Article Text

Abstract
Objective Colorectal cancer (CRC) is a common cancer and a leading cause of cancer deaths. Previous studies have identified a number of key steps in the evolution of CRC but our knowledge of driver mutations in CRC remains incomplete. Recognising the potential of studying different human populations to reveal novel insights in disease pathogenesis, we conducted genomic analysis of CRC in Saudi patients.
Design In the discovery phase of the study, we conducted whole genome sequencing of tumour and corresponding germline DNA in 27 patients with CRC. In addition to known driver mutations, we identified three MED12 somatic mutations. In the replication phase, we employed a next-generation sequencing approach to capture and sequence MED12 and other candidate genes in a larger sample of 400 patients with CRC and confirmed the enrichment for recurrent MED12 mutations.
Results In order to gain insight into a plausible biological mechanism for the potential role of MED12 mutations in CRC, we studied CRC cell lines that differ substantially in the expression level of MED12, and found the latter to be correlated inversely with transforming growth factor (TGF)-β signalling and directly with apoptosis in response to chemotherapeutic agents. Importantly, these correlations were replicated when MED12 expression was experimentally manipulated.
Conclusions Our data expand the recently described role of MED12 as a tumour suppressor in other cancers to include CRC, and suggest TGF-β signalling as a potential mediator of this effect.
- COLORECTAL CANCER
- MUTATION SCREENING
- COLORECTAL CANCER GENES
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Colorectal cancer is known to develop as a multistep process involving driver mutations.
MED12 mutations are known to drive several tumour types.
What are the new findings?
We show that MED12 is recurrently mutated in colorectal cancer, uniformly in the active (unmethylated) X chromosome.
We also show that MED12 deficiency confers resistance to chemotherapy at the tissue culture level.
How might it impact on clinical practice in the foreseeable future?
MED12 mutations identified through genomic sequencing of colorectal cancer may predict resistance to chemotherapy.
Introduction
Colorectal cancer (CRC) is a very common cancer that is only second to breast cancer in women, and lung and prostate cancer in men, and is the second leading cause of cancer death in Western countries.1 ,2 In the USA alone, it is estimated that 134 000 persons will be found to have CRC and that 49 000 will die from it. These alarming figures, however, are at least 40% lower than the mid-1980s thanks in large part to the implementation of preventive screening programmes that were conceived on the basis of improved understanding of CRC pathogenesis.3 ,4
The seminal proposal by Vogelstein et al in 1988 laid the ground for the contemporary view of the transformation of intestinal epithelium into invasive adenocarcinoma through a slow multistage mutational process.5 ,6 Efforts to identify mutations in tumour-suppressor genes and oncogenes that play a key role in the initial stages of CRC tumourigenesis were limited in scope due to technical challenges, although early attempts to sequence large sets of genes using Sanger sequencing have provided valuable insights.7–9 Recent advances in sequencing technology have propelled the implementation of genomic analysis of CRC, which greatly expanded the repertoire of driver mutations, both at the base pair (bp) and chromosomal scale.10
MED12 encodes a member of Mediator, a multiprotein complex involved in the transcriptional regulation of many genes by mediating the interaction of RNA polymerase with various transcriptional factors.11 Germline mutations in this X linked gene are known to cause intellectual disability in various syndromic forms.12 This was corroborated by mouse data that show a strict requirement for MED12 level in normal gastrulation and neurulation.13 More recently, somatic mutations in MED12 have been found to play a significant role in the aetiology of uterine leiomyoma, a finding that was reproduced in several cohorts.14 Subsequently, driver MED12 mutations were identified in other tumours including adrenocortical carcinoma, prostate cancer and breast fibroadenoma.15–17 Interestingly, the distribution of MED12 mutations differs in different types of tumour. In uterine leiomyoma, most of MED12 mutations were identified in exon 2 and lead to the activation of WNT pathway genes through interaction with β-catenin, while in prostate cancer MED12 mutations were mainly detected in the non-N-terminal region and seem to influence p53 and androgen signalling pathway.16 ,18–21
In this study, we implemented a hypothesis-free genomics approach in our search for relevant somatic mutations in CRC in Saudi Arabia, a country with a particularly high incidence of CRC with an unusually young age of onset.22 This allowed us to identify recurrent somatic MED12 mutations suggesting a potential role in CRC pathogenesis. Furthermore, functional analysis of relevant cell lines supports a previously reported role of MED12 deficiency in mediating drug resistance.23
Materials and methods
Human subjects
A total of 427 patient samples diagnosed with CRC at the King Faisal Specialist Hospital and Research Centre were collected from Department of Pathology. Detailed clinicopathological data were noted from case records and summarised in table 1. All tissue and blood samples were obtained from patients with approval from Institutional Review Board of the hospital. Informed consent was obtained from the patients from whom fresh cancer tissues were harvested for the study. For the study, waiver of consent was obtained for archived paraffin tissue blocks from Research Advisory Council (RAC) under project RAC#2140 005.
Clinicopathological variables for the patient cohort (n=427)
DNA isolation
DNA isolation from peripheral blood, fresh frozen and paraffin-embedded tissues was performed using Gentra DNA isolation kit (Gentra, Minneapolis, Minnesota, USA) following the manufacturer's recommendations.
Whole genome sequencing
Whole genome sequencing was performed on 27 patients with their corresponding normal blood. Paired-end libraries of approximately 500-bp inserts were sequenced as 150-bp paired-end reads. Raw sequencing data were processed by an ultrafast Isaac DNA sequence aligner designed to align next-generation sequencing data with low-error rates using human genome V.19. The Isaac Variant Caller was used to call single-nucleotide variants (SNVs) and small indels from the bam files.24 SnpEff was used for annotating the variants with dbSNP138, dbSNP142, 1000 Genomes project and ESP6500 database.25 We excluded common single-nucleotide polymorphisms (SNPs) with minor allele frequency of >0.001 as recorded in either dbSNP, National Heart, Lung, and Blood Institute exome sequencing project, 1000 Genomes and our in-house data from exome sequencing of over 730 normal samples. Non-coding variants, synonymous variants and variants present in highly repetitive regions were excluded for further analysis.
Candidate gene capture and sequencing
Recurrently mutated genes (mutated in at least three samples) from the 27 whole genome cases were selected for capture sequencing using SureSelect Target Enrichment Kit on Illumina HiSeq 2500 Sequencer. Fastq files were aligned to the human reference genome hg19 using burrows-wheeler aligner (BWA).26 Local realignment, PCR duplicates and base quality recalibration was performed using genome analysis toolkit (GATK) and Picard-tools.27 SNVs and small indels were called using GATK. The identified variants were annotated using SnpEff. Similar filters as applied to whole genome samples were used to get final mutation list.
PCR and Sanger sequencing
Primer 3 software was used to design the primers for the particular variants identified by whole exome or capture sequencing. PCR and Sanger sequencing were performed as described previously.28 Reference sequences were downloaded from GenBank. Sequencing traces were analysed using the Mutation Surveyor V.4.04 (Soft Genetics, State College, Pennsylvania, USA).
Allele-specific mutation analysis
Genomic DNA samples were digested using restriction endonuclease Mse I for 2 hours and then MethylCollector Ultra kit (Active Motif, Carlsbad, California, USA) was used to collect the pure/enriched methylated DNA and the counterpart unmethylated/methylated DNA following the manufacturer's recommendations as described previously.29 PCR and Sanger sequencing were then performed to examine which allele of X chromosome has the identified mutation in female mutation-positive cases.
Tissue culture experiments
All human CRC cell lines; CL34, Caco2, Colo-320, DLD1, HCT15, HCT116, HT29 and LOVO were obtained from American Type Culture Collection and cultured in Roswell Park Memorial Institute 1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 U/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2. All treatment experiments were performed in reduced FBS condition (5%). Apoptosis analysis was performed using annexin V/propidium iodide dual staining and measured by flow cytometry as previously described.30 Antibodies against MED12 (CST#14360), E-cadherin (CST#3195), N-cadherin (CST#14215), pERK1/2 (CST#4370), ERK1/2 (CST#4695) and β-actin (CST#3700) were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA). Antibodies against transforming growth factor (TGF)-β-R2 (sc#17799), vimentin (sc#5565) and GAPDH (sc#25778) were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA). Twist antibody (ab#175430) was purchased from Abcam (Cambridge, Massachusetts, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays
Initially, CRC cells were incubated at the concentration of 104 cells per well in triplicates in a 96-well format. Cells were then treated with various doses of 5-fluorouracil (5-FU) for 48 hours in a final volume of 0.2 mL. Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay, as previously described;31 6 wells for each dosage including vehicle control were analysed for each experiment.
Gene silencing using small interfering RNA
Two different sets of MED12 small interfering RNA (siRNA), and scrambled control siRNA were purchased from OriGene (Rockville, Maryland, USA). Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, California, USA) for 6 hours following which the lipid and siRNA complex was removed and fresh growth medium was added. After 48 hours of transfection cells were used for various experiments such as clonogenic assay, apoptosis analysis and immunoblotting.
Plasmid and transfection
Plasmid DNA encoding human MED12 was purchased from OriGene. Transfection was performed using Lipofectamine 2000 according to the manufacturer's protocol. Briefly, CRC cells were seeded in 6-well culture plates; when approximately 50% confluent, cells were transfected with 4 μg plasmid. After 48 hours of transfection cells were used for various experiments such as clonogenic assay, apoptosis analysis and immunoblotting.
Immunofluorescence analysis
CRC cells grown on coverslips in 6-well plates were fixed with ice-cold 100% methanol followed by permeabilisation with 0.2% Triton X-100, blocked with 5% horse serum in phosphate-buffered saline (PBS) solution, and incubated with antibodies to MED12 (1:200), TGF-β-R2 (1:50) or E-cadherin (1:200) in buffer A (1% bovine serum albumin, 0.1% Triton X-100, 10% horse serum in PBS solution) for 1 hour at 37°C. Cells were then incubated with Alexa Fluor 488 goat antirabbit or Alexa Fluor 594 goat antimouse antibody and mounted using DAPI. The cells were visualised using Olympus BX63 fluorescence microscope.
Results
Whole genome sequencing identified MED12 among other recurrently mutated genes in CRC
Our bioinformatics analysis of whole genome sequencing (WGS) performed on the tumour and corresponding blood-derived DNA from 27 patients with CRC identified 8356 non-synonymous somatic mutations. The average sequencing coverage for tumour samples was 36×, whereas for the corresponding normal, the coverage was 35×. All the samples were sequenced with an average read length of 150 bp. We categorised our cases into two groups based on microsatellite instability (MSI) status and mutations in mismatch excision repair (MMR) genes MLH1, MLH3, MSH2, MSH3, MSH6 and PMS2. Six samples were MSI or MMR mutation positive. The average non-synonymous somatic mutation rate for these cases was 1052. Interestingly, one of these six cases harbouring MSH3 and POLE mutations showed very high non-synonymous mutation density of 4157. The MSI-negative cases showed an average non-synonymous mutation density of 97 (table 2). The average synonymous somatic mutation rate for the six MSI-positive or MMR-positive cases was 373, while for the MSI low cases was 52. There were significantly more non-synonymous mutations compared with synonymous somatic mutations for the MSI-low cases (p<0.001), whereas no significance was found in the MSI-high positive cases (p=0.305, see online supplementary figure S1). We also found no significant difference between the number of non-synonymous and synonymous mutations per sample and patient age in both the groups (p=0.5922).
Comparison of non-synonymous mutation density with other exome studies
supplementary figure
In this discovery cohort, we prioritised genes that are somatically mutated in at least three patients as likely candidate ‘driver’ genes (see online supplementary table S1). Reassuringly, the list includes the well-established CRC driver genes APC, KRAS and TP53 (table 3). In addition, the list also includes the following candidate genes: RYR1, SYNE1 and MED12. Other well-known driver genes of CRC like BRAF and SMAD4 were not found in our initial small cohort of 27 samples, however, we have found PIK3CA mutated in just 1 case. MED12 was particularly interesting given its recently established role as a driver gene in a number of tumours and largely uncharacterised role in CRC.14–17 Furthermore, when recurrently mutated genes in our discovery cohort were ranked based on the relative mutation frequency by taking the protein size into account,34 ,35 MED12 ranked fourth to APC, KRAS and TP53 (table 4). The following likely pathogenic somatic variants were identified in MED12: c.110C>T;p.Thr37Met, c.130G>A;p.Gly44Ser and c.6369_6380dupCCAGCAGCAACA; p.His2123_Gln2126dup (table 5).
List of recurrent mutations found in 27 WGS and 400 capture sequencing cases
Ranking of recurrently mutated genes with bases on mutation rate
MED12 mutations with three different pathogenicity scores
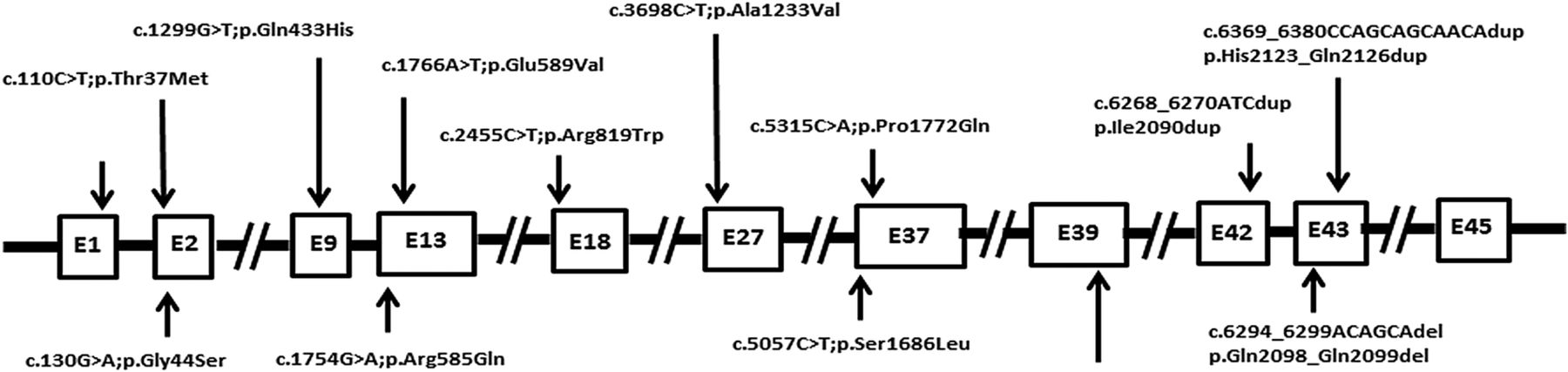
To further investigate a potential role of MED12 in CRC, we sequenced MED12 in a replication cohort of 400 patients with CRC using in-solution capture and next-generation sequencing. One sample was not sequenced due to quality control (QC) failure. Using a stringent filtering pipeline, we identified 15 likely pathogenic variants in MED12, 11 of which were confirmed by Sanger sequencing to be true somatic mutations in 9 cases (table 5; figure 1 and see online supplementary figure S2). Most of these mutations were pathogenic as predicted by three different computational algorithms.

Schematic diagram of distribution of 14 MED12 mutations identified in 12 colorectal cancer cases.
Therefore, by combining the discovery and replication cohort, the percentage of patients with somatic MED12 mutations is 2.8%, a small but comparable percentage to some of the known driver genes in CRC, for example, BRAF, PTEN, NRAS, SMAD4 and MUTYH.10 ,36–38 We found a total of 14 MED12 gene mutations in 12 CRC cases in this cohort. Out of 14 mutations, 11 were missense, 2 duplication and 1 deletion. Three mutations were seen in exon 1, two mutations were seen in exon 13, 37, 43 each and one mutation was found in exon 9, 18, 27, 39 and 42 each. The concurrent presence of nearly all of these MED12 somatic mutations with mutations in well-established driver genes suggests a potential role in tumour evolution past the initiation stage.28 Unexpected for an X linked gene, only 33% (4/12) patients with CRC with somatic MED12 mutations were males. This prompted us to investigate whether MED12-mutated females harboured their somatic mutations on the active allele X chromosome, essentially mimicking the effect of hemizygous male mutations. Indeed, in all eight females the presence of somatic MED12 mutation on the active allele of the chromosome X rather than inactive imprinted allele was confirmed (figure 2). Unfortunately, the relatively small number of MED12-mutated patients precluded formal testing for such associations as disease severity, response to chemotherapy and survival. However, it is worth noting that 50% of MED12-mutated patients with CRC who underwent chemotherapy (4/8) showed poor response to standard chemotherapy.
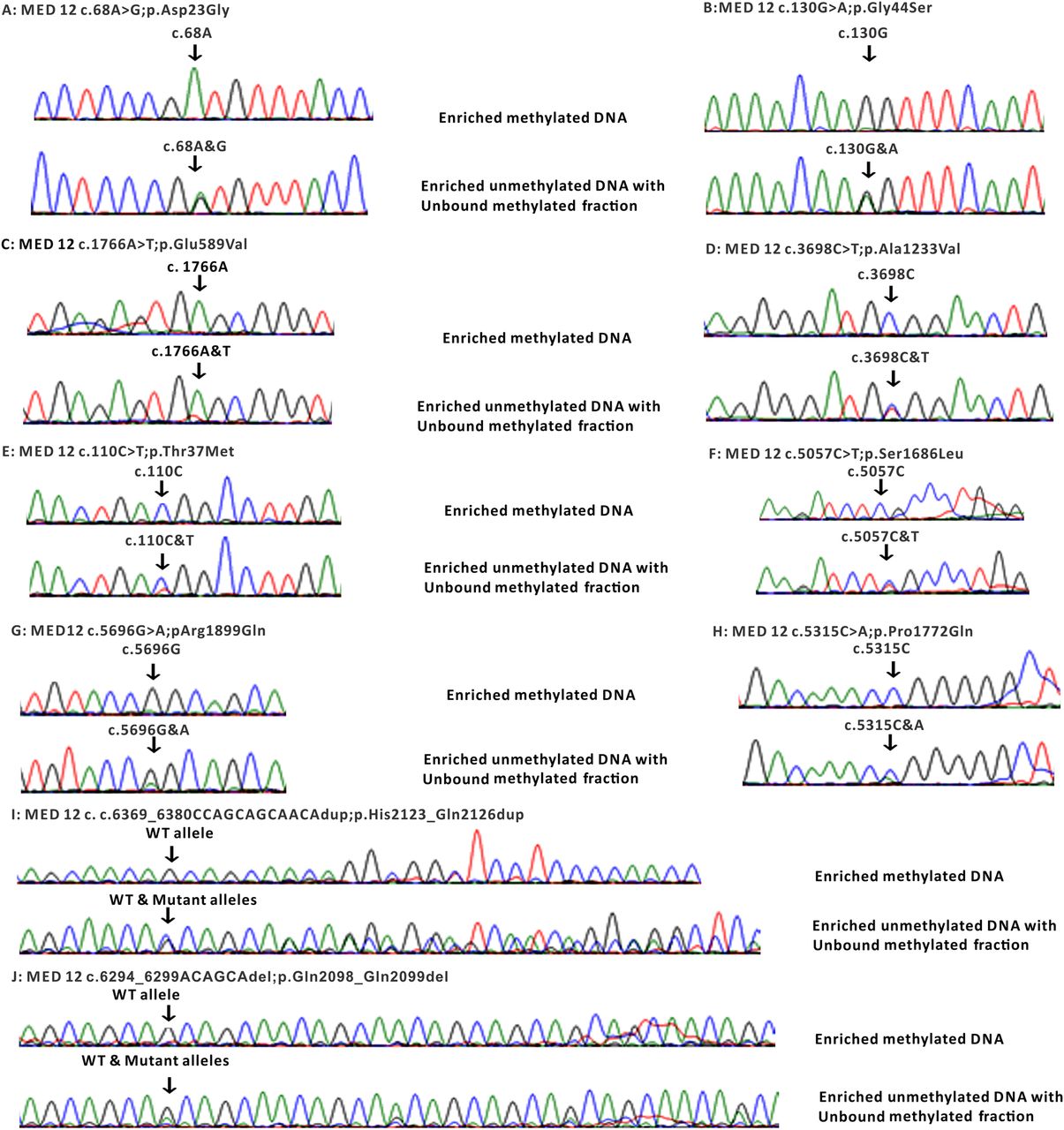
Sanger sequencing electropherogram of inactive/active allele of MED12 gene on chromosome X in eight female cases harbouring MED12 mutations. Top panel: sequencing electropherogram of enriched methylated DNA indicating no mutation was identified on the inactive allele caused by methylation. Lower panel: sequencing electropherogram of enriched unmethylated and unbounded methylated DNA. WT, wild type.
MED12 is a likely tumour suppressor that modulates TGF-β-induced epithelial-mesenchymal transition signalling in CRC cell lines
In order to gain insight into the biological plausibility of MED12 mutations in the pathogenesis of CRC, which would be suggested by our finding above, we set out to test its potential role in relevant CRC cell lines. To determine the role of MED12 expression in vitro, we assessed expression of MED12 in a panel of eight CRC cell lines by immunoblotting. As shown in figure 3A, there was differential expression of MED12 in these cell lines. On the basis of MED12 expression, we identified two CRC cell lines; DLD1 and LOVO that had overexpression of MED12 as well as two; Colo-320 and HT29 that had negligible expression, for further experimentation. MED12 abundance in these cells was also verified by immunofluorescence analysis (figure 3B). Using these cell lines, we determined the response to 5-FU treatment in inhibiting cell viability and inducing apoptosis. There was inhibition of cell viability and induction of apoptosis in both cell lines that had overexpression of MED12; however, 5-FU treatment had no effect on cell viability and induced minimal apoptotic response in the other two cell lines (figure 3C, D). Next, to confirm the role of MED12 in chemosensitivity, we downregulated the expression of MED12 using MED12-specific siRNA. As shown in figure 4A, B, silencing of MED12 significantly increased the proliferation of LOVO and DLD1 cells as confirmed by clonogenic assay. Following downregulation of MED12, we treated these cells with 5-FU for 48 hours and analysed the cells for apoptosis. As shown in figure 4C, when knocked down for MED12, DLD1 and LOVO cells became resistant to 5-FU treatment as compared with CRC cells transfected with non-specific scrambled siRNA. This was further confirmed using another set of MED12 siRNA that showed similar results (see online supplementary figure S3). MED12 has been shown to mediate response to alkalising agents and epidermal growth factor receptor inhibitors through the RAS pathway. Specifically, MED12 was found to suppress TGF-β signalling and that the latter is what mediates drug resistance.23 Therefore, we asked whether the effect we observed in CRC cell lines is similarly mediated by TGF-β. Indeed, we show that MED12 level is inversely correlated with TGF-β signalling as visualised by western blot analysis where MED12 knockdown led to upregulation of TGF-β-R2 and activation of ERK in both MED12 expressing cell lines (Figure 4D and see online supplementary figure S3). TGF-β is known to induce an epithelial-mesenchymal transition, leading to the loss of epithelial adhesion molecule E-cadherin and induction of several mesenchymal markers such as vimentin and N-cadherin.23 Silencing of MED12 decreased the E-cadherin expression with an accompanying upregulation of N-cadherin, vimentin and Twist expressions in MED12 expressing cells (figure 4D and see online supplementary figure S3). Similar results were observed by immunofluorescence where knockdown of MED12 induces TGF-β-R2 and decreases E-cadherin expressions in MED12 expressing cells (figure 4E). Conversely, forced expression of MED12 decreases cell proliferation in MED12 low expressing cells as shown by clonogenic assay (figure 5A, B). Forced expression of MED12 in Colo-320 and HT29 cells also sensitised them to 5-FU treatment as compared with cells transfected with empty vector (figure 5C). Moreover, MED12 overexpression in Colo-320 and HT29 cells markedly decreased TGF-β-R2 expression with an accompanying increased E-cadherin and decreased N-cadherin, vimentin and Twist expression compared with empty vector transfected cells (figure 5D). These results were further confirmed by immunofluorescence analysis (figure 5E). Taken together, these data suggest a potential role for MED12 in sensitising CRC cells to chemotherapy.

(A) Expression of MED12 in colorectal cancer (CRC) cell lines. Proteins were isolated from eight CRC cell lines and immunoblotted with antibodies against MED12 and GAPDH. (B) Representative images of fluorescence immunostaining for MED12 in four selected CRC cell lines and confirm results from western blot analysis. (C and D) CRC cells were treated with increasing doses of 5-fluorouracil (FU) for 48 hours and cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (C) and apoptosis by flow cytometry after staining the cells with annexin V/propidium iodide (D). Data presented in the bar graphs are the mean±SD of three independent experiments. *Indicates a statistically significant difference compared with control with p<0.05.

Silencing of MED12 increases the chemoresistance in colorectal cancer (CRC) cell lines. (A and B) CRC cells were transfected with either scrambled siRNA or MED12-specific small interfering RNA (siRNA) for 48 hours and clonogenic assay were performed. Cells (6×102) after post-transfection were seeded into each of three dishes (60 mm diameter), and grown for an additional 10 days, then stained with crystal violet (A). Colony numbers in the entire dish were counted (B). (C) CRC cells were transfected with either scrambled siRNA or MED12 siRNA and subsequently treated with 50 and 100 μM 5-fluorouracil (FU) for 48 hours. Following treatment, cells were analysed for apoptosis by flow cytometry. (D) LOVO and DLD1 cells were transfected with either scrambled siRNA or MED12-specific siRNA for 48 hours. Proteins were isolated and immunoblotted with antibodies against MED12, transforming growth factor (TGF)-β-R2, p-ERK1/2, ERK1/2, E-cadherin, N-cadherin, vimentin, Twist and β-actin for equal loading. (E) Representative images of fluorescence immunostaining for MED12, TGF-β-R2 and E-cadherin in LOVO and DLD1 cells after post-transfection with MED12 siRNA. Data presented in the bar graphs are the mean±SD of three independent experiments. *Indicates a statistically significant difference compared with control with p<0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forced expression of MED12 decreases the chemoresistance in colorectal cancer (CRC) cell lines. (A and B) CRC cells were transfected with either empty vector or MED12 pcDNA for 48 hours and clonogenic assay were performed. Cells (6×102) after post-transfection were seeded into each of three dishes (60 mm diameter), and grown for an additional 10 days, then stained with crystal violet (A). Colony numbers in the entire dish were counted (B). (C) CRC cells were transfected with either empty vector or MED12 pcDNA and subsequently treated with 50 and 100 μM 5-fluorouracil (FU) for 48 hours. Following treatment, cells were analysed for apoptosis by flow cytometry. (D) COLO-320 and HT29 cells were transfected with either empty vector or MED12 pcDNA for 48 hours. Proteins were isolated and immunoblotted with antibodies against MED12, transforming growth factor (TGF)-β-R2, p-ERK1/2, ERK1/2, E-cadherin, N-cadherin, vimentin, Twist and GAPDH for equal loading. (E) Representative images of fluorescence immunostaining for MED12, TGF-β-R2 and E-cadherin in COLO-320 and HT29 cells after post-transfection with MED12 pcDNA. Data presented in the bar graphs are the mean±SD of three independent experiments. *Indicates a statistically significant difference compared with control with p<0.05.
Discussion
Several studies have attempted to characterise the mutational landscape of CRC and several novel candidate driver genes have emerged as a result. We found a total of 14 MED12 gene mutations in 12 CRC cases in this cohort. Out of 14 mutations, 11 were missense, 2 duplication and 1 deletion. Three mutations were seen in exon 1, 2 mutations were seen in exon 13, 37, 43 each and 1 mutation was found in exon 9, 18, 27, 39 and 42 each. The Cancer Genome Atlas Network in their analysis on 224 CRCs found 15 MED12 mutations in 14 CRC cases.10 In their study they found, out of 15 mutations, 10 were missense, 4 were silent and 1 mutation was in-frame deletion. Mutation were seen in exon 36 (2 mutations), exon 42 (2 mutations) and 1 mutation each in exon 1, 2, 5, 7, 10, 18, 23, 29, 35, 40 and 43. Mutation in exon 2 of MED12 gene is the most common genetic aberration present in roughly 64% of leiomyomas.39 Although very rare, MED12 mutations in leiomyoma have also been reported in other regions of gene such as intron 140 and exon 1.41 In leiomyoma, codon 44 is the most common hotspot for MED12 mutation and both missense and in-frame insertion-deletion mutations are seen.42
The large study by the Cancer Genome Atlas Network did not highlight MED12 as a likely candidate even though 15 somatic mutations were identified, most likely because of the small percentage of affected samples.10 Indeed, Kämpjärvi et al43 specifically sequenced exon 2 MED12 in 392 patients with cancer and identified only two patients with somatic mutations. This is consistent with our data where we show that only 2.8% of patients with CRC have somatic MED12 mutations.
Genes in the RAS pathway are known to contribute to the pathogenesis of CRC, most notably KRAS and BRAF in which somatic mutations are observed in 30%–40% and 10%, respectively, of patients with CRC.44 ,45 These somatic mutations are typically activating in nature and are associated with the activation of the Erk1/2 kinase pathway.46 Thus, our finding that MED12 deficiency is also associated with the activation of the Erk1/2 kinase pathway would be consistent with the proposed oncogenic potential of MED12 somatic mutations in CRC. Although the numbers are small, we observed a trend where MED12-mutated patients with CRC have high likelihood of displaying resistance to alkalising chemotherapeutic agents. This appears to be consistent with our finding in CRC cell lines where we show that MED12 mediates sensitivity to 5-FU, likely via suppressing TGF-β signalling, a finding that was also obtained by Huang et al23 using different cell lines.
The location of MED12 on the X chromosome provides a unique advantage, where biallelic inactivation can be acquired as a one-step rather than the traditional two-step loss of heterozygosity (LOH) process.47 In other words, because females are functionally hemizygous for MED12, somatic mutations in the active X chromosome can result in complete inactivation of the gene. This has been shown in MED12-related breast fibroadenoma and leiomyoma.17 ,48 Our finding that all female patients with CRC with somatic MED12 mutations harboured the mutations on their active X chromosome serves as an additional line of evidence in support of these mutations playing a role in driving CRC tumourigenesis. The ‘single-hit’ requirement for inactivating MED12 in females, likely explains the lack of male predominance as expected for an X linked gene and represents a hypothetical opportunity for treatment through reactivating (demethylating) the normal X chromosome in these patients to restore normal level of MED12. We will pursue this as a future research direction.
In conclusion, we show that MED12 is recurrently mutated in Saudi patients with CRC and propose this corroborates previously proposed tumour suppresser role. Our data also suggest that such a role may be mediated through antagonising TGF-β signalling.
Acknowledgments
We thank Wael Haqawi, Zeeshan Qadri, Dionne Rae Rala, Ingrid Francesca Victoria, Maha Al Rasheed, Khadija Al Obaisi, Roxanne Melosantos, Rafia Begum, Sarah Siraj and Saravanan Thangavel for technical assistance.
References
Footnotes
Contributors We have added as a coauthor a team member who helped generate the additional data in functional analyses (PP).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Research ethics committee, King Faisal Specialist Hospital and Research Centre.
Provenance and peer review Not commissioned; externally peer reviewed.