subjected to intrahepatic FFA measurement (left panel), intrahepatic TG measurement (middle panel) or intrahepatic TC measurement (right panel). (D–F) MPST+/+ and MPST+/- mice were fed an HFD for 8 weeks. (D) Liver tissues were subjected to western blot analyses of MPST protein level (upper panel) or real-time qPCR for MPST mRNA level determination (lower panel). (E) Representative images of H&E and Oil Red O staining of liver sections in HFD-fed MPST+/+ and MPST+/- mice. Original magnification ×400. (F) Liver tissues were subjected to intrahepatic FFA measurement (left panel), intrahepatic TG measurement (middle panel) or intrahepatic TC measurement (right panel). The results are expressed as the mean ±SD of 3–5 mice per group from two independent experiments. *p<0.05, **p<0.01, ***p<0.001. FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HFD, high fat diet; MPST, 3-mercaptopyruvate sulfurtransferase; qPCR, quantitative PCR; TC, total cholesterol; TG, triglyceride.
To further investigate the effect of partial MPST deficiency in NAFLD, heterozygous MPST+/– mice and their wild-type (WT) controls (MPST+/+) were placed on an HFD for 8 weeks. As illustrated in figure 2D, hepatic MPST expression was decreased both at the protein and mRNA level in HFD-fed MPST+/- mice compared with WT controls. In accordance with the effect of adenovirus-mediated MPST partial knockdown in HFD-fed mice, the partial deletion of MPST in MPST+/- mice markedly ameliorated the fatty liver phenotype (figure 2E); reduced hepatic FFAs and TG and TC contents (figure 2F); and downregulated the plasma level of TC, particularly very-low-density lipoprotein cholesterol (online supplementary table 3). Plasma alanine transaminase and aspartate transaminase levels also showed a decreased tendency in HFD-fed MPST+/- mice compared with WT controls (online supplementary table 3). Taken together, these results indicate that MPST is an important factor that facilitates the development of NAFLD.
Partial knockdown of MPST markedly attenuates while the overexpression of MPST exacerbates FFA-induced fat accumulation in hepatocytes in vitro
To investigate the effect of MPST on FFA-induced fat accumulation in hepatocytes in vitro, MPST expression was knocked down by MPST small interfering RNA (siRNA) in cultured hepatocytes (figure 3A), and the knockdown significantly ameliorated fat accumulation (Oil Red O staining in figure 3B) and intracellular TG and TC contents in the FFA-stimulated L02 cells (figure 3C). In contrast, the overexpression of MPST via the transfection of the pIRES2-GFP-MPST plasmid (figure 3D) markedly increased lipid accumulation (figure 3E) and elevated TG and TC levels in the FFA-stimulated L02 cells (figure 3F).

Partial knockdown of MPST markedly attenuates while overexpression of MPST exacerbates FFA-induced fat accumulation in hepatocyte in vitro. (A–C) After transfection with scrambled siRNA (negative control) and MPST siRNA for 24 hours, L02 cells were treated with FFAs for 24 hours. (A) Western blot and qPCR analyses were performed to determine MPST expression. (B) Representative images of Oil Red O staining of L02 cells is shown. Original magnification ×400. (C) Intracellular TG and TC contents were determined. (D–F) After transfecting with plasmid containing GFP with or without full-length MPST DNA for 24 hours, L02 cells were treated with FFAs for 24 hours. (D) Western blot and qPCR analyses were performed to determine MPST expression. (E) Representative images of Oil Red O staining of L02 cells is shown. Original magnification ×400. (F) The intracellular TG and TC contents were determined. The results are expressed as the mean±SD of three independent experiments. *p<0.05, **p<0.01, ***p<0.001. FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; MPST, 3-mercaptopyruvate sulfurtransferase; mRNA, messenger RNA; qPCR, quantitative PCR; siRNA, small interfering RNA; TC, total cholesterol; TG, triglyceride.
Partial knockdown of hepatic MPST significantly increases H2S production
MPST is an important generator of H2S, which plays a critical role in the preservation of normal lipid metabolism under physiological or pathophysiological conditions.12 25 26 Therefore, we hypothesized that MPST regulated hepatic steatosis via the regulation of H2S production. First, we measured H2S levels in HFD-fed mice and FFA-treated L02 cells. As illustrated in figure 4, HFD feeding markedly downregulated hepatic H2S levels (figure 4A), and FFA treatment reduced supernatant H2S levels of L02 cells (figure 4B). Second, we examined the effects of partial knockdown of hepatic MPST on hepatic H2S levels in HFD-fed mice and in FFA-treated L02 cells. Unexpectedly, partial knockdown of MPST via adenovirus-mediated short hairpin RNA (shRNA) delivery or heterozygous deletion markedly upregulated hepatic H2S levels in HFD-fed mice (figure 4C, D). Similar to the in vivo results, siRNA-mediated partial knockdown of MPST significantly elevated H2S levels in the supernatants of FFA-treated L02 cells (figure 4E), whereas the overexpression of MPST reduced H2S levels in the supernatants of FFA-treated L02 cells (figure 4F).

Partial knockdown of MPST significantly increases H2S levels. (A) Liver tissues from 8-week HFD-fed and SCD-fed mice were subjected to intrahepatic H2S measurements. (B) L02 cells were treated with FFAs (1 mM) for 24 hours, and the fresh supernatants were collected to measure H2S levels. (C) HFD-fed C57/BL6 mice were given 1×109 plaque-forming units of AD-GFP or adenovirus shMPST (AD-shMPST) via the tail vein every 2 weeks from the beginning of experiment and were sacrificed after 8 weeks of HFD feeding. Liver tissues were subjected to intrahepatic H2S measurement. (D) Quantification of intrahepatic H2S levels in HFD-fed MPST+/+ and MPST+/- mice. (E) After transfection with scrambled siRNA (negative control) and MPST siRNA for 24 hours, L02 cells were treated with FFAs (1 mM) for 24 hours. The fresh supernatants were collected to measure H2S levels. (F) After transfection with plasmid containing GFP with or without full-length MPST DNA for 24 hours, L02 cells were treated with FFAs for 24 hours. The fresh supernatants were collected to measure H2S levels. The results are expressed as the mean±SD of 3–5 mice per group from two independent experiments or three independent experiments. *p<0.05, **p<0.01. FFA, free fatty acid; GFP, green fluorescent protein; H2S, hydrogen sulfide; HFD, high fat diet; MPST, 3-mercaptopyruvate sulfurtransferase; siRNA, small interfering RNA.
Partial knockdown of hepatic MPST upregulates CSE through a direct MPST–CSE interaction
MPST is a generator of H2S in the liver, but unexpectedly, our above data revealed that partial knockdown of MPST elevated hepatic H2S levels in vivo and in vitro. To elucidate the molecular mechanisms of MPST on H2S metabolism in NAFLD, we searched potential MPST-interacting proteins in the STRING database27 (online supplementary figure 3A) and found that MPST has a potential functional connection with CSE (also known as CTH), a main H2S-generating enzyme that is predominantly expressed in the vasculature and liver (online supplementary figure 3B). The score of prediction for the MPST–CSE interaction is 0.977, ranking the first place among the top 10 functional MPST-centric protein-interaction networks (online supplementary figure 3C). Interestingly, partial knockdown of MPST via either adenovirus-mediated shRNA delivery or heterozygous deletion significantly upregulated hepatic CSE expression in the HFD-fed mice (figure 5A, B). The in vitro results also showed that partial knockdown of MPST significantly enhanced CSE expression (figure 5C), while the overexpression of MPST decreased CSE expression in FFA-treated L02 cells (figure 5D).

Partial knockdown of MPST upregulates CSE by interacting with CSE. (A) HFD-fed C57/BL6 mice were given 1×109 plaque-forming units of AD-GFP or AD-shMPST via the tail vein every 2 weeks from the beginning of experiment and were sacrificed after 8 weeks of HFD feeding. Liver tissues were subjected to western blot analyses of hepatic CSE protein expression. (B) Western blot analyses of hepatic CSE expression in HFD-fed MPST+/+ and MPST+/- mice. (C) After transfection with scrambled siRNA (negative control) and MPST siRNA for 24 hours, L02 cells were treated with FFAs (1 mM) for 24 hours. Western blot analyses were performed to determine CSE protein expression. (D) After transfection with plasmid containing GFP with or without full-length MPST DNA for 24 hours, L02 cells were treated with FFAs (1 mM) for 24 hours. Western blot analyses were performed to determine CSE protein expression. (E) Immunoblot analysis of the interaction between MPST and CSE by co-immunoprecipitation. FFA-treated L02 cells were transfected with Flag-MPST and subjected to immunoprecipitation with an anti-Flag monoclonal antibody. Co-immunoprecipitated CSE was detected with an anti-CSE antibody. The results are expressed as the mean±SD of 3–5 mice per group from two independent experiments or three independent experiments. *p<0.05, **p<0.01. CSE, cystathionine γ-lyase; FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; HFD, high fat diet; IB, immunoblot; IP, immunoprecipitation; MPST, 3-mercaptopyruvate sulfurtransferase; siRNA, small interfering RNA.
To verify the protein–protein interaction prediction, we prepared stably expressing Flag-MPST L02 cells and performed co-immunoprecipitation (co-IP) assays (online supplementary figure 3D). Our results revealed a marked interaction between MPST and CSE after FFA treatment (figure 5E).
MPST attenuates H2S production and subsequently promotes fat accumulation in hepatocytes through a CSE-dependent mechanism
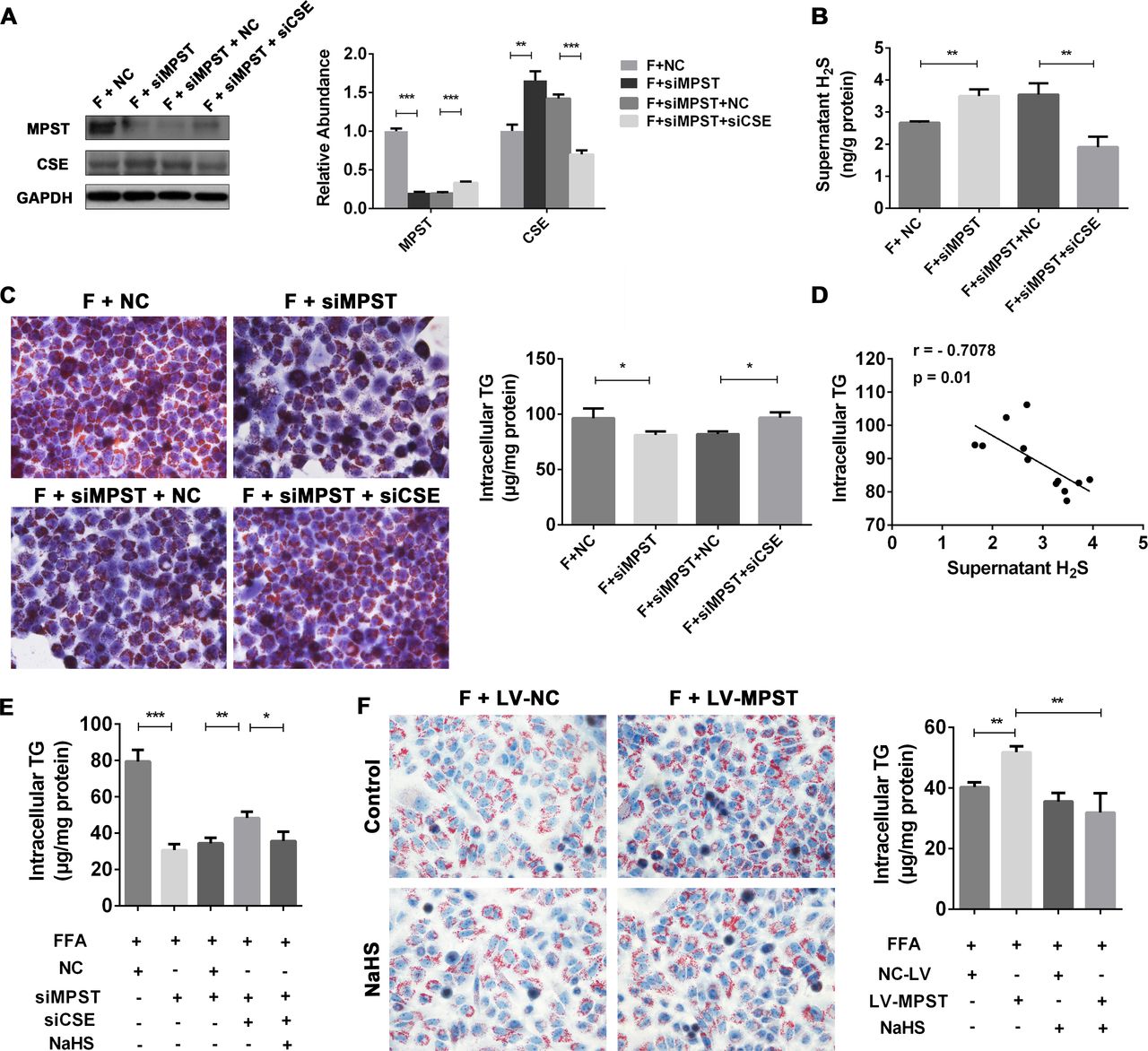
The above data suggest that MPST downregulates hepatic expression of CSE. Recent reports have identified a role of CSE/H2S in promoting fatty liver development.25 28 Thus we hypothesized that the MPST-induced inhibition of H2S production is facilitated via the compensatory inhibition of CSE. To test this hypothesis, we first measured hepatic CSE levels. In contrast to the upregulation of MPST in HFD-fed mice and FFA-treated L02 cells as described above, hepatic expression of CSE was downregulated under these conditions (online supplementary figure 4A, B). Second, we used siRNA to knockdown CSE expression (online supplementary figure 4C, D) to define the role of CSE in the MPST-mediated regulation of H2S. As illustrated in figure 6A, inhibition of MPST upregulated hepatic expression of CSE protein, while inhibition of CSE increased hepatic expression of MPST. Figure 6B, C show that inhibition of MPST elevated supernatant H2S levels but decreased intracellular TG levels; both of these effects were reversed by CSE siRNA. Interestingly, a negative correlation between H2S levels and intracellular TG contents was observed (figure 6D). This observation prompted us to investigate whether H2S is responsible for the protection from lipid deposition within hepatocytes. We found that treatment with NaHS (H2S donor) reduced lipid accumulation in FFA-stimulated L02 cells (online supplementary figure 5A, B) and suppressed the expression of both MPST and CSE (online supplementary figure 5C). Furthermore, exogenous H2S supplementation decreased intracellular TG contents in L02 cells treated with both MPST and CSE siRNAs (figure 6E). Finally, the incubation of FFA-treated cells with NaHS prevented the exacerbation of fat accumulation induced by MPST overexpression (figure 6F). These data suggest that MPST regulated H2S production through a CSE-dependent mechanism and that H2S mediates the potent anti-fatty liver effects in the MPST regulation of hepatocyte steatosis.

MPST attenuates H2S production and subsequently promotes fat accumulation in hepatocytes through CSE dependent-mechanism. (A–C) After transfection with scrambled siRNA (negative control) and MPST siRNA for 24 hours, L02 cells were transfected with or without CSE siRNA for 24 hours. Then the cells were treated with FFAs (1 mM) for another 24 hours and subjected to western blot analyses (A). The fresh supernatants were collected to measure H2S levels (B). Representative Oil Red O staining images and measurements of intracellular TG content are shown in (C). Original magnification ×400. (D) The correlation between supernatant H2S levels and intracellular TG contents in FFA-treated L02 cells was analysed. (E) L02 cells transfected with MPST siRNA and CSE siRNA were pretreated with NaHS (1 mM) for 2 hours and then treated with FFAs (1 mM) with or without NaHS for 24 hours. Intracellular TG contents were determined. (F) L02 cells were pretreated with NaHS (1 mM) for 2 hours and then treated with FFAs (1 mM) with or without NaHS for 24 hours. Representative Oil Red O staining images and measurements of intracellular TG contents were shown. The results are expressed as the mean±SD of three independent experiments. *p<0.05, **p<0.01, ***p<0.001. CSE, cystathionine γ-lyase; FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H2S, hydrogen sulfide; MPST, 3-mercaptopyruvate sulfurtransferase; NaHS, sodium hydrosulfide; siRNA, small interfering RNA; TG, triglyceride.
SREBP-1 pathway is involved in the MPST regulation of fat accumulation in hepatocytes
De novo lipogenesis (DNL) can be amplified by increases in sterol regulatory element binding protein 1c (SREBP-1), the central transcription factor that induces the expression of enzymes involved in fatty acids DNL, hepatic TG and cholesterol synthesis.29 The overexpression of SREBP-1 in animal models has been identified as a contributor to lipotoxic liver injury.30
Interestingly, previous studies suggested that antisteatogenic effects of H2S are mediated via the modulation of SREBP-1 expression.17 31 Here we also demonstrated that the upregulated SREBP-1 expression levels in FFA-treated L02 cells were counteracted by exogenous H2S supplementation, which suggests that H2S regulated lipid metabolism mainly by modulating SREBP-1 (online supplementary figure 5C). Therefore, we hypothesized that MPST regulates fat accumulation via regulation of H2S production and SREBP-1 expression. As illustrated in figure 7A, inhibition of MPST markedly downregulated hepatic expression of SREBP-1 protein and its downstream proteins including fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC) in HFD-fed mice. In parallel with in vivo results, knockdown of MPST in the FFA-stimulated L02 cells markedly inhibited the protein expressions of SREBP-1 and its downstream proteins (figure 7B). In contrast, the overexpression of MPST significantly upregulated SREBP-1, ACC and FAS expressions in the FFA-stimulated L02 cells (figure 7C). Furthermore, MPST siRNA-mediated downregulation of SREBP-1, ACC and FAS was restored by CSE siRNA treatment in FFA-treated L02 cells (figure 7D). Finally, co-IP assay results demonstrated the protein–protein interaction between MPST and SREBP-1.

SREBP-1 pathway is involved in MPST-regulated fat accumulation in hepatocytes. (A) HFD-fed C57/BL6 mice were given 1×109 plaque-forming units of AD-GFP or AD-shMPST via the tail vein every 2 weeks from the beginning of experiment and were sacrificed after 8 weeks of HFD feeding. Liver tissues were subjected to western blot analyses. (B) After transfection with scrambled siRNA (negative control) and MPST siRNA for 24 hours, L02 cells were treated with FFAs (1 mM) for 24 hours. Western blot analyses were performed to determine SREBP-1, FAS and ACC protein expressions. (C) After transfecting with plasmid containing GFP with or without full-length MPST DNA for 24 hours, L02 cells were treated with FFAs (1 mM) for 24 hours. Western blot analyses were performed. (D) After the transfection with NC and MPST siRNA for 24 hours, L02 cells were transfected with or without CSE siRNA for 24 hours. Then the cells were treated with FFAs (1 mM) for another 24 hours and subjected to western blot analyses. (E) Immunoblot analysis of the interaction between MPST and SREBP-1 by co-immunoprecipitation. FFA-treated L02 cells were transfected with Flag-MPST and subjected to immunoprecipitation with an anti-Flag monoclonal antibody. Co-immunoprecipitated SREBP-1 was detected with an anti-SREBP-1 antibody. The results are expressed as the mean±SD of five mice per group or three independent experiments. *p<0.05, **p<0.01, ***p<0.001. ACC, acetyl-CoA carboxylase; CSE, cystathionine γ-lyase; FAS, fatty acid synthase; FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; HFD, high fat diet; IB, immunoblot; IP, immunoprecipitation; MPST, 3-mercaptopyruvate sulfurtransferase; siRNA, small interfering RNA; SREBP-1, sterol regulatory element-binding protein 1c.
Disturbances in fatty acid oxidation also account for excess lipid storage in the liver. The key enzymes of fatty acid oxidation pathways in the liver are regulated by peroxisome proliferator-activated receptor α (PPARα), the expression of which has been found to be modulated in H2S-regulated hepatic steatosis.17 31 However, in this study, we found that partial knockdown of MPST did not affect the expression of genes involved in fatty acid β-oxidation that were mainly regulated by PPARα in HFD-fed mice (online supplementary figure 6A, B). The overexpression of MPST did not affect the expression of PPARα targeted downstream genes in FFA-treated L02 cells either (online supplementary figure 6C). Moreover, neither the reduction of endogenous H2S nor the exogenous H2S supplementation affected CPT1A, FGF21, medium-chain acyl-CoA dehydrogenase or long-chain acyl-CoA dehydrogenase protein levels in FFA-treated L02 cells (online supplementary figure 6D, E). These data indicate that fat oxidation pathways may not be involved in the effect of MPST regulation on NAFLD.
Knockdown of MPST weakens JNK phosphorylation and ameliorates hepatic oxidative stress
H2S was also reported to protect against liver injury by suppressing the C-Jun N-terminal kinase (JNK) signalling pathway,32 which is activated in steatotic hepatocytes and regulates oxidative stress and subsequent inflammation.33 34 Thus, we evaluated the effect of MPST on JNK signalling, oxidative stress and inflammation. As illustrated in figure 8A, B, knockdown of hepatic MPST remarkably inhibited JNK phosphorylation in the livers of HFD-fed mice (figure 8A) and in the FFA-treated L02 cells (figure 8B). In contrast, overexpression of MPST significantly enhanced JNK phosphorylation in the FFA-stimulated L02 cells (figure 8C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Knockdown of MPST weakens JNK phosphorylation, ameliorates hepatic oxidative stress and suppresses the release of MCP-1. (A–C) Western blots and the quantifications of the ratio of JNK phosphorylation over total JNK, as quantified by densitometry from the immunoblots after normalisation to GAPDH expression, from HFD-fed mice with AD-shMPST or AD-GFP treatment (A), from FFA-treated L02 cells with siMPST or siNC transfection (B) or from FFA-treated L02 cells with GFP-MPST or GFP transfection (C). Transfections with short hairpin RNA or vector and treatment with FFA are the same as described in other figures. (D) Liver tissues from HFD-fed mice after adenovirus-mediated knockdown of MPST were subjected to intrahepatic MDA measurements (left panel). L02 cells were transfected with NC or MPST siRNA for 24 hours and then were treated with FFAs (1 mM) for 24 hours. Intracellular MDA contents and SOD activity were measured. (E) After transfecting with NC and MPST siRNA for 24 hours, L02 cells were transfected with or without CSE siRNA for 24 hours. Then the cells were treated with FFAs (1 mM) for another 24 hours and subjected to intracellular MDA content measurements. (F) Liver tissues from HFD-fed mice after adenovirus-mediated knockdown of MPST were subjected to real-time quantitative PCR for MCP-1 mRNA level determination (upper panel). Plasma samples were collected to measure MCP-1 levels with an ELISA kit (lower panel). The results are expressed as the mean±SD of five mice per group or three independent experiments. *p<0.05, **p<0.01. (G) Proposed model by which FFA promotes NAFLD via the dysregulation of the MPST/H2S pathway. In NAFLD, elevated FFA flux upregulates hepatic MPST expression and then downregulates CSE expression via a direct MPST–CSE interaction, which subsequently reduces H2S production. The decreased H2S levels result in increased expression of the lipogenic enzyme SREBP-1 and its downstream genes including ACC and FAS, leading to the increased de novo synthesis of fatty acids and the subsequent increase of TG as well as cholesterol contents, triggering the malignant cycle of aggravated lipid accumulation in the liver accompanied by increased secretion of VLDL. On the other hand, the reduced H2S levels result in the activation of JNK phosphorylation signalling and aggravate hepatic oxidative stress as well as provoke MCP-1 release, synergistically contributing to the exacerbation of NAFLD. ACC, acetyl-CoA carboxylase; CSE, cystathionine γ-lyase; FAS, fatty acid synthase; FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; HFD, high fat diet; JNK, C-Jun N-terminal kinase; MDA, malondialdehyde; MPST, 3-mercaptopyruvate sulfurtransferase; mRNA, messenger RNA; siRNA, small interfering RNA; SOD, superoxide dismutase; SREBP-1, sterol regulatory element-binding protein 1c; TG, triglyceride; VLDL, very-low-density lipoprotein.
Increased intrahepatic levels of FFAs provide a source of oxidative stress, and consequently, lipid peroxidation promotes NAFLD progression.35 We found that knockdown of hepatic MPST markedly reduced intrahepatic malondialdehyde (MDA) contents in the HFD-fed mice and in L02 cells, while superoxide dismutase activity in L02 cells was significantly enhanced by MPST inhibition (figure 8D). Furthermore, the additional knockdown of CSE by siRNA restored the MDA content in siMPST-treated L02 cells (figure 8E). Among the inflammatory genes, hepatic gene expression and circulating level of MCP-1 were significantly suppressed after partial knockdown of hepatic MPST in HFD-fed mice (figure 8F, online supplementary figure 7), while most of the other inflammatory gene profiles and circulating inflammatory biomarkers were not altered after MPST inhibition (online supplementary figure 7).
Discussion
In this study, we provided evidence that the FFA-induced pathologic upregulation of MPST in hepatocytes is involved in the pathogenesis of NAFLD via inhibition of the CSE/H2S pathway and consequent upregulation of SREBP-1, JNK and oxidative stress pathways. We have integrated each of these findings in a model (summarised in figure 8G) that depicts a novel important pathway by which FFAs promote NAFLD. The rational for this model is presented in the following discussion.
Impaired endogenous H2S synthesis was reported to be associated with hepatic fibrosis and cirrhosis13 36 37 and fatty liver induced by HFD feeding16 26 or methionine and choline-deficient (MCD) diet feeding.17 In this study, we also confirmed that H2S levels were reduced in cellular and mouse models of NAFLD. However, the molecular explanation for why H2S synthesis is impaired in NAFLD remains unclear. The CBS/CSE system is considered to be responsible for the majority of endogenous H2S production, while MPST, the maximal activity of which is attained under non-physiologic alkaline conditions using 3-mercaptopyruvate as substrate, plays a quite different role in the generation of H2S.38 Previous studies reported that the deficiency of CSE in mice decreased endogenous H2S level,10 25 which is in agreement with an important role of CSE in promoting H2S production. Theoretically, MPST also stimulates H2S production from a persulfurated acceptor substrate.21 However, surprisingly, in the current study, we found that inhibition of MPST via either administration of adenovirus-mediated shRNA or heterozygous deletion of the Mpst gene markedly increased H2S production and attenuated HFD-induced hepatic steatosis. Our mechanistic studies suggest that MPST inhibits H2S production via the downregulation of CSE. In addition, we provided several lines of evidence suggesting that MPST downregulates CSE via the protein–protein interaction. First, based on the prediction from the protein–protein interaction database, we uncovered a direct and functional interaction between MPST and CSE. An analysis of the STRING database revealed that the score of prediction for the MPST–CSE interaction ranks the first place among the top 10 functional MPST-centric protein-interaction networks. Second, hepatic CSE expression was significantly upregulated in response to a moderate reduction in MPST expression, while the overexpression of MPST induced significant downregulation of CSE expression. Finally, co-IP analyses showed that MPST directly interacted with CSE, establishing a molecular mechanism underlying the feedback regulations between these two H2S-generating enzymes. In addition, by inhibiting the upregulated CSE expression in the MPST-knockdown FFA-treated L02 cells, we found that H2S levels were significantly decreased compared with the siMPST alone groups, validating a CSE dependency of MPST-mediated inhibition of H2S production. These results imply a negative feedback between MPST and CSE, which cooperates with each other to maintain H2S-metabolism homeostasis. Interestingly, a similar compensatory regulation was also found between CBS and CSE in the livers of HFD-fed mice.16 Analogously, another recent study reported that DL-propargylglycine, an inhibitor of CSE, increases CBS-mediated desulfhydration in hepatocytes, implying a compensatory mechanism of H2S production.39
Up to date, the impact of H2S on liver lipid metabolism remains unclear. A recent study found that H2S treatment reduced serum TG levels and counteracted steatosis in HFD-fed mice.26 Previous studies have demonstrated the impact of altered endogenous H2S levels on the plasma lipid profile in CSE-deficient or CBS-deficient mice.25 36 In this study, we found that elevated intrahepatic H2S levels, induced by MPST partial knockdown, were associated with attenuated hepatic steatosis in HFD-fed mice. In cultured cells, H2S levels also negatively correlated with the intracellular TG contents. Additionally, we provided several lines of conclusive evidence supporting H2S-mediated amelioration of steatosis. First, inhibition of CSE eliminated the compensatory upregulation of CSE in the MPST-knockdown cells, resulting in rebounded hepatocyte steatosis. Second, administration of NaHS improved lipid accumulation and prevented the exacerbation of fat accumulation induced by MPST overexpression in FFA-stimulated L02 cells. Finally, exogenous H2S supplementation reversed the augmentation of intracellular TG contents in hepatocytes that exhibited a reduced H2S decline after the double inhibition of MPST/CSE. Collectively, impaired H2S synthesis may be an important cause for NAFLD, and MPST promotes fat accumulation in hepatocytes via the inhibition of H2S production.
Another important finding from the current study is the identification of the mechanism by which H2S regulates lipid metabolism. Hepatic lipid accumulation results from an imbalance between lipid synthesis and lipid disposal. We found that elevated H2S levels were associated with the significant suppression of SREBP-1, FAS and ACC in cellular and mouse model of NAFLD after MPST inhibition. In contrast, MPST overexpression reduced H2S production and promoted SREBP-1 expression. Further study showed that exogenous H2S supplementation suppressed the expression of SREBP-1 activated by FFA stimulation, while the double inhibition of MPST and CSE decreased H2S production and enhanced the SREBP-1 pathway. These results suggest a significant role of H2S in the regulation of hepatic lipid metabolism via the modulation of the SREBP-1 pathway, a critical factor for lipid synthesis. β-Oxidation is another important step in the control of lipid metabolism, in which PPARα plays a central role in the regulation of the expression of genes that encode several mitochondrial fatty acid-catabolising enzymes in addition to mediating inducible mitochondrial and peroxisomal fatty acid β-oxidation. However, our results showed that the alteration of H2S level caused by the regulation of MPST in vivo and in vitro did not affect the expressions of PPARα-regulated genes involved in fatty acid β-oxidation. Furthermore, neither reduced H2S production due to double inhibition of MPST/CSE nor exogenous H2S supplementation altered the expression of β-oxidation pathway-associated genes. Therefore, the effect of MPST regulation on hepatic steatosis is largely dependent on SREBP-1-mediated lipid synthesis processing and independent from the fatty acid oxidation pathway.
H2S may also regulate the development of NAFLD by a SREBP-1-independent mechanism. A recent study reported that H2S protected against hepatic ischemia/reperfusion injury partly by weakening hepatocyte apoptosis through the suppression of the JNK signalling pathway.32 As a major effector of lipotoxic injury in the liver, the JNK pathway is activated in steatotic hepatocytes and inhibition of JNK protects against hepatocyte injury by ameliorating oxidative stress and subsequent inflammation.33 34 In this study, we found that elevated H2S levels induced by MPST silencing were associated with the significant inhibition of hepatic JNK phosphorylation in HFD-fed mice and in FFA-stimulated L02 cells, whereas reduced H2S levels induced by MPST overexpression caused opposite effects. In addition, hepatic lipotoxicity is known to generate oxidative stress and inflammation,40 and H2S supplementation has been shown to prevent MCD-induced NASH in rats via the inhibition of oxidative stress and suppressing inflammation.17 In the current study, we also found that elevated H2S levels were associated with significantly ameliorated oxidative stress and the marked suppression of MCP-1 expression, suggesting that MPST-regulated H2S is an important mediator in the JNK signalling pathway and oxidative stress as well as inflammation involved in the pathogenesis of NAFLD.
The findings of this study provide novel clues for developing potential therapeutic strategies for NAFLD. The increased endogenous H2S, derived from compensatory generation by CSE after partial knockdown of MPST, can easily diffuse across cell membranes in a receptor-independent manner and can activate various cellular targets, making it an attractive pharmacological agent.17 Further studies are needed to clarify the detailed regulatory mechanism of MPST on H2S production and the subsequent impact on NAFLD.
In conclusion, our study provides new insights into the mechanism of hepatic lipotoxicity that FFA-induced upregulation of MPST is implicated in the pathogenesis of NAFLD via regulation of H2S in a CSE-dependent manner, identifying that MPST may be a potential therapeutic target for NAFLD.
Acknowledgments
We thank Huatuo Zhu, Rongliang Tong, Xiang Yin, Shengwen Song, Mei Yang, Beng Yang and the teachers as well as students of the Key Lab of Combined Multi-Organ Transplantation, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, the First Affiliated Hospital, College of Medicine, Zhejiang University, for their outstanding technical assistance. We thank Professor Bin Gao (National Institutes of Health, USA) for critically revising and editing the manuscript.
References
Footnotes
ML, CX and JS contributed equally.
Contributors ML and CX designed and performed the study, analysed the data and wrote the paper. JS collected the patient data, analysed the data and critically revised the manuscript. JD, XW, JG, DC, CL, JZ, YL, ZT and XK performed the study. YL and CY designed the study and critically revised the manuscript.
Funding This work was supported by the National Natural Science Foundation of China (No 81230012 to YL, 81470838 to CX, and 81570523 to CY). The funders did not play any role in the study design, data collection and analysis, decisions regarding data release or manuscript preparation.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University.
Provenance and peer review Not commissioned; externally peer reviewed.