Article Text

Abstract
Background Inflammatory bowel disease (IBD) can arise from genetic mutations that compromise intestinal epithelial cell integrity or immune regulation. SHIP has previously been shown to play a pivotal role in limiting the number of immunoregulatory cells and their function.
Aim To determine whether SHIP plays a pivotal role in control of immune tolerance in the gut mucosa.
Methods Gastrointestinal pathology was assessed in three separate strains of SHIP-deficient mice and their respective wild-type (WT) littermates. Gastrointestinal pathology was analysed in SHIP-deficient hosts reconstituted with WT haematopoietic cell grafts, and WT hosts reconstituted with SHIP-deficient haematopoietic cell grafts including whole splenocytes, purified T cells or natural killer (NK) cells. Major immune cell populations were also analysed in the small intestine of SHIP-deficient mice and WT controls.
Results SHIP-deficient mice developed segmental, transmural pyo-granulomatous ilietis that recapitulated classical features of Crohn's disease enteric pathology. Analysis of haematopoietic chimeras showed that WT bone marrow reconstitution of SHIP−/− hosts corrects ileitis. Reconstitution with SHIP−/− splenocytes transferred ileitis to WT hosts. Adoptive transfer of purified SHIP−/− T cells or NK cells to WT hosts did not transfer ileitis. There was a paucity of both CD4 and CD8 T cells in the small intestines of SHIP-deficient mice; however, neutrophil numbers were significantly increased.
Conclusions SHIP plays a pivotal role in immune function in the intestine; further scrutiny of this pathway in IBD patients is warranted. It is proposed that SHIP-deficient ileitis results from a local deficit in mucosal T cell immunity that promotes a damaging granulocyte–monocyte inflammation of the distal ileum.
- SHIP
- immunoregulation
- inflammatory bowel disease
- Crohn's disease
- bone marrow chimera
- cell signalling
- crohn's colitis
- genetics
- mucosal immunity
- signal transduction
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- SHIP
- immunoregulation
- inflammatory bowel disease
- Crohn's disease
- bone marrow chimera
- cell signalling
- crohn's colitis
- genetics
- mucosal immunity
- signal transduction
Significance of this study
What is already known about this subject?
Immune regulation is critical for the integrity and function of the small intestine.
SHIP controls the homeostasis of immunoregulatory myeloid and T lymphoid cells in peripheral lymphoid tissues such as spleen and lymph node.
Myeloid consolidation of the lungs is a significant cause of morbidity and mortality in SHIP-deficient mice.
What are the new findings?
Despite enhanced immune regulation in the peripheral lymphoid tissues of SHIP-deficient mice, immune function and regulation is compromised in the small intestine of these mice.
Normal immune function and homeostasis in the small intestine requires enzymatically active SHIP.
Expression of SHIP by intestinal epithelial cells is not required for the integrity of the small intestine.
SHIP-deficient haematolymphoid cells are sufficient to transfer ileitis to wild-type hosts.
There is a paucity of both CD4 and CD8 T cells in the small intestine of SHIP-deficient mice.
There is local neutrophilia in the small intestines of SHIP-deficient mice.
How might it impact on clinical practice in the foreseeable future?
These findings suggest that detailed analysis of the SHIP gene is warranted in human IBD patients and may also lead to a better understanding of immune cell signalling pathways required for immune function and regulation in the human gut.
Introduction
Crohn's disease (CD) is a chronic, relapsing idiopathic inflammatory bowel disease (IBD) that can affect various sites within the gastrointestinal tract, but classically the ileum, which leads to abdominal cramping, diarrhoea and gastrointestinal bleeding.1–4 CD histopathological lesions begin with the formation of multiple aphthoid ulcers infiltrated by neutrophils, which progress and coalesce into sharply delimited, regional, transmural inflammations with granuloma formation, thickening of the muscularis propria, strictures, fissures and fistulas.1–4 Although genome-wide association studies have identified more than 30 susceptibility loci for IBD that disturb either intestinal epithelial cell (IEC) barrier homeostasis or immune effector cell functions and interactions with luminal flora and antigens,5–7 many susceptibility genes possess differing functions in IEC and haematopoietic cells, and primary IEC or immunological defects in genetically susceptible individuals often remain enigmatic.8 9
Primary immunological alterations proposed to contribute to the development of CD pathology include increased expression of Th1-polarising cytokines interleukin (IL)-12 and interferon γ by cells in the lamina propria,10 11 aberrant IL-23-initiated Th17-associated immune responses,12 13 defects in regulatory T cell (Treg) modulation of effector functions via transforming growth factor β or IL-10 dependent mechanisms,14 aberrant B-cell specificity and serum autoreactivity15 and myeloid cell dysfunction.16 That primary defects in immune cell function contribute to CD in some patients is supported by reports of disease remission following allogeneic bone marrow transplantation.17
IEC barrier defects that may contribute to CD development include the inability of a Paneth cell-derived NOD2 variant to expel luminal bacteria,18 diminished IEC autophagy and antimicrobial granule exocytosis,19 altered IEC toll-like receptor expression with potentially dysregulated responses to common bacterial motifs,20 altered IEC cytokine expression21 and increased IEC paracellular permeability to enteric luminal contents, perhaps due to inadequate formation of IEC tight junctions.22
Although animal models of IBD have been described, most model systems affect the large intestine, do not manifest CD-specific histopathological features, and are often induced rather than spontaneously developing models.5 7 23 Only two animal models spontaneously develop chronic inflammation of the ileum, the SAMP1/Yit mouse24 and the TNF∆ARE mouse.25 SAMP1/Yit mice spontaneously develop terminal ileitis by 10 weeks of age driven by Th1- and Th2-type immune responses, the severity of which can be decreased by anti-tumour necrosis factor (TNF) antibodies.23 26 27 Although SAMP1/Yit mesenteric lymph node CD4+ T cells are capable of adoptively transferring ileitis to severe combined immunodeficient (SCID) mice, increased IEC paracellular permeability appears to be the primary susceptibility factor in this model.22 The TNF∆ARE mouse model was generated by deletion of 69bp within the AU-rich region (ARE) of the gene that encodes TNF, resulting in increased systemic TNF levels associated with expansion of CD8+ effector cell subpopulations that mediate chronic ileitis and arthritis in both heterozygous and homozygous mice.25 28 Haematopoietic cells of neither the SAMP1/Yit nor the TNF∆ARE mouse are capable of adoptively transferring CD-like disease to an immunocompetent host.
The Src homology 2 (SH2)-containing inositol-5-phosphatase (SHIP) protein becomes tyrosine phosphorylated in haematopoietic cells following activation of surface receptors for various cytokines including erythropoietin, IL-3, GM-CSF, M-CSF, or in response to B cell antigen receptor cross-linking, or T cell activation.29 30 SHIP signalling plays a pivotal role in limiting the number and function of immunoregulatory cells in the peripheral lymphoid system.31–37 We recently found that SHIP limits the number of immunoregulatory T cells present in the spleen and mesenteric lymph nodes.35 SHIP performs this role by controlling the number of Treg cells in the periphery either by limiting the survival of FoxP3+ Treg cells or by preventing the inappropriate acquisition of FoxP3 expression by naïve CD4 T cells.35 Consistent with this finding, the p110 subunit of an enzyme that SHIP opposes, PI3K, is critical for the formation and survival of Treg cells in the periphery.38 39 SHIP-deficiency also promotes the inappropriate expansion of myeloid immunoregulatory (MIR) cells in spleen and lymph nodes.32 33 Thus, in major peripheral lymphoid tissues like spleen and lymph node, SHIP-deficiency leads to profound and functional expansion of immunoregulatory capacity. In aggregate these studies establish that inositol phospholipid signalling plays a pivotal role in the formation, homeostasis and function of both myeloid and lymphoid immunoregulatory cell populations.
SHIP, through its 5′-inositol phosphatase domain, hydrolyses the second messenger PI(3,4,5)P3 and thus limits PI3K activation of key downstream immune effector pathways such as Akt and NF-κB.30 40 SHIP-deficient mice are smaller than their WT and heterozygous littermates and exhibit a greatly reduced lifespan with survival beyond 10–12 weeks of life observed only rarely,32 41 attributable in part to the development of an eosinophilic crystalline pneumonia with infiltration by mixed inflammatory cells resulting in patchy lung consolidation.41 Eosinophilic crystalline pneumonia is a cause of shortened life span in various genetically engineered mouse models.42 43 Granulocytes are less susceptible to apoptotic signals in SHIP-deficient mice, and granulocyte–monocyte infiltrations can be found in the liver, kidney, heart, skeletal muscle, pancreas lymph nodes and thymus.44 The increased capacity of SHIP-deficient T cells to differentiate into Treg cells is accompanied by a deficiency in Th17 cell differentiation and a reduced ability to cause colitis after adoptive transfer of CD4+ T cells to T cell deficient hosts.35 36
Although SHIP expression and function has generally been thought to be confined to cells in the haematopoietic system, recent evidence indicates that SHIP is also expressed by some parenchymal cell types, including osteoblasts45 and endothelial cells.46 In fact, we recently identified the first known function for SHIP outside the haematolymphoid compartment as SHIP is required for support of haematopoietic stem cell function and quiescence by bone marrow (BM) niche cells.45
Given its role in immune regulation and lymphoid function we sought to examine whether SHIP-deficiency might have consequences at enteric mucosal surfaces where greater stress is placed on both immune effector and regulatory processes due to the close and constant interaction with commensal microflora and potential pathogens at this organ site. Surprisingly, our analysis reveals a paucity of both CD4 and CD8 T cells in the small intestine with a concomitant increase in neutrophils at this site. Consistent with altered immune status we find spontaneous development of ileitis with features that closely recapitulate those of CD pathology. In addition, the susceptibility factor for this CD-like ileitis of SHIP-deficient mice appears to be in the haematopoietic compartment, and likely granulocyte–monocyte, as the CD-like ileitis was transferable to an immunocompetent host following irradiation and reconstitution with SHIP splenocytes, but not following transfer of either T cells or natural killer (NK) cells. Consequently, the SHIP-deficient mouse is the first CD animal model capable of adoptively transferring CD-like disease to an immunocompetent host.
Materials and methods
Mice
The development and production of SHIP−/− mice has been described previously. In brief SHIP−/− mice were generated by deletion of the promoter and first exon of SHIP via a Cre-LoxP strategy and then backcrossed to the C57BL6/J background. SHIPΔIP/ΔIP mice were a kind gift of Dr Jeffery Ravetch (Rockefeller University, New York).47 MxCreSHIPflox/flox mice were described previously.33 SHIP expression is ablated in these mice following three intraperitoneal injections of polyI/C at 625μ/dose. MxCreSHIPflox/flox mice were sacrificed for tissue harvest 4–5 weeks after the last polyI/C injection. The PI3K haploinsufficient SHIP−/− mice analysed in this study possess a mutation in the p85 regulatory subunit of PI3K48 and a SHIP mutation created by Helgason et al.41 All mice were between 6 and 13 weeks of age at time of sacrifice. All mice were maintained in an accredited, barrier facility confirmed free of a comprehensive list of potential parasites and microbial pathogens including Citrobacter rodentium, Pseudomonas aeruginosa, Salmonella spp., and Clostridium perfringens, viral Antibodies to murine norovirus (MNV), parainfluenza virus type I (Sendai), coronavirus (MHV), Mycoplasma pulmonis, paramyxovirus (PVM), parvovirus (MPV, MMV), poliovirus (TMEV) reovirus type 3 (Reo), arterivirus (LCM), adenovirus (MAD1, MAD2), poxvirus (Ectro), rotavirus (EDIM), papovavirus (Poly), and free of Helicobacter sp. by PCR screening. Further, microorganisms were not identified in any tissue using WarthineStarry, Giemsa, Ziehle Neelsen and PAS stains.
Adoptive transfer experiments
BM cells were flushed from intact femur and tibia and collected in tissue media (TM) consisting of RPMI, 3% fetal bovine serum (FBS) and 10 mM HEPES (Invitrogen, Carlsbad, California, USA). Spleens were crushed with a 10 ml syringe plunger. The single cell suspension was then filtered through a 70 mm strainer (BD Bioscience, San Jose, CA) and red blood cell (RBC) lysis performed at room temperature for 5 min in 1× RBC lysis buffer (eBioscience, San Diego, California, USA). Cells were centrifuged and resuspended in 1× Dulbecco phosphate-buffered saline (D-PBS). C57BL/6 recipients were given antibiotic water prior to receiving a split dose of 1100 Rads (600+500) from an x-ray irradiator. Irradiated recipients were then transplanted with 5×105 BM cells or splenocytes as indicated via retro-orbital injection. For adoptive transfer of T and NK cells, CD3+NKp46− T and NKp46+CD3− NK cells were simultaneously sorted from spleens of SHIP−/− mice and adoptively transferred into irradiated C57BL6 hosts (550 Rads) via retro-orbital injection. Each C57BL/6 host received 2.4×105 SHIP−/− T cells and 7.5×103 SHIP−/− NK cells.
Histopathological analysis
Necropsies were performed in a systematic, comprehensive manner. The entire alimentary tract, including the oesophagus, stomach, duodenum, jejunum, ileum, caecum, and colon and associated mesentery and mesenteric lymph nodes was evaluated, and the alimentary tract was insufflated with 10% neutral-buffered formalin and rolled in segments to fit as Swiss rolls into cassettes for histological processing and microscopic evaluation. The skin, subcutis, skeletal muscle, inguinal lymph node and mammary fat pad, cervical lymph node, salivary glands, reproductive tract and associated glands, liver, gall bladder, spleen, pancreas, kidneys and adrenal glands, were evaluated. The larynx, trachea with attached thyroid and parathyroid glands, heart, thymus, and lungs insufflated with 10% neutral-buffered formalin were collected. Tissues were fixed in 10% neutral-buffered formalin, dehydrated, embedded in paraffin, sectioned at 3 μm and stained with H&E. Histological sections of each segment of the alimentary tract were masked, assessed and assigned an inflammatory grade of 0–6, with grade 6 representing the most progressed, severe lesion. Inflammatory grades assigned were as follows: inflammatory grade 0=no significant abnormalities; inflammatory grade 1=mild predominantly polymorphonuclear (PMN) leucocyte infiltrations (<25 PMN/hpf) of the lamina propria and/or enteric lymph nodule; inflammatory grade 2=moderate predominantly PMN leucocyte infiltrations (>25 cells/hpf) of the lamina propria and/or lymph nodule; inflammatory grade 3=marked inflammatory cell infiltrations with extension below the muscularis mucosa causing architectural distortion of the mucosa and submucosa with attendant crypt hyperplasia; inflammatory grade 4=marked infiltrations with extension into the tunica muscularis; inflammatory grade 5=marked transmural leucocyte infiltrations; inflammatory grade 6=marked transmural leucocyte infiltrations with extension into the mesentery and/or other organs. In each section the presence of granuloma, crypt abscess, stricture, fissure, and whether dissemination of inflammation to other sites had occurred was noted. Inflammatory scores were compared between cohorts by one-way ANOVA. All reported p-values are two-sided with p<0.01 considered significant. The sections were scored by a single, board certified veterinary pathologist. The sections were scored by a veterinary pathologist with confirmation of scoring system and subsequent gradations by a clinical pathologist.
Flow cytometry
The antibodies used for staining of cells prepared from small intestines included CD3ε, CD4, CD8, CD16/32, CD62L, CD62E and Ly6G and were obtained from BD Pharmingen (San Jose, California, USA). Samples were acquired on a FACSCalibur and analysed using FlowJo8. Dead cells were excluded from the analysis following cytometer acquisition of staining data based on exclusion of the DAPI dye.
Results
Ileitis in SHIP-deficient mice
Seventy-nine 6–8 week old mice, including 26 SHIP−/− mice, 26 SHIPΔIP/ΔIP mice, one PI3K+/−SHIP−/− and 26 wild type littermates, each of both sexes were submitted to systematic, comprehensive necropsies, with histopathological examination of the entire insufflated alimentary tract. Primary gastrointestinal lesions were present only within the ileum of SHIP-deficient mice, and consisted of a segmental ileitis. Lesions were graded according to the extent of inflammation as grade 0–6 (figures 1–6). Of the 53 SHIP-deficient mice evaluated, only 3 of the 53 (6%) of SHIP-deficient mice lacked ileitis, while 50 of the 53 (94%) of SHIP-deficient mice had some degree of ileitis, significantly more than the absence of ileitis in WT littermates (p<0.001). Both SHIP−/− and SHIPΔIP/ΔIP that harbour deletions in exons encoding different regions in the SHIP1 locus developed ileitis with comparable frequency with 25 of 26 (96%) of SHIP−/− mice, and 24 of 26 (92%) of SHIPΔIP/ΔIP mice affected with comparable mean ileitis inflammatory grades of 4.2±2.1 and 4.0±2.2, respectively. Ileitis was also observed in a PI3K+/−SHIP−/− mouse (grade 4), indicating that a third independent SHIP mutant strain develops this pathology and that PI3K haploinsufficiency does not protect from development of ileitis. PI3K+/−SHIP+/+ and PI3K+/−SHIP+/− controls showed no evidence of ileitis (grade 0). Inflammatory lesions of the ileum in all SHIP-deficient mice analysed were present on a background of otherwise unaffected, uninflamed gastrointestinal tract.

Grade 1 Crohn's disease (CD)-like ileitis of SHIP-deficient mice. Longitudinal sections through Swiss rolls of small intestine from SHIP-deficient mice with early manifestations of CD-like disease, interpreted as grade 1, with mild predominantly polymorphonuclear (PMN) leucocyte infiltration of the lamina propria (B, arrows). This PMN infiltration of the ileum was typically present in the mucosa overlying and within lymph nodules (C–F). Adjacent ileum and more proximal small intestine (C, asterisk) were unaffected with normal villous mucosal architecture. Higher magnifications show a predominantly PMN leucocyte infiltration of the villous lamina propria (F), and within the underlying lymph nodule, in this case accompanied by histiocytes and multinucleated giant cells (E).

Grade 2 Crohn's disease-like ileitis of SHIP-deficient mice was comprised of a moderate, mixed, predominantly polymorphonuclear leucocyte infiltration of the ileum lamina propria (D) and underlying lymph nodule (C), with attendant crypt hyperplasia (B, upper bracketed arrows) with elongation, crowding, and numerous mitotic figures (D), compared to the unaffected mucosa of the adjacent small intestine (A, asterisk) with normal villous architecture and typical crypt depth (B, lower bracketed arrows).

Grade 3 Crohn's disease-like ileitis of SHIP-deficient mice was comprised of a marked, mixed, often predominantly polymorphonuclear (PMN) inflammatory cell infiltration of the ileum with expansion of the mucosa and submucosa (A, bracketed arrows), loss of normal villous architecture, and crypt elongation (A, arrow). Higher magnifications of the affected ileum (B, C) showed a mixed, predominantly PMN leucocyte infiltration.

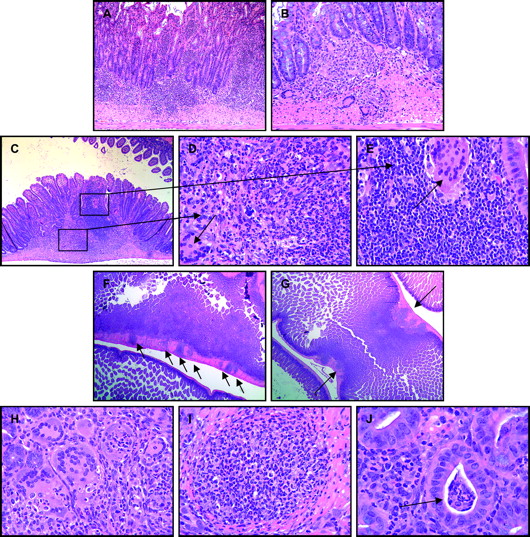
Grade 4 Crohn's disease-like ileitis of SHIP-deficient mice was comprised of a marked, mixed inflammatory cell infiltration of the ileum extending through the mucosa, submucosa, and into the tunica muscularis (A, B, C, F, G). Such inflammatory cell infiltrations varied in composition, were in some areas of affected ileum predominantly polymorphonuclear (D), while in other areas were predominantly mononuclear with lymphocytes, histiocytes and numerous multi-nucleated giant cells present (B, E). Grade 4 ileitis was typically sharply delimited aggregates of pyo-granulomatous inflammatory cell infiltrations, at times in rosary-bead arrays (F, arrows), that caused thickening of the bowel wall (G, arrows) and lumen narrowing. Multinucleated giant cells (H), and pyo-granulomas (I) were frequently present in affected segments, but crypt abscesses (J, arrow) were less frequently observed.

Grade 5 Crohn's disease-like ileitis of SHIP-deficient mice consisted of marked, mixed, transmural inflammatory cell infiltrations that extended to the serosa, some of which due to bowel wall thickening and stricture formation had developed fissures that penetrated into and ended blindly within the tunica muscularis (A; B, arrow).

Most SHIP-deficient mice presented with grade 6 Crohn's disease-like ileitis which consisted of marked, mixed, transmural inflammatory cell infiltrations that extended by way of fissures through the ileum serosa into the mesentery (A–C, arrows), the inflammation of which was comprised of mixed inflammatory cells and fibroblasts in aggregates and whirls (E), and pyo-granuloma, some with central necrosis (F, arrow).
The earliest manifestation of CD-like ileitis in SHIP-deficient mice, assigned grade 1, consisted of a mild (<25 PMN/hpf), predominantly PMN leucocyte infiltration of the lamina propria and frequently underlying lymph nodule (figure 1). The ileum of 9 of the 53 (17%) of the SHIP-deficient mice had grade 1 lesions. This mild, earliest lesion of the SHIP-deficient ileum had progressed in other individual SHIP−/− and SHIPΔIP/ΔIP mice to a moderate (>25 PMN/hpf), mixed, but still predominantly PMN leucocyte infiltration of the expanded lamina propria and underlying lymph nodule, assigned grade 2, with attendant crypt regenerative hyperplasia manifest as IEC crowding, and elongation of crypt depth (figure 2). Grade 2 ileitis was present in 2 of the 53 (4%) of SHIP-deficient mice. Grade 3 inflammatory cell infiltrations were mixed and penetrated below the muscularis mucosa with distortion of villous architecture by the expanded mucosa and submucosa, and attendant crypt hyperplasia (figure 3). Five of the 53 (9%) of SHIP-deficient mice had grade 3 inflammatory lesions within the ileum.
Marked, mixed inflammatory cell infiltrations of the ileum of SHIP-deficient mice extending from the mucosa, submucosa, and into the tunica muscularis were assigned grade 4 (figure 4). Six of the 53 (11%) of SHIP-deficient mice had grade 4 inflammatory lesions within the ileum. Composition of grade 4 inflammation varied from predominantly PMN in some segments to predominantly mononuclear in others, often as a mix of granulocytes, lymphocytes, histiocytes and multinucleated giant cells (figure 4D,E). Inflammatory cell infiltrations that extended into the tunica muscularis were segmental, sharply delimited, and arranged either randomly or as rows of aggregates in rosary-bead arrays (figure 4F). In some areas of grade 4 inflammation the overlying mucosa was focally ulcerated. Granulomas and pyo-granulomas were present in the mucosa, submucosa and/or tunica muscularis. Thickening of the bowel wall due to grade 4 inflammation resulted in lumen narrowing and stricture formation in some mice (figure 4G).
Marked, mixed inflammatory cell infiltrations that extended transmural, deep into the bowel wall to the serosa, assigned grade 5 (figure 5), were present in 5 of the 53 (9%) of SHIP-deficient mice. In some cases, grade 5 ileitis thickened the bowel wall, narrowed the lumen, led to stricture formation and the development of fissures that penetrated into and ended blindly within the tunica muscularis (figure 5A,B).
SHIP-deficient mice with marked, mixed, transmural inflammatory cell infiltrations of the ileum that extended by way of fissures through the thickened tunica muscularis and serosa into the mesentery (figure 6A–C) were assigned grade 6. More SHIP-deficient mice were assigned inflammatory grade 6 than any other grade with 23 of the 53 (43%) of SHIP-deficient mice assigned ileitis grade 6. Inflammation extending into the mesentery consisted of mixed inflammatory cells, granuloma, fibrosis, and rarely thrombosis and vasculitis (figure 6D–F).
Ileitis of SHIP-deficient mice of all grades 1–6 was segmental, presenting either as focal, discontinuous inflammatory cell infiltrations or as broad bands of inflammation within the ileum (figure 7A–C). Neighbouring segments of ileum and those of more proximal small intestine or distal large intestine were unaffected. Strictures were observed in 12 of 53 (23%) of SHIP-deficient mice (figure 4G), granuloma were present in 15 of 53 (28%) (figure 4I), and crypt abscesses were observed in 5 of 53 (9%) of SHIP-deficient mice (figure 4J). Pyloric metaplasia of the ileum, and fistula formation, other characteristics of CD histopathology in humans, were not evident in SHIP-deficient mice.

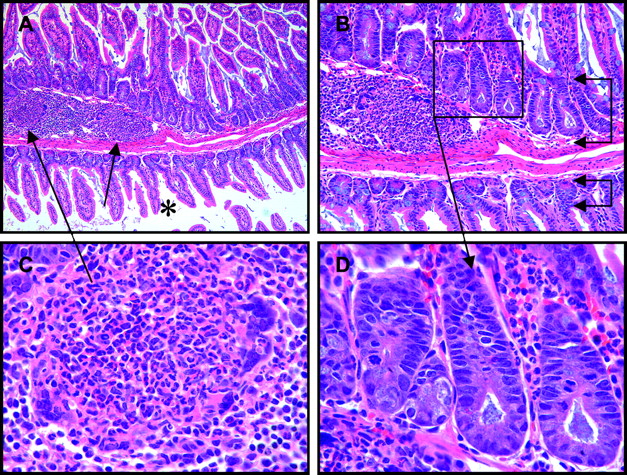
Segmental nature of Crohn's disease-like ileitis of SHIP-deficient mice comprised of either discrete, discontinuous foci of inflammation (A, arrows), or broad bands of inflammation (B, bracketed arrows), each with expansion of the mucosa and submucosa, and thickening of the tunica muscularis (C, left arrow) compared to neighbouring unaffected segments (C, right arrow; A, B, C, asterisks) with normal villous architecture and bowel wall thickness.
Rarely, ileitis spread by direct extension to other gastrointestinal sites, including in two mice where inflammation of the ileum extended via the mesentery to the colon wall (figure 8A,B), and in a third SHIP-deficient mouse where inflammation had spread to the gastro-oesophageal juncture (figure 8C,D). Draining mesenteric lymph nodes were often enlarged and inflamed in 33 of 53 (62%) of SHIP-deficient mice (figure 8E,F). An individual SHIP−/− mouse had inflammatory cellular thrombi in mesenteric veins and a granulomatous pulmonary consolidation comparable in composition to the ileitis in this case, suggesting that vascular thrombi and emboli may rarely develop as a consequence of CD-like ileitis in SHIP-deficient mice and may disseminate inflammation via haematogenous routes (figure 8G–J). This pulmonary consolidation was distinct from that which occurs routinely in SHIP−/− and SHIPΔIPΔIP mice as an eosinophilic crystalline pneumonia (figure 9A–F).
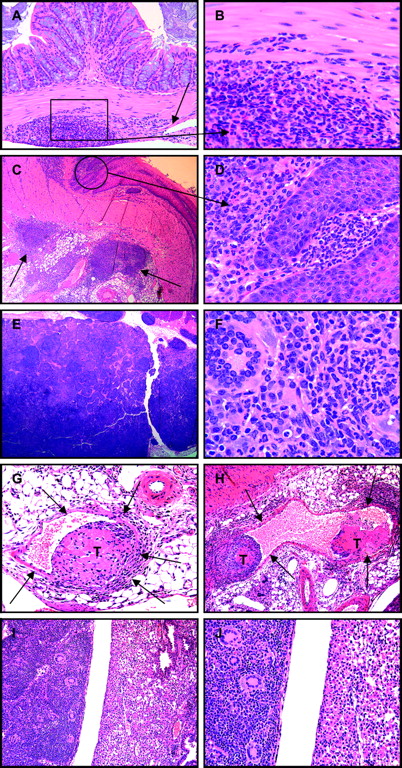
Inflammation extended beyond the ileum in a few SHIP-deficient mice where mixed leucocyte infiltrations of the mesentery infiltrated into the colon wall (A, arrow; B), or the juncture of the oesophagus and stomach (C, arrows; D). Draining mesenteric lymph nodes were frequently enlarged and consisted of mixed inflammatory cells with numerous multinucleate cells (E, F). Rarely, inflammatory cellular thrombi (T) were present within mesenteric veins (G, H, arrows), and as emboli may have lodged in pulmonary microvasculature resulting in a granulomatous pulmonary consolidation comprised of mixed inflammatory cells with numerous multinucleate cells (I, J, left) that contrasted sharply with the eosinophilic crystalline pneumonia of neighbouring lobes (I, J, right) typical of SHIP-deficient mice.
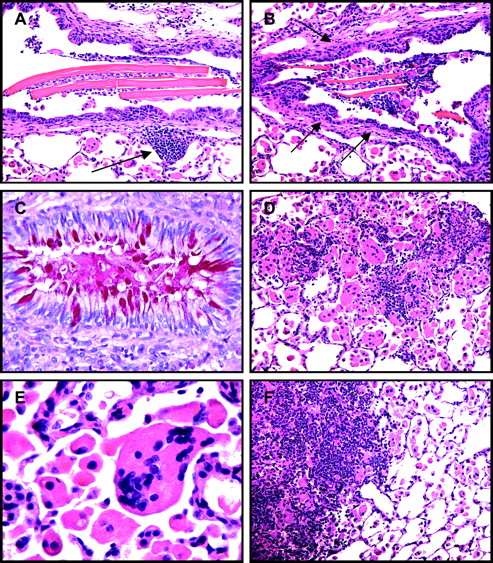
SHIP-deficient mice develop an eosinophilic crystalline pneumonia that consists of large crystals in bronchiolar airways, foci of mixed inflammatory cell infiltrations (A, arrow), bronchiolar subepithelial fibrosis (B, arrows), hypertrophy and mucous metaplasia of bronchiolar epithelium (C), numerous alveolar macrophages and multinucleate cells (D, E), and infiltrating leucocytes resulting in patchy pulmonary consolidation (F, left).
Ilietis results following ablation of SHIP expression in adulthood
The above studies show that ileitis occurs in germline SHIP mutant mice, indicating this CD-like pathology may have a developmental origin. To test this possibility we analysed intestinal pathology in MxCreSHIPflox/flox mice where SHIP expression is normal in fetal, neonatal and adolescent life, but can be ablated during adulthood following polyI/C administration.33 This approach results in eosinophilic crystalline pneumonia in adult mice rendered SHIP deficient.49 For this analysis twelve 6–8 week old MxCreSHIPflox/flox mice and twelve SHIPflox/flox controls, each of both sexes were submitted to comparable histopathological analyses. Primary gastrointestinal lesions were present only within the ileum of MxCreSHIPflox/flox mice, while SHIPflox/flox mice were unaffected 4–5 weeks after the last injection of polyI/C. Of the 12 MxCreSHIPflox/flox mice evaluated, only 1 (8%) lacked ileitis, while 11 of 12 (92%) developed ileitis with a mean inflammatory grade of 3.5±1.9. Comparable to ileitis of SHIP−/− and SHIPΔIP/ΔIP mice, ileitis of MxCre SHIP flox/flox mice was segmental in nature with mixed inflammatory cell infiltrations. Granuloma formation was evident in 2 of 12 (17%) of MxCreSHIPflox/flox mice. Thus, CD-like ileitis develops with selective deletion in adult mice, illustrating that SHIP expression is required to maintain normal mucosal immune homeostasis and regulation in the adult ileum.
SHIP-deficient parenchymal cells do not cause ileitis
To assess the possibility that SHIP-deficiency might alter the development or function of parenchymal cell types in the ileum such as the IEC, we created bone marrow (BM) chimeras where six SHIP−/− hosts were reconstituted with WT C57BL/6 BM and analysed histopathologically 4 months post-transplantation. The gastrointestinal tract of the six SHIP−/− hosts reconstituted with WT BM lacked any abnormalities including all features of the CD-like ileitis observed in germline SHIP−/− and SHIPΔIP/ΔIP mice. We also analysed the lungs of these reconstituted mice. Although components of a moderate eosinophilic crystalline pneumonia were present in the SHIP−/− hosts reconstituted with WT C57BL/6 BM, including crystal formation, and an increase in the number of alveolar macrophages, the marked myeloid infiltration and pulmonary consolidation observed in SHIP−/− and SHIPΔIP/ΔIP mice was not present in lungs of SHIP−/− hosts reconstituted with WT C57BL/6 BM.
SHIP-deficient haematolymphoid cells transfer ileitis to WT hosts
To directly test whether SHIP-deficient haematolymphoid cells are responsible for the ileitis observed in SHIP-deficient strains, we adoptively transferred 5×105 SHIP−/− splenocytes to each of six WT C57BL/6 hosts. The lungs and intestines were then analysed 1 month later for evidence of pathology. Segmental ileitis similar to that observed in germline SHIP-deficient mice was present in 5 of 6 WT mice that received SHIP−/− splenocytes with a mean ileitis score of 1.8 (the range of scores was 0–5). This analysis demonstrates that SHIP-deficiency of the haematolymphoid compartment is sufficient to alter enteric immune cell homeostatic responses in the small intestine resulting in a CD-like phenotype. Interestingly, adoptive transfer of FACS-purified CD3+ T cells, NK1.1+CD3− NK cells prepared from the spleens of SHIP−/− mice was unable to transfer any detectable ileitis to WT hosts (three hosts analysed per cell type). Thus, SHIP-deficient T cells or NK cells are sufficient for transfer of ileitis. We also observed that SHIP−/− splenocytes transfer a mild to moderate eosinophilic crystalline pneumonia to WT hosts with progression to patchy pulmonary consolidation in some cases as is observed in both germline SHIP-deficient mice41 and MxCreSHIPflox/flox mice following gene ablation.49
Neutrophilia combined with T cell paucity in the SHIP-deficient small intestine
Our pathology findings and adoptive transfer studies described above suggest that a granulocyte–monocyte lineage cell may underlie the ileitis that we observe in SHIP-deficient mice. To further evaluate this possibility, we conducted multi-parametric cytometry assays of major immune cell populations including neutrophils, dendritic cells, CD4/CD8 T cells and Treg cells present in small intestines of SHIP-deficient and WT mice. In support of a role for neutrophils in ileitis, we find there is an absolute increase in total number of neutrophils present in the small intestine of SHIP−/− mice (figure 10). This applied to neutrophils of the Ly6G+CD16/32+ phenotype as well as the CD62L+ subset (figure 10 A,B). These findings of ileal neutrophilia are consistent with our histopathology studies described above. Surprisingly, we find a profound reduction in the number of both CD4 (figure 11A) and CD8 (figure 11B) T cells present in SHIP-deficient small intestines. When paired with the T cell adoptive transfer studies described above, these findings suggest that the CD-like ileitis observed in SHIP-deficient mice is not attributable to an autoimmune attack by SHIP-deficient T cells. Analysis of CD11c+CD11b+ mature dendritic cells and CD4+CD25+ Treg cells did not show a significant difference in the numbers of these immune cells between SHIP−/− and WT small intestines.

Neutrophilia in SHIP-deficient small intestine. FACS detection of CD16/32+Ly6G+ neutrophils (A) and their CD62L/E surface staining (B) in small intestine of SHIP-deficient mice and wild-type (WT) littermates. (C) Absolute total CD16/32+Ly6G+ neutrophils and CD62L+ neutrophils in the small intestine of SHIP-deficient and WT mice as indicated. ***p<0.001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Profound T cell deficit in SHIP-deficient small intestine. FACS detection of CD3+CD4+ (A) and CD3+CD8+ (B) T cells in the small intestine of SHIP-deficient mice and their wild-type (WT) littermates. (C) Absolute CD4 and CD8 T cell numbers in the small intestine of SHIP-deficient and WT mice as indicated. **p<0.01.
Discussion
CD pathology begins with the formation of multiple aphthoid ulcers infiltrated by neutrophils, which progress and coalesce into sharply delimited, regional, transmural inflammations with formation of strictures, fissures and fistulas.1–4 Here we show that mutation of the SHIP1 locus leads to a comparable ileitis dominated by neutrophils. Although elements of CD pathology are lacking in the SHIP-deficient mouse described herein, including development of multiple aphthoid ulcers, pyloric gland metaplasia, granulation tissue, and fistula formation, many CD histopathological features are present. SHIP-deficient ileitis is not precipitated by an apparent dysfunction on the part of SHIP-deficient parenchymal elements as reconstitution of SHIP−/− hosts with SHIP-competent haematolymphoid cells prevents development of ileitis. However, these same hosts still retain some facets of eosinophilic crystalline pneumonia indicating that SHIP−/− parenchymal elements may contribute to the lung pathology found in germline SHIP-deficient mice. Conversely, we find that transfer of SHIP-deficient haematolymphoid cells from the spleen is sufficient to trigger CD-like ileitis in an immunocompetent host. Transfer of cells capable of a cell-mediated immune attack, T cells and NK cells, do not elicit this CD-like ileitis. These findings suggest that a SHIP-deficient myeloid cell may promote the CD-like enteritis that occurs in SHIP-deficient mice. Granulocytes are less susceptible to apoptotic signals in SHIP-deficient mice, granulocyte–monocyte infiltrations can be found in various tissues of SHIP-deficient mice,44 and mononuclear phagocytes are essential for maintenance of intestinal homeostasis.50
Retention of components of eosinophilic crystalline pneumonia in the SHIP−/− hosts reconstituted with WT BM indicates that aspects of this lung pathology are of a parenchymal, cell autonomous origin. Mass spectrometry analyses have identified Ym1 as the principal crystalline protein of this pneumonia, perhaps secreted by activated alveolar macrophages, and proposed to be involved in eosinophil recruitment, immune activation and tissue repair.34 However, since SHIP-deficient mice that undergo allogeneic BM transplant have a normal lifespan,32 these retained features of pulmonary pathology are apparently not sufficient to progress to the fatal lung consolidation that occurs in germline, SHIP-deficient mice.41 Further, with the present observations, CD-like enteritis should be included among the pathologies that develop and contribute to morbidity and mortality in germline, SHIP-deficient mice.
The absence of enteric pathology in SHIP−/− hosts reconstituted with WT BM indicates that SHIP-deficiency does not cause an IEC functional barrier defect leading to ileitis. Conversely, that the CD-like phenotype can be transferred to an immunocompetent host by SHIP-deficient haematopoietic cells suggests that SHIP-deficient ileitis is due to an alteration in the regulation of innate immune functions (eg, granulocyte–monocyte responses), a failure of T cell mediated mucosal barrier functions or a combination of such potential alterations. Ileitis in SHIP-deficient mice may result from a deficit in mucosal T cell function that permits inappropriate granulocyte–monocyte responses to commensal organisms and luminal antigens culminating in the inflamed states observed in the ileum of SHIP-deficient mice.
Although the SAMP1/Yit mouse24 and the TNF∆ARE mouse25 spontaneously develop ileitis, the present SHIP-deficient mouse is unique in that it recapitulates the CD phenotype apparently through perturbation of immune cell populations that can be transferred to a WT host. There are a number of possible candidates for the enteritis-inducing cell. Because we observed neutrophilic infiltrates in the distal ileum lamina propria of most SHIP-deficient mice, we speculate that inappropriate responses by neutrophils to commensal microflora in the lumen of the ileum may underlie or trigger the immune attack. SHIP plays a pivotal role in preventing neutrophilia, as granulocyte numbers in peripheral blood are profoundly increased in SHIP−/− mice41 as well as mice treated with a SHIP1 selective inhibitor, 3AC.49 Inappropriate effector function by SHIP−/− neutrophils at organ sites has not been examined to date, and thus our findings represent the first indication of neutrophil-mediated tissue damage in SHIP-deficient hosts. Conversely, it seems unlikely that the enteritis results from inappropriate T cell attack in the small intestine, as SHIP-deficiency causes a profound local deficit in both CD4 and CD8 T cell numbers in the small intestine, while normal numbers of Treg cells are still present at this site.
The CD-like pathological findings described above are routinely observed in the ileum of both SHIP−/− strains31 41 and the SHIPΔIPΔIP strain47 that harbour different mutations of the SHIP1 gene indicating this is a highly penetrant phenotype. Moreover, because the phenotype occurs in the SHIPΔIPΔIP strain where only the inositol phosphatase domain encoding exons of SHIP are excised, we conclude that the phenotype is dependent on loss of SHIP enzymatic activity. SHIP through its 5′-inositol phosphatase domain hydrolyses the second messenger PI(3,4,5)P3 and thus limits PI3K activation of key downstream survival and effector pathways promoted by Akt and NF-κB.30 40 However, recent findings indicate that SHIP, via generation of PI(3,4)P2, can also promote cellular survival by increasing activation of Akt,49 consistent with the demonstration that Akt binds to and is more effectively activated by the SHIP product, PI(3,4)P2, than by its substrate, PI(3,4,5)P3.51 Tiwari et al found that SHIP can also promote immune effector function via synthesis of PI(3,4)P2.52 It remains to be determined whether SHIP-deficiency is causing ileitis due to a lack of inhibitory or activating signalling in immune cells. This may be dependent on cellular context. For instance, one could envision that SHIP may be required for efficient mucosal trafficking and survival of T cells via generation of PI(3,4)P2, but by decreasing PI(3,4,5)P3 in neutrophils SHIP is also limiting their numbers and function at this enteric site. Future lineage specific SHIP ablation studies using SHIPflox/flox mice and appropriate Cre transgenes combined with biochemical analysis PI3K effector pathways in these models may elucidate the molecular mechanism underlying SHIP's role in preserving normal immune function/regulation in the ileum.
Genetic analysis of mice and genome wide association studies have led to the identification of mutations that can predispose to CD with varying probability of incidence and timing of onset. Our analysis suggests that in the laboratory mouse, SHIP is a strong genetic determinant of CD-like pathology, suggesting that a similar situation may also be revealed by close examination of SHIP expression and function in CD patients. Tantalising evidence for a role of SHIP1 in human IBD is suggested by the recent demonstration that a chromosome 2 polymorphism located at 2q37 is associated with a significantly increased and early onset of IBD.53 The SHIP1 locus is also found in the 2q37 region of human chromosome 2. This potential association merits comparison of SHIP enzymatic activity in IBD patients and normal controls using a recently developed fluorogenic assay that measures SHIP1 activity.49 Future investigations of cellular and molecular mechanisms responsible for ileitis in SHIP-deficient mice may also provide insights into how immunological alterations contribute to IBD.
References
Footnotes
Linked articles 225664.
WGK and MYP contributed equally.
Funding This work was supported in part by grants from the NIH (RO1 HL72523, R01 HL085580) and the Paige Arnold Butterfly Run. WGK is the Murphy Family Professor of Children's Oncology Research and a SUNY Empire Scholar.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.