
Abstract
In the past decades, continuous effort has been paid to deeply understanding the pathophysiology of inflammatory bowel diseases (IBD), such as ulcerative colitis or Crohn’s disease. As the disease typically arises as chronic inflammation of the gastrointestinal mucosa, research has been focused on how such an uncontrolled, deleterious immune response may arise and persist in a certain cohort of patients. Based on those immunologic analyses, the establishment of anti-TNF-α therapy, and the following series of biologic agents achieved great success and dramatically changed the therapeutic strategy of IBD patients. However, to guarantee long-term remission of the disease, the therapeutic standard has been raised to achieve “mucosal healing”, which requires complete repair of the gastrointestinal mucosa. Recent studies have revealed the unexpected importance of epithelial cells in the pathophysiology of IBD. The general barrier function as well as the cell lineage-specific functions have been deeply attributed to the development of chronic intestinal inflammation. Also, the groundbreaking establishment of the in vitro intestinal stem cell culture system has opened up a way of developing stem cell transplantation therapy to treat otherwise refractory ulcers that may persist in IBD patients. In this review, we would like to focus on the role of epithelial cells in the pathophysiology of IBD, and also give a perspective to the upcoming development of regenerative therapies that may become one of the therapeutic choices to achieve mucosal healing in refractory patients of IBD.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD) is an idiopathic gastrointestinal disease where chronic inflammation dominates the mucosa, and thereby destructs both the structure and function of the gastrointestinal tract. An increasing number of patients are found not only in Western countries but also in newly developing Asian countries.
As the disease manifests as an “inflammatory” disease, research regarding IBD has long been focused on the immunologic aspect of the disease. One of the most successful outcomes of these studies is the development of biologics such as anti-TNF-α antibody therapy, which has dramatically changed the therapeutic strategy of IBD in a very short time [1, 2]. However, a certain subset of patients is still clearly refractory to those newly developed treatments, and also a higher standard of treatment achievement is required to maintain remission for a longer period in an increasing number of patients.
A recent series of clinical studies have shown that complete regeneration of the intestinal mucosa, called “mucosal healing”, predicts long-term remission and low risk of surgical treatment in IBD patients [3]. Therefore the importance of intestinal epithelial cells (IECs) in IBD is sharply increasing, and thus gathering much attention of many researchers.
Regarding the pathogenesis of IBD, a series of genome-wide association (GWAS) studies have identified many genes that may predispose patients to IBD [4–7]. Surprisingly, those studies showed that not only the immune cell-related genes, but also those genes that are closely related to IEC-specific functions may well determine the susceptibility to IBD. Accordingly, following functional studies of genes such as ATG16L1 has proven that at least a part of those genes may play a certain role in IECs, and thereby promote the onset or the persistence of the disease [8].
In the treatment of IBD, newly emerging techniques such as the culture of intestinal stem cells (ISCs) in vitro, and its transplantation to repair refractory ulcers, may develop to an alternative therapeutic method in the near future.
In this review, we would like to address the emerging role of IECs in the pathogenesis of IBD, and further discuss IEC-oriented or IEC-based therapies that are currently under development, including the transplantation of ISCs.
Homeostasis of the normal intestinal epithelia
The intestinal epithelium covers the whole inner surface of the gastrointestinal tract, which may count up to at least 32 m2 per body in humans [9]. Such a broad lining of the body surface largely exceeds that of skin, and constitutes the largest frontline to the outer-body environment. To confront and cope with the continuous but dynamic change of the surrounding environment, the intestinal epithelia is completely renewed within a short period, reaching up to every 4–5 days in the small intestine [10].
Such a rapid renewal is basically supported by the function of ISCs. These cells maintain their ability to self-renew, and also to differentiate into the five distinct lineages of differentiated IECs [11]. ISCs reside at the bottom of the crypt region, and are strictly regulated to maintain a certain number of ISCs per crypt [12]. An increasing number of studies have identified the molecular signatures as well as intracellular signals that are specific for ISCs [13]. Among those molecular pathways, Wnt signaling has been most intensively studied, and identified as one of the most indispensable pathways [14]. Studies have shown that LGR5 is an ISC-specific gene [15], which encodes a receptor for R-Spondin-1 [16]. It functionally acts as a co-enhancer of Wnt signaling, and co-operates with Wnt-mediated canonical pathway activation in ISCs through regulation of the E3-ligase Rnf43 [17, 18]. Accordingly, a number of genes regulated downstream of the canonical Wnt pathway, such as ASCL2 [19, 20], have been assigned and proved to be ISC-specific genes as well.
The Notch pathway is another pathway that has an indispensable role in maintenance of the intestinal epithelium [21–23]. In general, it is trans-activated through membrane-bound Notch receptors, by cell-to-cell contact with adjacent cells that express Notch ligands [24]. In the intestine, Notch1 and Notch2 had been identified as the indispensable receptors to maintain proper proliferation and differentiation of progenitor cells [25]. For the ligands, loss of Dll1 and Dll4 results in silencing of Notch activation in IECs, and subsequently leads to secretory cell hyperplasia [26]. Our recent study has identified the distinct distribution of Dll1-positive or Dll4-positive IECs, and showed that Dll1 expression is restricted to Atoh1-positive cells in the crypt region [27]. From these studies, activation of Notch in progenitor cells is suggested to direct their fate towards absorptive cell lineages, whereas cis-inhibition of Notch directs those cells towards one of the four secretory lineage cells (Goblet, Enteroendocrine, Paneth or Tuft), depending on the pro-secretory transcription factor Atoh1 [28, 29]. However, studies have shown that activation of this pathway is also required for the maintenance of ISCs [30]. Thus, finely regulated proliferation and differentiation of cells arising from ISCs, and also the maintenance of ISC itself is under the concerted regulation of these molecular pathways.
The intestinal epithelium exists as the crossroad of IBD susceptibility factors
The pathogenesis of IBD has been a challenging subject for many researchers, and has gathered great interest from both immunologists and geneticists. The most recent model built as a result of this intensive research has been described elsewhere [31–33]. In summary, it is suggested that the onset of IBD is based on the following four major factors: genetic susceptibility, immune dysregulation, microbial flora (dysbiosis) and environmental factors (food, smoking, drugs, etc.). Especially the role of the intestinal microbiota has been featured, as it can be modulated through its interplay with dietary factors [32], or by the genetic predisposition of the host [33]. Accordingly, a multi-hit model of IBD has been suggested [34]. In such a model, genetic factors, mucosal barrier function, and dysbiosis constitute the most basal factors that support the onset and perpetuation of the disease. However, we would like to emphasize that the intestinal epithelium constitutes the interface between these elements, and thus should be considered to have a key role in governing the interactions between microbial flora, environmental factors, genetic factors, and the host immune response (Fig. 1). The intestinal epithelium constitutes the border of the inner and outer body, and therefore directly confronts the commensal as well as pathogenic bacteria, and environmental factors such as food antigens. On the other hand, the intestinal epithelium interacts bi-directionally with the underlying immune cells through direct contact or by various cytokines. In addition, genetic factors may affect the function of epithelial cells, in addition to the immune cells residing at the lamina propria [35]. Thus, the intestinal epithelial tissue exists as a crossroad of the four factors that constitute the pathogenesis of IBD.

The intestinal epithelium at the crossroad of IBD pathogenesis factors. The intestinal epithelium links the IBD pathogenesis factors such as microbial flora, environmental factors, or the host immune response by directly interacting with those factors. Also, several susceptibility genes of IBD may directly affect their function
Role of IECs in the pathogenesis of IBD
Recent studies have further emphasized the importance of epithelial cells in the pathogenesis of IBD. As one of the most important functions of the epithelial tissue is to keep the inner-body space apart from the outer environment, disruption of this barrier function may lead to the invasion of the pro-inflammatory bacteria, or the immunogenic environmental antigens. The term “mucosal barrier” may include the physical barrier function that is maintained by the cell-to-cell junction between epithelial cells, and also the antimicrobial functions that are mediated by specific lineages of IECs. For the former case, several lines of knockout mice studies, such as knockout mice of Gai2 [36] or JAM-A [37], suggest that disruption in the assembly of the tight junction or the adherens junction may lead to spontaneous development of intestinal inflammation. Our recent study has identified that TNF-α-TNFR2 signaling in IECs may increase the expression of MLCK, and thereby disrupt the assembly of the tight junction [38]. Therefore, proinflammatory cytokine signaling may further enhance the “leakiness” of the epithelial layer, which may promote the perpetuation of the local inflammation. However, mice expressing the constitutive active form of MLCK show increased epithelial permeability but does not develop spontaneous colitis in vivo [39], suggesting that leakiness of the epithelial layer alone may not lead to spontaneous development of colitis but requires another “deleterious hit” for the onset of the disease [40].
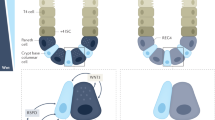
Also, specific cell lineages had been assigned to the pathogenesis of IBD. Goblet cells produce and secrete mucin (Fig. 2), and thereby protect the mucosa from invasion of the pathogenic bacteria [41]. However, “depletion of goblet cells” is a common pathologic finding in IBD [42]. Loss of proper mucin secretion may directly lead to chronic inflammation, as it has been shown that MUC2 deficient mice spontaneously develop chronic colitis [43]. Besides mucin production, goblet cells secretes cytokines such as IL-7 [44] and thus may exert immunoregulatory functions, and can also deliver luminal antigens to the underlying DCs [45]. Recent studies have shown that a newly identified Cl− channel, bestrophin2 (BEST2), is expressed specifically by colonic goblet cells [46, 47]. Mice lacking BEST2 develops spontaneous colitis, and are more susceptible to DSS challenge, suggesting that this channel may play a distinct role to maintain goblet cell-dependent barrier functions [46]. We have further identified that BEST2 expression is down-regulated in the inflamed colonic mucosa of UC patients [47]. Accordingly, BEST2 gene is located at the formerly identified disease susceptibility locus [48]. As BEST2 may also function as a HCO3 − transporter [49], it may have an important role in normal mucus formation, which requires finely regulated HCO3 − secretion [50]. Also, as BEST2 expression is clearly absent in the small intestine, it would be interesting to see whether such a colon-specific gene function may determine the colon-restricted distribution of ulcerative colitis (UC).
Another lineage that has been featured in recent studies of IBD pathogenesis is Paneth cells [51]. Paneth cells form intracellular vesicles (Fig. 3), and secrete antimicrobial peptides such as defensins, PLA2G2A, or RegIIIγ [52, 53]. At the same time, it resides just next to the ISCs [54], where it sends Wnt, EGF, or Notch signal to the ISCs and constitutes the stem cells niche [55].

Mucosal barrier function of intestinal goblet cells. a Goblet cells are the dominant cell population in the colon. Alcian Blue staining of a human colon tissue shows mucin-producing goblet cells (blue). b Goblet cells produce MUC2 or TFF3 and maintain the mucosal layer to sequester gut microbes from the surface of the epithelial layer [39]. Expression of specific ion channels such as Bestrophin2 may support the proper function of the mucus layer [44, 45]. Goblet cells can also interact with the lamina propria immune cells through antigen delivery or cytokine production [42, 43]. c Mucus layer covers the mice colonic surface and constitutes the goblet cell-mediated barrier function. The mucus layer can be visualized in a tissue fixed by Carnoy’s solution, either by Alcian Blue staining, MUC2 immunostaining (green), or fluorescence-labeled lectins such as UEA1 or WGA (red). Note that the mucus layer has the inner and the outer layer

Specific location and function of Paneth cells. a Paneth cells reside at the bottom of the small intestinal crypt. Immunostaining of a human small intestinal tissue shows PLA2G2A-positive Paneth cells (red) at the bottom of the crypt. Msi-1-positive crypt base columnar cells (stem cells, green) are interspersed between those Paneth cells [52]. b Dual task of Paneth cells and its role in IBD pathogenesis. Paneth cells produce antimicrobial peptides such as α-defensins, PLA2G2A, or REGIIIγ and play an important part in mucosal barrier function. At the same time, these cells produce factors such as Wnt or EGF and constitute the stem cell niche [53]. Several functional defects or dysregulation of the intracellular pathway has been assigned to the pathogenesis of CD [54–60]
The functional relevance of several IBD susceptibility genes has been identified in Paneth cells. NOD2 encodes an intracellular receptor for bacterial LPS, and its risk allele for IBD is associated with less α-defensin production by Paneth cells [56]. ATG16L1 encodes a key molecule for autophagy. Cadwell et al. has shown that the risk allele of ATG16L1 for Crohn’s disease (CD) predisposes Paneth cells to lose their ability to form normal intracellular granules [57, 58]. This risk allele may induce a low level of autophagy in Paneth cells by caspase-3-dependent protein processing [59]. Also, Blumberg et al. has shown that XBP1, a transcription factor that is induced upon ER stress, is another susceptibility gene that leads to Paneth cell dysfunction [60]. By combining functional deletion of both ATG16L1 and XBP1 in mice models, they identified that ileal CD may arise solely from Paneth cell dysfunction [61]. Another study has shown that in Crohn’s disease, a Paneth cell-specific cell death is promoted by a mechanism called “Necroptosis” [62]. Our recent studies have shown that RIPK, a key molecule of necroptosis, can regulate autophagy through the regulation of p62-LC3 complex formation [63]. Also, the ubiquitin-editing enzyme A20 appears to promote autophagy through its complex formation with MTOR, and regulate the survival in CD4 T-cells [64]. Further studies focusing on the molecular interactions linking autophagy, necroptosis, and ER stress may provide new insights to the pathophysiology of Paneth cell-driven ileal CD. However, it would be also interesting to see if other epithelial cell linage counterparts may play such a role in other types of IBD.
Mechanism of epithelial repair in IBD
Under the active phase of IBD, it is a common finding that multiple ulcers appear in the gastrointestinal tract as a result of inflammation-induced tissue damage. We know that an intrinsic repair system can usually regenerate and heal those ulcers, once the inflammation has been controlled. However, we also know that some ulcers are certainly refractory, and are difficult to cure even under the control of overt inflammation. In IBD, tissue regeneration must be carried out within the inflamed local environment, and thus can be modified by various inflammatory signaling.
The epithelial repair response is a sequential process that can be divided into at least two phases; the acute phase for a process called “restitution”, and a later phase for cell proliferation and crypt division [65, 66]. Restitution is an acute phase response that does not require cell proliferation, but requires the dynamic re-distribution of the existing cells to cover the open area of the ulcer and restore the integrity of the epithelial layer. TGF-β is recognized as the key growth factor that governs this process [67], while factors such as trefoil peptides seem to regulate this process through other pathways [68].
At a later phase, cell proliferation is promoted by various growth factors such as EGF, FGF, KGF, or HGF, which are presumably produced by the local mucosal environment [69]. Also, several cytokines that are produced by the pro-inflammatory lymphocytes, such as IL-1β or IL-6, may modulate this phase of response. Of note, IL-22 is a unique cytokine that is produced by Th17 cells or NK cells, but targeted exclusively to IECs in the intestinal mucosa [70]. It has been shown that the IL-22-STAT3 pathway promotes epithelial regeneration through cell proliferation and increased secretion of factors such as RegIIIβ, RegIIIγ, or S100A9 [71–73]. Mizoguchi et al. has shown that local production of IL-22 may promote tissue repair in a mice colitis model [74]. However, our studies have shown that activation of the Notch pathway is also indispensable during this phase of tissue repair. Both the expression of Notch ligands and activated form of Notch1 (NICD) is increased in the IECs of the inflamed colonic mucosa [27, 75]. Blocking the global Notch activation induced severe loss of the proliferative response in the IECs residing at the colitic mucosa, which resulted in significant exacerbation to the DSS colitis [75]. Further studies have shown that at the downstream of Notch, the transcription factor Hes1 may interact and enhance IL-22-STAT3 signaling in IECs, and thereby promote the anti-microbial response of those cells [76]. Thus, the intracellular signaling activated by the pro-inflammatory cytokines, growth factors, or by the intrinsic proliferative signals may interact and co-operate to efficiently conduct the repair process. Accordingly, a recent study has shown that gp130, a co-receptor of IL-6, may activate both YAP and Notch and contribute to the tissue repair of the colitic mucosa [77].
At the later phase of the tissue repair, re-building of the crypt structure may appear as a result of the effective restore in number of IECs. This process may take place in a deep relationship with the underlying mesenchymal tissues. Miyoshi et al. showed that Wnt5a is secreted by the mesenchymal cells of the injured colonic mucosa, and plays a critical role in restoring the crypt structure [78]. In their model, Wnt5a seems to suppress the local cell proliferation through activation of the TGF-β signaling. However, whether the same mechanism also works in the inflamed mucosa of IBD patients remains to be elucidated.
Colitis-associated cancer represents another aspect of IEC-driven pathogenic manifestation of IBD
Another relatively rare but life-threatening phenotype arising from IECs is colitis-associated cancer (CAC). UC patients are at risk of developing CAC when the disease duration reaches up to 10 years or more [79]. Also, extra-intestinal manifestations such as primary sclerosing cirrhosis (PSC) add to the risk of developing CAC [80]. As the development of CAC is frequently fatal in IBD patients, challenges in endoscopic survey to find the earliest primary lesions are underway [81, 82].
It is well known that sporadic colon cancers develop by sequential mutation of cancer-driver genes, such as APC, KRAS, and p53 [83]. The process is called the “adenoma-carcinoma sequence”, which suggests that accumulation of mutations in cancer-driver genes promotes a more malignant phenotype, starting from adenoma and finally to carcinoma [84]. However, CAC has been shown to develop in a completely different pathway, which usually lacks constitutive activations of the APC-β-catenin-TCF axis, but frequently acquires mutation of p53 at the early stage of development [85]. Factors such as oxidative stress, and chronic exposure to pro-inflammatory cytokines are thought to be key players in the initiation of CAC. ROS or NOS production may increase in the inflammatory mucosal environment as part of the protective response, which frequently gives DNA damages to cells [86]. Cytokines such as TNF-α or IL-6 have been deeply implicated in the development of CAC. TNF-α is a pro-inflammatory cytokine that activates the NF-κB pathway in IECs through its specific receptor [87]. IEC-specific deletion of IKKβ, a key molecule in the NF-κB pathway, reduced the incidence of colitis-induced tumorigenesis, suggesting that IEC-intrinsic NF-κB activation is a major regulator of CAC [88]. Recent studies have suggested that TNF-α may also induce constitutive expression of the transcription factor ATOH1, and direct tumor cells to acquire highly proliferative, and invasive character [89, 90]. Also, loss of the gp130-STAT3 axis in IECs reduces the growth and multiplicity of colitis-induced tumors [91]. Thus, both TNF-α-NF-κB pathway and IL-6-STAT3 pathway seem to play a key role in CAC initiation and promotion. Therefore, both pathways have been considered as the potential target of CAC prevention or treatment [92–95].
What kind of IEC has the potential to be the origin of CAC? Studies have shown that the most relevant origin of sporadic cancer is the ISC [96]. However, recruitment of a certain population of differentiated cells into the stem cell pool is observed in several disease models, where the intrinsic ISCs are severely impaired or lost due to inflammation-induced tissue damage [97, 98]. Accordingly, activation of NF-κB signaling may de-differentiate villus non-stem cells and guide those cells to initiate “Top-down” type of tumorigenesis [99]. In addition, tuft cells seem to have the potential to initiate tumors exclusively after exposure to the inflammatory environment [98]. Thus, the intestinal inflammation may recruit certain population of IECs that are otherwise insusceptible to tumor initiation, to acquire tumorigenic potency, and subsequently promote formation of CACs arising from a distinct cell origin compared to sporadic cancers.
IECs as target cells for the treatment of IBD
The importance of repairing inflammation-induced ulcers and thereby restoring the normal epithelial architecture (as well as its integrity) has lead to the concept termed as “mucosal healing”, which has become the standard treatment goal of IBD [3, 100, 101]. Many clinical studies have shown that achievement of mucosal healing in UC or CD patients predicts both long-term remission and low risk for surgery [102, 103]. Conversely, failure to achieve mucosal healing may predict a poor prognosis [104].
So how can we achieve mucosal healing? Studies have shown that several current treatments may have protective or regenerative effect on the damaged epithelium, and thus promote mucosal healing. For example, mesalazine has been shown to promote mucosal healing [105–108]. Also anti-TNF-α therapy seems to have a favorable effect on mucosal repair [109–112]. However, whether those biologic agents have any direct effect on IECs remains uncertain.
A more direct approach to facilitate the intrinsic epithelial repair system may be adding the growth factors to promote proliferation of the stem-progenitor population [113]. Accordingly, several growth factors have been challenged for its use in treatment of IBD patients. EGF has been applied to mild-to-moderate UC patients and suggested to have some potential to promote mucosal repair [114]. Daily subcutaneous injection of Teduglutide, an analogue of GLP-2, has been tried in moderate-to-severe CD patients, which showed potential effect upon mucosal healing [115]. Also, recombinant HGF has been shown to facilitate mucosal repair in a rat colitis model [116]. Human recombinant R-Spondin-1 is another candidate that has been proven to promote proliferation of crypt cells [117].
Although the deep involvement of specific cell lineages such as Paneth cells or goblet cells has been clearly proved in the pathogenesis of IBD, lineage-specific treatments are poorly established. A recent study has shown that an EP4 agonist, an analogue that may partially mimic signals of prostaglandins, may have regenerative effect on IBD patients presumably through its effect on goblet cells [118].
Regenerative medicine as the emerging therapeutic approach to achieve mucosal healing
Besides those challenges to promote tissue repair by enhancing the intrinsic repair response, technical advances have brought about the idea of in vitro tissue engineering and its application to regenerate the damaged gastrointestinal mucosa of IBD patients.
One of the most outstanding advances in this area is the establishment of the long-term, in vitro culture system for ISCs. An initial report by Ootani et al. has shown that co-culture with mesenchymal cells in a 2D matrix enables long-term maintenance of ISCs [119]. However, Sato et al. has further developed and sophisticated the culture system, and showed that 3D culture is effective in maintaining the continuous and robust expansion of ISCs in vitro, without the support of mesenchymal cells [120]. Instead of the mesenchymal cells, the IECs are embedded in a extracellular matrix consisted of collagens and laminins, and cultured in a media supplemented with growth factors such as Wnt3a, R-spondin-1, EGF, or Noggin. Currently, slightly different culture protocols are established and used by various research groups. However, the culture condition established by Yui et al. has a quite unique feature as it uses completely defined media contents and a purified type I collagen for the extracellular matrix [121]. In those in vitro culture methods, ISCs form a 3D structure called as “organids” or “enteroids”. The organoid is composed not only by stem cells but also by proliferative cells and differentiated cells, and thus is also described as “mini-gut” [122].
It has recently been shown that those in vitro cultured cells have the capacity to reconstitute the damaged epithelial layer in vivo by trans-anal transplantation [121]. When those organoids were transplanted into a DSS-colitis model, donor cells clearly found their place on the damaged surface of the colitis-induced ulcer, and started to re-constitute the crypt structure. The integrated donor stem cells showed sustained function as resident stem cells for over 6 months. Importantly, clinical index of DSS colitis was significantly improved by the transplantation. Thus, these results clearly show the proof-of-principle that ISC transplantation may provide clinical benefit for IBD patients. Further studies have shown that organoids of fetal intestinal or small intestinal origin shares their capacity to reconstitute the damaged colonic mucosa [123, 124].
Also, other groups have shown that pluripotent cells may be an alternative choice of engineering ISCs in vitro. Spence et al. have shown that a sequential differentiation program can guide both mice ES cells and iPS cells to endoderm progenitors, and subsequently to IECs [125]. Such an in vivo induction of pluripotent cells towards intestinal tissues is also possible in human ES cells or iPS cells, and those engineered intestinal tissues are validated to have a satisfying level of intestine-specific functions [126]. A comprehensive search for transcription factors that are required for endodermal differentiation has revealed that so-called “iHep cells” are able to form intestinal organoids, and can be transplanted onto the damaged colonic epithelium [127]. These findings open up the way to use pluripotent cell-derived intestinal organoids as a resource to repair damaged intestinal tissues [128]. As the first therapeutic use of iPS-derived cells was launched last year in Japan for the treatment of macular degeneration [129], application of iPS-derived cells in other organs, such as in the intestine, is highly expected. However, risks and benefits exist in both somatic stem cell based- and pluripotent stem cell-based transplantation therapy [130, 131].
What kind of problem should we need to solve to establish ISC-based transplantation therapy in IBD? First, as no one has experienced this kind of therapy for gastrointestinal diseases, several issues regarding the safety of using ex vivo cultured cells must be validated. It is not clear whether an in vitro culture of human ISCs may predispose these cells to develop tumors. Such a risk may be an important issue especially for transplantations using ES or iPS cell-derived cells [132]. In addition, we currently do not know much about what kind of pathogen may exist in the donor intestinal mucosa. Thus upon somatic stem cell transplantation, a comprehensive survey of pathogens for both the donor mucosa and the cultured cells may be required. Secondly, we need to develop an appropriate way to deliver the cultured cells to the target region, possibly through gastrointestinal endoscopic devices. The method must be optimized to deliver 3D-cultured cells exclusively to the target region, and also must facilitate those cells to remain at the target site for a certain time period that is enough for them to fix to the underlying recipient mucosa. Finally, we need to validate the beneficial clinical effect of ISC transplantation through a designed prospective study, and optimize its use in combination with currently available therapies or with other transplantation therapies [133].
Future directions
As an increasing number of studies now feature the IECs as a major player in development and persistence of IBD, future studies may further uncover unexpected mechanisms by which IECs regulate the pathophysiology of IBD. Such a deep understanding of IECs-dependent pathogenesis of IBD may support the development of a new category of IBD drugs that are targeted to epithelial cell barrier functions, or to their innate immune functions.
On the other hand, an advance in therapy may be expected by the development of ISC-based transplantation. Such a therapy may provide a beneficial effect not only for the short-term maintenance of remission, but also for the long-term remission, and presumably have the potential to dramatically reduce the risk of developing CACs by “refreshing” the patient mucosa through the replacement of the resident “exhausted” epithelial cells.
References
van Dullemen HM, van Deventer SJ, Hommes DW, et al. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology. 1995;109:129–35.
Ford AC, Sandborn WJ, Khan KJ, et al. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. 2011;106:644–59.
Neurath MF, Travis SPL. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. 2012;61:1619–35.
Rioux JD, Daly MJ, Silverberg MS, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–8.
Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604.
Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25.
Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11.
Cadwell K. Crohn’s disease susceptibility gene interactions, a NOD to the newcomer ATG16L1. Gastroenterology. 2010;139:1448–50.
Helander HF, Fändriks L. Surface area of the digestive tract ––revisited. Scand J Gastroenterol. 2014;49:681–9.
van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60.
Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer. 2014;14:468–80.
Snippert HJ, van der Flier LG, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–44.
oz JMN, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent +4 cell markers. EMBO J. 2012;00:1–13.
Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346:1248012.
Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7.
Glinka A, Dolde C, Kirsch N, et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 2011;12:1055–61.
Koo B-K, Spit M, Jordens I, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–9.
de Lau W, Peng WC, Gros P, et al. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305–16.
Schuijers J, Junker JP, Mokry M, et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell. 2015;16:158–70.
van der Flier LG, van Gijn ME, Hatzis P, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–12.
Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology. 2011;141:448–59.
Sancho R, Cremona CA, Behrens A. Stem cell and progenitor fate in the mammalian intestine: notch and lateral inhibition in homeostasis and disease. EMBO Rep. 2015;16:571–81.
Noah TK, Shroyer NF. Notch in the intestine: regulation of homeostasis and pathogenesis. Annu. Rev. Physiol. 2013;75:263–88.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Riccio O, van Gijn ME, Bezdek AC, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9:377–83.
Pellegrinet L, Rodilla V, Liu Z, et al. Dll1- and Dll4-Mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011;140:1230–7.
Shimizu H, Okamoto R, Ito G, et al. Distinct expression patterns of Notch ligands, Dll1 and Dll4, in normal and inflamed mice intestine. PeerJ. 2014;2:e370.
van Es JH, de Geest N, van de Born M, et al. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat Commun. 2010;1:18.
Jensen J, Pedersen EE, Galante P, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;1:36–44.
VanDussen KL, Carulli AJ, Keeley TM, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–97.
Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621.
Goldsmith JR, Sartor RB. The role of diet on intestinal microbiota metabolism: downstream impacts on host immune function and health, and therapeutic implications. J Gastroenterol. 2014;49:785–98.
Sheehan D, Moran C, Shanahan F. The microbiota in inflammatory bowel disease. J Gastroenterol. 2015;50:495–507.
Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306.
Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17.
Rudolph U, Finegold MJ, Rich SJ, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;10:143–50.
Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–76.
Suzuki M, Nagaishi T, Yamazaki M, et al. Myosin light chain kinase expression induced via tumor necrosis factor receptor 2 signaling in the epithelial cells regulates the development of colitis-associated carcinogenesis. PLoS One. 2014;9:e88369.
Su L, Shen L, Clayburgh DR, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–63.
Pastorelli L, De Salvo C, Mercado JR, et al. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280.
Birchenough GMH, Johansson MEV, Gustafsson, et al. New developments in goblet cell mucus secretion and function. Mucosal Immunol 2015;8:712–19.
Surawicz CM, Haggitt RC, Husseman M, et al. Mucosal biopsy diagnosis of colitis: acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology. 1994;107:755–63.
Vander sluis M, De Koning BAE, De Bruijn ACJM, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–29.
Oshima S, Nakamura T, Namiki S, et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol Cell Biol. 2004;24:6298–310.
McDole JR, Wheeler LW, McDonald KG, et al. Goblet cells deliver luminal antigen to CD103 + dendritic cells in the small intestine. Nature. 2012;483:345–9.
Yu K, Lujan R, Marmorstein A, et al. Bestrophin-2 mediates bicarbonate transport by goblet cells in mouse colon. J. Clin. Invest. 2010;120:1722–35.
Ito G, Okamoto R, Murano T, et al. Lineage-specific expression of bestrophin-2 and bestrophin-4 in human intestinal epithelial cells. PLoS ONE. 2013;8:e79693.
Willliams CN, Kocher K, Lander ES, et al. Using a genome-wide scan and meta-analysis to identify a novel IBD locus and confirm previously identified IBD loci. Inflamm Bowel Dis. 2002;8:375–81.
Qu Z, Hartzell HC. Bestrophin Cl− channels are highly permeable to HCO3. AJP: cell. Physiology. 2008;294:C1371–7.
Yang N, Garcia MAS, Quinton PM. Normal mucus formation requires cAMP-dependent HCO3- secretion and Ca2+-mediated mucin exocytosis. J Physiol. 2013;591:4581–93.
Sandow MJ, Whitehead R. Paneth cell. Gut. 1979;20:420–31.
Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75:289–311.
Shipra V, Yamamoto M, Severson KM, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–8.
Murayama M, Okamoto R, Tsuchiya K, et al. Musashi-1 suppresses expression of Paneth cell-specific genes in human intestinal epithelial cells. J Gastroenterol. 2009;44:173–82.
Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2012;469:415–8.
Wehkamp J, Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–64.
Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63.
Cadwell K, Patel KK, Komatsu M, et al. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009;5:250–2.
Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014;506:456–62.
Kaser A, Blumberg RS. Cell biology: stressful genetics in Crohn’s disease. Nature. 2014;506:441–2.
Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–6.
Günther C, Martini E, Wittkopf N, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9.
Matsuzawa Y, Oshima S, Nibe Y, et al. RIPK3 regulates p62-LC3 complex formation via the caspase-8-dependent cleavage of p62. Biochem. Biophys. Res. Commun. 2015;456:298–304.
Matsuzawa Y, Oshima S, Takahara M, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 2015. doi:10.1080/15548627.2015.1055439.
Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77.
Okamoto R, Watanabe M. Cellular and molecular mechanisms of the epithelial repair in IBD. Dig Dis Sci. 2005;50(Suppl 1):S34–8.
Dignass AU, Podolsky DK. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor beta. Gastroenterology. 1993;105:1323–32.
Dignass A, Lynch-Devaney K, Kindon H, et al. Trefoil peptides promote epithelial migration through a transforming growth factor beta-independent pathway. J. Clin. Invest. 1994;94:376–83.
Okamoto R, Watanabe M. Molecular and clinical basis for the regeneration of human gastrointestinal epithelia. J Gastroenterol. 2004;39:1–6.
Wolk K, Witte E, Witte K, et al. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31.
Sekikawa A, Fukui H, Suzuki K, et al. Involvement of the IL-22/REG Ia axis in ulcerative colitis. Lab Invest. 2010;90:496–505.
Mizoguchi A. Healing of intestinal inflammation by IL-22. Inflamm Bowel Dis. 2012;18:1777–84.
Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–72.
Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 2008;118:534–44.
Okamoto R, Tsuchiya K, Nemoto Y, et al. Requirement of Notch activation during regeneration of the intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2009;296:G23–35.
Murano T, Okamoto R, Ito G, et al. Hes1 promotes the IL-22-mediated antimicrobial response by enhancing STAT3-dependent transcription in human intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2014;443:840–6.
Taniguchi K, Wu L-W, Grivennikov SI, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519:57–62.
Miyoshi H, Ajima R, Luo CT, et al. Wnt5a potentiates TGF––signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–13.
Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2012;35:229–44.
Loftus EV. Epidemiology and risk factors for colorectal dysplasia and cancer in ulcerative colitis. Gastroenterol Clin North Am. 2006;35:517–31.
Matsumoto T, Iwao Y, Igarashi M, et al. Endoscopic and chromoendoscopic atlas featuring dysplastic lesions in surveillance colonoscopy for patients with long-standing ulcerative colitis. Inflamm Bowel Dis. 2008;14:259–64.
Pellisé M. Overcoming challenges in IBD management: management of colonic dysplastic lesions. Dig Dis. 2013;31:244–7.
Walther A, Johnstone E, Swanton C, et al. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–99.
Cho KR, Vogelstein B. Genetic alterations in the adenoma–carcinoma sequence. Cancer. 1992;70:1727–31.
Foersch S, Neurath MF. Colitis-associated neoplasia: molecular basis and clinical translation. Cell Mol Life Sci. 2014;71:3523–35.
Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85.
Onizawa M, Nagaishi T, Kanai T, et al. Signaling pathway via TNF-alpha/NF-kappaB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G850–9.
Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96.
Kano Y, Tsuchiya K, Zheng X, et al. The acquisition of malignant potential in colon cancer is regulated by the stabilization of Atonal homolog 1 protein. Biochem. Biophys. Res. Commun. 2013;432:175–81.
Fukushima K, Tsuchiya K, Kano Y, et al. Atonal homolog 1 protein stabilized by tumor necrosis factor α induces high malignant potential in colon cancer cell line. Cancer Sci. 2015. doi:10.1111/cas.12703.
Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91.
Waldner MJ, Foersch S, Neurath MF. Interleukin-6-A key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012;8:1248–53.
Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–9.
Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59.
Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–9.
Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11.
van Es JH, Sato T, van de Wetering M, et al. Dll1 + secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–104.
Westphalen CB, Asfaha S, Hayakawa Y, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 2014;124:1283–95.
Schwitalla S, Fingerle AA, Cammareri P, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38.
Rutgeerts P, Vermeire S, Van Assche G. Mucosal healing in inflammatory bowel disease: impossible ideal or therapeutic target? Gut. 2007;56:453–5.
Importance of mucosal healing in ulcerative colitis. Inflamm Bowel Dis. 2010;16:338–46.
Peyrin-Biroulet L, Ferrante M, Magro F, et al. Results from the 2nd Scientific Workshop of the ECCO. I: Impact of mucosal healing on the course of inflammatory bowel disease. J Crohns Colitis. 2011;5:477–83.
Frøslie KF, Jahnsen J, Moum BA, et al. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology. 2007;133:412–22.
Ardizzone S, Cassinotti A, Duca P, et al. Mucosal healing predicts late outcomes after the first course of corticosteroids for newly diagnosed ulcerative colitis. Clin. Gastroenterol. Hepatol. 2011;9:483.
Lichtenstein GR, Ramsey D, Rubin DT. Randomised clinical trial: delayed-release oral mesalazine 4.8 g/day vs. 2.4 g/day in endoscopic mucosal healing––ASCEND I and II combined analysis. Aliment Pharmacol Ther. 2011;33:672–8.
Bokemeyer B, Hommes D, Gill I, et al. Mesalazine in left-sided ulcerative colitis: efficacy analyses from the PODIUM trial on maintenance of remission and mucosal healing. J Crohns Colitis. 2012;6:476–82.
Baumgart DC, Vierziger K, Sturm A, et al. Mesalamine promotes intestinal epithelial wound healing in vitro through a TGF-beta-independent mechanism. Scand J Gastroenterol. 2005;40:958–64.
Probert CSJ, Dignass AU, Lindgren S, et al. Combined oral and rectal mesalazine for the treatment of mild-to-moderately active ulcerative colitis: rapid symptom resolution and improvements in quality of life. J Crohns Colitis 2014;8:200–7.
D’haens G, Van Deventer S, Van Hogezand R, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn’s disease: A European multicenter trial. Gastroenterology. 1999;116:1029–34.
Colombel JF, Rutgeerts P, Reinisch W, et al. Early mucosal healing with infliximab is associated with improved long-term clinical outcomes in ulcerative colitis. Gastroenterology. 2011;141:1194–201.
Rutgeerts P, Van Assche G, Sandborn WJ, et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: data from the EXTEND trial. Gastroenterology. 2012;142:1102.
Kierkus J, Dadalski M, Szymanska E, et al. The impact of infliximab induction therapy on mucosal healing and clinical remission in Polish pediatric patients with moderate-to-severe Crohn’s disease. Eur J Gastroenterol Hepatol. 2012;24:495–500.
Krishnan K, Arnone B, Buchman A. Intestinal growth factors: potential use in the treatment of inflammatory bowel disease and their role in mucosal healing. Inflamm Bowel Dis. 2011;17:410–22.
Sinha A, Nightingale J, West KP, et al. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med. 2003;349:350–7.
Buchman AL, Katz S, Fang JC, et al. Teduglutide Study Group. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm Bowel Dis. 2010;16:962–73.
Numata M, Ido A, Moriuchi A, et al. Hepatocyte growth factor facilitates the repair of large colonic ulcers in 2,4,6-trinitrobenzene sulfonic acid-induced colitis in rats. Inflamm Bowel Dis. 2005;11:551–8.
Kim K-A, Kakitani M, Zhao J, et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science. American Associ Adv Sci. 2005;309:1256–9.
Nakase H, Fujiyama Y, Oshitani N, et al. Effect of EP4 agonist (ONO-4819CD) for patients with mild to moderate ulcerative colitis refractory to 5-aminosalicylates: a randomized phase II, placebo-controlled trial. Inflamm Bowel Dis. 2010;16:731–3.
Ootani A, Li X, Sangiorgi E, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15:701–6.
Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5.
Yui S, Nakamura T, Sato T, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat Med. 2012;18:618–23.
Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–4.
Fordam RP, Yui S, Hannan N, et al. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell. 2013;13:734–44.
Fukuda M, Mizutani T, Mochizuki W, et al. Small intestinal stem cell identity is maintained with functional Paneth cells in heterotopically grafted epithelium onto the colon. Genes Dev. 2014;28:1752–7.
Spence JR, Mayhew CN, Rankin SA, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–9.
Watson CL, Mahe MM, Múnera J, et al. An in vivo model of human small intestine using pluripotent stem cells. Nat Med. 2014;20:1310–4.
Morris S, Cahan P, Li H, et al. Dissecting engineered cell types and enhancing cell fate conversion via Cell Net. Cell. 2014;158:889–902.
Watanabe M. Adult tissue stem cell therapy for gastrointestinal diseases. J. Gastroenterol. Hepatol. 2014. doi:10.1111/jgh.12555.
Aaltonen LA, Reardon S. Japan stem-cell trial stirs envy. Nature. 2014;513:287–8.
Herberts CA, Kwa MSG, Hermsen HPH. Risk factors in the development of stem cell therapy. J Transl Med. 2011;9:29.
Heslop JA, Hammond TG, Santeramo I, et al. Concise review: workshop review: understanding and assessing the risks of stem cell-based therapies. Stem Cells Transl Med. 2015;4:389–400.
Lee AS, Tang C, Rao MS, et al. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19:998–1004.
Nagaishi K, Arimura Y, Fujimiya M. Stem cell therapy for inflammatory bowel disease. J Gastroenterol. 2015;50:280–6.
Acknowledgments
The authors would like to thank Ms. Junko Morio-Akiyama and Dr. Shu Kato for providing excellent histologic images.
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Okamoto, R., Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J Gastroenterol 51, 11–21 (2016). https://doi.org/10.1007/s00535-015-1098-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-015-1098-4