





Abstract
Molecular and cultivation approaches were employed to study the phylogenetic richness and temporal dynamics of Streptococcus and Veillonella populations in the small intestine. Microbial profiling of human small intestinal samples collected from four ileostomy subjects at four time points displayed abundant populations of Streptococcus spp. most affiliated with S. salivarius, S. thermophilus, and S. parasanguinis, as well as Veillonella spp. affiliated with V. atypica, V. parvula, V. dispar, and V. rogosae. Relative abundances varied per subject and time of sampling. Streptococcus and Veillonella isolates were cultured using selective media from ileostoma effluent samples collected at two time points from a single subject. The richness of the Streptococcus and Veillonella isolates was assessed at species and strain level by 16S rRNA gene sequencing and genetic fingerprinting, respectively. A total of 160 Streptococcus and 37 Veillonella isolates were obtained. Genetic fingerprinting differentiated seven Streptococcus lineages from ileostoma effluent, illustrating the strain richness within this ecosystem. The Veillonella isolates were represented by a single phylotype. Our study demonstrated that the small intestinal Streptococcus populations displayed considerable changes over time at the genetic lineage level because only representative strains of a single Streptococcus lineage could be cultivated from ileostoma effluent at both time points.
Introduction
The human body is populated with complex microbial communities, which vary in composition between body sites (Costello et al., 2009). The microbiota in the human gastrointestinal (GI) tract, for example, have adapted to the different conditions in the specific GI habitats [for a recent review see (Walter & Ley, 2011)] and are impressive not only because of its very high population density, but also because of its high phylogenetic diversity (Rajilić-Stojanović et al., 2007; van den Bogert et al., 2011a, b) and its extensive functional capabilities that complement the human genetic potential (Qin et al., 2010). The composition and dynamics of the bacterial community in the lower GI tract have been well described (Booijink et al., 2007; Rajilić-Stojanović et al., 2009; Tap et al., 2009; Turnbaugh et al., 2009), whereas the upper GI tract microbiota in healthy humans are less well characterized as a consequence of sampling difficulties (Booijink et al., 2007; Leser & Molbak, 2009; Cotter, 2011). Nevertheless, the different sections of the upper GI tract encompass distinct bacterial groups that interact with the host (Lawson & Coyle, 2010). Furthermore, the small intestine represents the first region where food components and the intestinal bacteria encounter each other, and it is also the region of the intestine that is predominantly involved in primary nutrient digestion and absorption (Booijink et al., 2007; Leser & Molbak, 2009). Therefore, the small intestinal microbiota are expected to be of great importance to the host by playing a prominent role in the primary carbohydrate metabolism (Zoetendal et al., 2012) and have an important influence on host physiology and health status [for a recent review see (Cotter, 2011)] by, for example, immune system modulation through luminal sampling and handling of bacteria (see Duerkop et al., 2009 for a review).
Recently, high-throughput 16S ribosomal RNA (rRNA) gene profiling was employed to characterize the human small intestinal microbiota in samples obtained from healthy individuals using an orally introduced catheter as well as samples collected from ileostomy subjects (Hartman et al., 2009; Booijink et al., 2010; Zoetendal et al., 2012). The latter group of individuals underwent surgical removal of their colon, and as a consequence, luminal content of their terminal ileum is excreted from an abdominal stoma and can repetitively be collected in a noninvasive manner. These studies revealed that the bacterial community in ileostoma effluent is also encountered in the small intestine of healthy subjects with Streptococcus and Veillonella spp. as predominant components. Interestingly, both bacterial populations are abundant not only in the small intestine but also in the microbiota of the stomach (Bik et al., 2006), esophagus (Pei et al., 2004), throat (Andersson et al., 2008), and oral cavity (Keijser et al., 2008).
The fact that members of Streptococcus and Veillonella are frequently co-occurring at these body sites may be partially attributable to their potential for metabolic interaction that has been shown to occur in the oral cavity (Egland et al., 2004) and postulated for the small intestine (Zoetendal et al., 2012). Streptococcus spp. are involved in the fermentation of sugars, yielding lactic acid as their predominant fermentation end product. In turn, Veillonella are renowned for their capacity to use lactic acid as a carbon and energy source (Ng & Hamilton, 1971).
Because no small intestinal Streptococcus and Veillonella isolates have to our knowledge been described, this study focuses on in-depth assessment of their phylogenetic richness and population dynamics as well as the streptococcal carbohydrate metabolic capacities through a combination of cultivation and molecular typing methodologies.
Materials and methods
Ethics statement
The study was approved by the University Hospital Maastricht Ethical Committee and was conducted in full accordance with the principles of the ‘Declaration of Helsinki’ (52nd WMA General Assembly, Edinburgh, Scotland, October 2000). Volunteers were informed about the study orally and in writing. Moreover, the volunteers signed a written informed consent before participation.
Profiling small intestinal populations
In total, 16 ileostoma effluent samples were collected from four ileostomy subjects (Table 0001) who were colectomized at least 5 years prior to testing, are clinically considered to be healthy, and have a normally functioning small intestine: patients did not report any complaints related to GI functioning for at least 3 years prior to testing and were not following any treatment for GI-related symptoms. The subjects donated four samples each, collected on two distinct time points of the day (morning and afternoon), on two separate days (at least 2 days apart). The subjects collected the ileostoma effluent in a clean, empty ileostoma bag, which was emptied in centrifuge bottles (Nalgene, Rochester, NY) containing 100 mL RNAlater® (Ambion, Austin, TX), immediately after the bulk of the effluent flowed into the bag. Samples were stored for 4–10 h at room temperature, after which the samples were frozen by transferring the tubes to dry ice. Frozen samples were transported to the laboratory, where they were kept at −80 °C until further analysis.
Characteristics of subjects included in this study
| Subject | Gender | Age |
| S1 | Male | 79 |
| S2 | Female | 65 |
| S3 | Male | 60 |
| S4 | Female | 60 |
| Subject | Gender | Age |
| S1 | Male | 79 |
| S2 | Female | 65 |
| S3 | Male | 60 |
| S4 | Female | 60 |
Characteristics of subjects included in this study
| Subject | Gender | Age |
| S1 | Male | 79 |
| S2 | Female | 65 |
| S3 | Male | 60 |
| S4 | Female | 60 |
| Subject | Gender | Age |
| S1 | Male | 79 |
| S2 | Female | 65 |
| S3 | Male | 60 |
| S4 | Female | 60 |
DNA was extracted using a method as described previously (Zoetendal et al., 2006), with minor modifications. In short, 1 mL ileostoma effluent suspension was mixed with 4 mL PBS followed by centrifugation at 4600 g at 4 °C for 10 min. The cell pellet was resuspended in 0.5 mL ice-cold TE buffer (Tris–HCl pH 7.6, EDTA pH 8.0), after which the mixture was transferred to a microfuge tube containing 0.18 g Macaloid suspension (Zoetendal et al., 2006), 0.1 mm zirconium beads, and 50 μL 10% SDS (Invitrogen, Carlsbad, CA). The solution was mixed with 500 μL acid phenol (Invitrogen), followed by three Fastprep (Bertin Technologies, Montigny le Bretonneux, France) treatments at 5.5 m s−1 for 45 s with cooling on ice for 90 s between treatments. The sample was centrifuged at 13 400 g at 4 °C for 15 min, after which the nucleic acids in the aqueous phase were purified by consecutive extraction with phenol/chloroform/isoamyl alcohol (25 : 24 : 1) and chloroform/isoamyl alcohol (24 : 1). Phases were separated by centrifugation (13 400 g at 4 °C, 5 min) using Phase Lock Gel tubes (5 Prime, Hamburg, Germany). Subsequently, 300 μL of the aqueous phase was treated with 3 μL RNAse A (10 mg mL−1; Qiagen GmbH, Hilden, Germany) and incubated at 37 °C for 15 min. Subsequent steps employed a modified version of the QIAamp DNA Stool Mini Kit protocol: 22.5 μL proteinase K (20 mg mL−1; Ambion) and 300 μL buffer AL were added to the sample followed by incubation at 70 °C for 10 min. After addition of 300 μL ethanol (VWR, Amsterdam, The Netherlands), the sample was transferred to a QIAamp column and centrifuged (13 000 g, 1 min). DNA pellets were washed with AW1 and AW2 buffers according to manufacturer's instructions. Finally, the DNA was eluted with 30 μL nuclease-free water (Promega, Leiden, The Netherlands).
For 16S rRNA gene–based microbial composition profiling, barcoded amplicons from the V1-V2 region of 16S rRNA genes were generated by PCR using the 27F-DegS primer (van den Bogert et al., 2011a) that was appended with the titanium sequencing adaptor A and a 8-nt sample-specific barcode (Hamady et al., 2008) at the 5′-end, and an equimolar mix of two reverse primers (338R I and II (Daims et al., 1999); Table 0002) that carried the titanium adaptor B at the 5′-end.
Adaptors and primers used in this study
| Primer | Primer sequence (5′-3′) | Reference |
| Adaptor A | CCATCTCATCCCTGCGTGTCTCCGACTCAG | Provided by GATC-Biotech |
| Adaptor B | CCTATCCCCTGTGTGCCTTGGCAGTCTCAG | |
| 27F-DegS | GTTYGATYMTGGCTCAG | van den Bogert et al. (2011a) |
| 338R-I | GCWGCCTCCCGTAGGAGT | Daims et al. (1999) |
| 338R-II | GCWGCCACCCGTAGGTGT | |
| 27F | GTTTGATCCTGGCTCAG | Lane (1991) |
| 1492R-rev | CGGCTACCTTGTTACGAC | |
| 357F | CTCCTACGGGAGGCAGCAG | |
| (GTG)5 | GTGGTGGTGGTGGTG |
| Primer | Primer sequence (5′-3′) | Reference |
| Adaptor A | CCATCTCATCCCTGCGTGTCTCCGACTCAG | Provided by GATC-Biotech |
| Adaptor B | CCTATCCCCTGTGTGCCTTGGCAGTCTCAG | |
| 27F-DegS | GTTYGATYMTGGCTCAG | van den Bogert et al. (2011a) |
| 338R-I | GCWGCCTCCCGTAGGAGT | Daims et al. (1999) |
| 338R-II | GCWGCCACCCGTAGGTGT | |
| 27F | GTTTGATCCTGGCTCAG | Lane (1991) |
| 1492R-rev | CGGCTACCTTGTTACGAC | |
| 357F | CTCCTACGGGAGGCAGCAG | |
| (GTG)5 | GTGGTGGTGGTGGTG |
Primer names may not correspond to original publication.
M = A or C; R = A or G; W = A or T; Y = C or T.
Adaptors and primers used in this study
| Primer | Primer sequence (5′-3′) | Reference |
| Adaptor A | CCATCTCATCCCTGCGTGTCTCCGACTCAG | Provided by GATC-Biotech |
| Adaptor B | CCTATCCCCTGTGTGCCTTGGCAGTCTCAG | |
| 27F-DegS | GTTYGATYMTGGCTCAG | van den Bogert et al. (2011a) |
| 338R-I | GCWGCCTCCCGTAGGAGT | Daims et al. (1999) |
| 338R-II | GCWGCCACCCGTAGGTGT | |
| 27F | GTTTGATCCTGGCTCAG | Lane (1991) |
| 1492R-rev | CGGCTACCTTGTTACGAC | |
| 357F | CTCCTACGGGAGGCAGCAG | |
| (GTG)5 | GTGGTGGTGGTGGTG |
| Primer | Primer sequence (5′-3′) | Reference |
| Adaptor A | CCATCTCATCCCTGCGTGTCTCCGACTCAG | Provided by GATC-Biotech |
| Adaptor B | CCTATCCCCTGTGTGCCTTGGCAGTCTCAG | |
| 27F-DegS | GTTYGATYMTGGCTCAG | van den Bogert et al. (2011a) |
| 338R-I | GCWGCCTCCCGTAGGAGT | Daims et al. (1999) |
| 338R-II | GCWGCCACCCGTAGGTGT | |
| 27F | GTTTGATCCTGGCTCAG | Lane (1991) |
| 1492R-rev | CGGCTACCTTGTTACGAC | |
| 357F | CTCCTACGGGAGGCAGCAG | |
| (GTG)5 | GTGGTGGTGGTGGTG |
Primer names may not correspond to original publication.
M = A or C; R = A or G; W = A or T; Y = C or T.
PCRs were performed using a thermocycler GS0001 (Gene Technologies, Braintree, UK) in a total volume of 100 μL containing 1× HF buffer (Finnzymes, Vantaa, Finland), 2 μL PCR Grade Nucleotide Mix (Roche, Diagnostics GmbH, Mannheim, Germany), 2 U of Phusion® Hot Start II High-Fidelity DNA polymerase, 500 nM of a forward and the reverse primer mix (Biolegio BV, Nijmegen, The Netherlands), and 0.2–0.4 ng μL−1 of template DNA. The amplification program consisted of an initial denaturation at 98 °C for 30 s; 30 cycles of denaturation at 98 °C for 10 s, annealing at 56 °C for 20 s, and elongation at 72 °C for 20 s; and a final extension at 72 °C for 10 min. The size of the PCR products (~375 bp) was confirmed by gel electrophoresis using 5 μL of the amplification reaction mixture on a 1% (w/v) agarose gel containing 1× SYBR® Safe (Invitrogen). PCR products were purified with the High Pure Cleanup Micro Kit (Roche) using 10 μL nuclease-free water for elution and quantified using a NanoDrop ND-1000 spectrophotometer (Nano-Drop Technologies, Wilmington, DE). Purified PCR products were mixed in approximately equimolar amounts and electrophoresed on an agarose gel, followed by excision and purification using the DNA gel extraction kit (Millipore, Billerica, MA). Purified amplicon pools were pyrosequenced using a Genome Sequencer FLX in combination with titanium chemistry (GATC-Biotech, Konstanz, Germany).
The pyrosequencing data analysis was carried out with a workflow employing the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (Caporaso et al., 2010) using settings as recommended in the QIIME 1.2 tutorial with the following exceptions: reads were filtered for chimeric sequences using Chimera Slayer (Haas et al., 2011), and OTU clustering was performed with an identity threshold of 97%, using parameters as recommended in the QIIME newsletter of December 17, 2010 (http://qiime.wordpress.com/2010/12/17/new-default-parameters-for-uclust-otu-pickers/). Additional data handling was carried out using in-house developed Python and Perl scripts. The Ribosomal Database Project (RDP) classifier version 2.2 (Wang et al., 2007) was used for taxonomic classifications up to the genus level. The most likely species was determined by comparing sequences against the RDP reference set using NCBI blast (Altschul et al., 1990), tentatively classifying an OTU as a specific when the blast score of the OTU–reference sequence pair was higher than that of the lowest-scoring reference sequence–reference sequence pair for that species in the RDP set. Results of the tentative assignments were evaluated by generating maximum-likelihood phylogenetic trees containing a representative sequence from each OTU and reference sequences from the RDP database (Cole et al., 2009). Multiple sequence alignments were carried out with Muscle (Edgar, 2004), the part of the alignment matching the amplicon was retrieved using JalView (Waterhouse et al., 2009), and maximum-likelihood phylogenetic trees were generated using Phyml (Guindon & Gascuel, 2003).
Sample collection for cultivation
An ileostoma effluent sample was obtained in the evening from subject 1 (Table 0001; t = 0) by transferring approximately 20 mL ileostoma effluent from the ileostoma bag to a 50 mL-tube containing 20 mL phosphate-buffered saline (PBS)–cysteine solution [8 g L−1 NaCl, 0.2 g L−1 KCl, 1.44 g L−1 Na2HPO4, 0.24 g L−1 KH2PO4 (Sigma, St. Louis, MO) and 1 g L−1 cysteine–HCl (Sigma), pH 6.8]. The PBS–cysteine solution was flushed with N2 to enhance survival of anaerobic bacteria such as Veillonella. Because large fluctuations on phylotype and function level were observed over a time spam of 1 year (Zoetendal et al., 2012), a second ileostoma effluent sample was collected in the morning from the same ileostomy subject (t = 1) 1 year after the first sampling to determine the population dynamics at the genetic lineage level.
All samples were placed in sealed plastic bags with an anoxic atmosphere, generated by an Anaerocult® A mini sachet (Merck, Darmstadt, Germany), and stored at home in the refrigerator. Samples were transported to the laboratory and processed within 24 h after collection.
Cultivation
Of all samples, serial dilutions were prepared in PBS–cysteine solution (101–105) and plated on Mitis Salivarius (MS) agar (Becton Dickinson, Breda, The Netherlands) supplemented with Tellurite solution 1% (Becton Dickinson) according to manufacturer's instructions, and Veillonella selective agar (VSA) (Rogosa, 1956; Rogosa et al., 1958) to facilitate selective isolation of Streptococcus and Veillonella, respectively. MS agar plates were incubated aerobically, whereas VSA plates were incubated in anaerobic jars with an anaerobic atmosphere generated by an Anaerocult® A sachet (Merck). All plates were incubated at 37 °C for 18–48 h. Emerging colonies were randomly picked and grown in liquid media at 37 °C for 18–48 h.
The MS isolates were grown in MS medium, based on the MS agar, but lacking trypan blue, crystal violet, and agar. The VSA isolates were cultivated under anoxic atmosphere, generated by the Anaerocult® A mini system in Veillonella medium described in the DSMZ catalogue (Medium 136), which contains lactate as its main carbon source for growth. Bacterial isolates were stored at −80 °C in these same media to which 15% glycerol was added.
16S rRNA gene sequencing and analysis
Near-full-length 16S rRNA gene fragments from the bacterial isolates were PCR amplified using a PCR protocol described previously (van den Bogert et al., 2011a) with the 27F and Uni-1492-rev primers (Table 0002) and either a single colony or 2.5 μL of bacterial suspension/glycerol stock as a template source. Amplicon size was verified by electrophoresis on a 1.0% (w/v) agarose gel. PCR products were purified and subsequently sequenced from the 27F, 357F, and Uni-1492-rev (Lane, 1991) priming sites (GATC-Biotech, Konstanz, Germany; Table 0002). The obtained sequence reads per amplicon were assembled using Clone Manager 9 Professional Edition (Scientific & Educational Software, Cary, NC), yielding near-full-length 16S rRNA gene sequences, which were taxonomically classified using a locally installed version of the RDP classifier version 2.2 (Wang et al., 2007) with a default confidence threshold of 80%.
The 16S rRNA gene sequences were aligned using the SINA Alignment Service (http://www.arb-silva.de/aligner/) (Pruesse et al., 2007) and subsequently imported into ARB (Ludwig et al., 2004) and merged with the SILVA reference database release 106. A neighbor-joining distance matrix employing no correction was calculated using ARB to group sequences into distinct phylotypes based on a threshold of 97% sequence identity.
The near-full-length 16S rRNA gene sequences were deposited in the GenBank database and are available under accession numbers JQ680047–JQ680145 and JQ680199–JQ680348.
Typing of bacterial isolates
Bacterial isolates were classified into genetic lineages using amplified fragment length polymorphism (AFLP) and Rep-PCR genetic fingerprinting. AFLP was performed as described previously (Kutahya et al., 2011). Based on analysis of replicates with the AFLP protocol, 90% of similarity was used as threshold for the separation of individual genetic lineages (data not shown).
Rep-PCR fingerprinting analyses were performed using a thermocycler GS0001 with an amplification program described by Matsheka et al. (2006). Each reaction was performed in a total volume of 25 μL composed of 1× PCR buffer (Promega), 1 μM of the (GTG)5 primer (Biolegio BV; Table 0002), 200 μM of each deoxyribonucleotide triphosphate (Roche, Diagnostics GmbH), 1.25 U GoTaq® DNA polymerase (Promega), and 2.5 μL of glycerol stock. Rep-PCR products were separated by electrophoresis on a 1.5% (w/v) agarose gel and stained by in gel 1× SYBR® Safe DNA stain. After standardized electrophoresis (75 min, 100 V, 1× TAE), banding patterns were visualized under UV light and digitally captured using the Gel Doc XR System (Bio-Rad) with Quantity One software version 4.6.6 build 102. Comparative analysis of the resulting fingerprints was performed using the BioNumerics suite (version 4.6.1; Applied Maths, St Martens Latem, Belgium). Similarities among profiles were calculated using the Pearson correlation coefficient, and cluster analyses were performed applying the unweighted-pair group method with arithmetic averages algorithm (UPGMA) with an optimization of 0.69%. Based on analysis of replicates with the Rep-PCR protocol, 84% of similarity was used as threshold for the separation of individual genetic lineages (data not shown). Rep-PCR-based groupings for which no near-full-length 16S rRNA gene sequences were obtained, or which were inconsistent with 16S rRNA gene classification results, were excluded from further analysis.
The validity of using glycerol stocks as template source for rep-PCRs was verified by comparing the resulting profiles with those obtained from Rep-PCRs using isolated genomic DNA of three randomly selected Streptococcus isolates, yielding identical profiles (data not shown).
Substrate conversion capacity of individual Streptococcus isolates was evaluated using API 50 CH strips in combination with API CHL medium (Biomerieux, Marcy l'Etoile, France). To this end, isolates were grown overnight in MS medium and washed twice with 0.9% NaCl prior to inoculation of the strips, incubation at 37 °C, and assessment of the reactions after 24 and 48 h according to manufacturer's instructions. To determine whether fermentation patterns were consistent for isolates belonging to the same genetic lineage, two isolates from each genetic lineage (if available) were tested and compared.
Growth of the isolates on different carbon sources was assessed using carbohydrate-free MS medium (MSBasal), supplemented with l-arabinose, D-(+)-glucose monohydrate, d-mannitol, N-acetylglucosamine, sucrose, d-trehalose, d-raffinose, soluble starch, or glycogen [Sigma] at a standard concentration of 1% (w/v). Bacteria suspended in 0.9% NaCl were diluted to an OD600 of 0.002 in the different MS-derived media and incubated at 37 °C for 18 h. Growth was assessed by OD600 determination.
MALDI-TOF MS analysis was performed with a Microflex mass spectrometer (Bruker Daltonics, Bremen, Germany) using FlexControl software (version 3.0). Spectra were recorded in the positive linear mode (laser frequency, 20 Hz; ion source 1, voltage at 20 kV; ion source 2, voltage at 18.4 kV; lens voltage, 9.1 kV; mass range, 2000–20 000 Da). Spectra were internally calibrated using Escherichia coli ribosomal proteins. The spectra were imported into the integrated Biotyper software (version 2.0) and analyzed by standard pattern matching with default settings. The spectrum of each isolate was compared with those in the database containing nine Veillonella and 61 Streptococcus spp. (see Supporting Information, Data S1). Identification was provided with an accompanying score (log score 0–3) of reliability. This score is based on (1) matching of the spectrum in general; (2) matching of the locus of the peaks; and (3) matching of the height of the peaks. Scores < 1.7 represent no reliable identification. A score ≥1.7 and < 2.0 is considered identification at the genus level, and scores ≥2.0, identification at the species level (Veloo et al., 2011).
Colonies of each isolate were directly spotted on the MALDI plate and were overlaid with 1 μL of matrix solution (α-cyano-4-hydroxy-cinnamic acid in 50% acetonitrile and 2.5% trifluoroacetic acid) and air-dried. Measurements were performed as described previously (van Veen et al., 2010). If an identification score was below a 1.7 cutoff, the isolate was again spotted on a MALDI plate and pretreated with 1 μL of 70% formic acid before being overlaid with the matrix solution. The highest of all the scores per isolate was considered the final result.
Results
Multiple Streptococcus and Veillonella spp. consistently co-occur in ileostoma effluent
To establish the Streptococcus and Veillonella spp. relative abundances in ileostoma effluent, we characterized the microbial communities collected from four ileostomy subjects at four different time points through 16S rRNA gene pyrosequencing. In total, 162 785 quality-filtered sequences with 10 174 (±standard deviation of 3855) sequences per sample were obtained. From all sequences, 39 812 and 1986 were assigned to the genera Streptococcus and Veillonella, respectively. Both genera were detected in all samples, with Streptococcus relative abundances ranging from 0.4 to 88.3% (Fig. 1a). Veillonella was generally present at lower relative abundance with variable abundances per sample ranging from < 0.1% to 10.1% (Fig. 0001a).
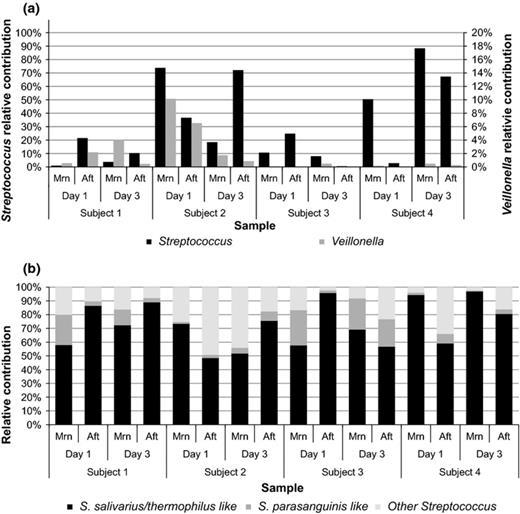
Relative contribution of Streptococcus (primary axis) and Veillonella (secondary axis; a) and Streptococcus spp. (b) as detected with pyrosequencing in morning (Mrn) and afternoon (Aft) ileostoma samples. Because discrimination of S. salivarius and S. thermophilus was not possible based on partial 16S rRNA gene pyrosequencing, relative abundances for phylotypes assigned to these species were combined to ‘S. salivarius/thermophilus like’.
Interestingly, out of the different Streptococcus phylotypes detected in the whole dataset, phylotypes most closely related to the species S. salivarius, S. thermophilus (S. salivarius species group), and S. parasanguinis (S. mitis species group) were of high relative abundance in all samples (Figs 0001b and S1a). The Veillonella population in ileostoma effluent was represented by phylotypes that most closely resembled the species V. dispar, V. parvula, V. rogosae, and V. atypica. Because discrimination of these species is not possible based on partial 16S rRNA gene sequences generated by pyrosequencing (Fig. S1b), relative abundances for all phylotypes were combined (Fig. 0001a). The clustering in Fig. S1b does show that we can specify that most of the OTUs detected in the small intestine are closely related to these four species and not to other Veillonella spp. (e.g. V. ratti and V. criceti).
These findings demonstrate that the same Streptococcus and Veillonella spp. are detected in ileostoma effluent samples collected at different time points as well as from different subjects, indicating that these species are typical small intestinal commensals. To further characterize the small intestinal Streptococcus and Veillonella population richness as well as its temporal dynamics at the strain level, we applied an integrated approach that combined cultivation and physiological characterization (substrate utilization assays) with molecular typing, including genetic fingerprinting and MALDI-TOF MS-typing of bacterial isolates of these bacterial genera from ileostoma effluent of one of the subjects.
Phylotype diversity of ileostoma effluent–derived bacterial isolates
A total of 272 bacterial isolates collected from two ileostoma effluent samples at two time points (t = 0 and, 1 year later, at t = 1; Table 0003) were classified on basis of a combination of 16S rRNA gene sequencing and genetic fingerprinting by AFLP and Rep-PCR. These isolates were classified as the genera Streptococcus (160), Enterococcus (66), Veillonella (37), Bacteroides (5), and Lactobacillus (4) (Table 0003). While Streptococcus was exclusively isolated using MS agar and Veillonella, Lactobacillus, and Bacteroides were only isolated from VSA, Enterococcus isolates were recovered from both media (Table 0003). In the remainder of the paper, we focus on analysis of the Streptococcus and Veillonella isolates as typical commensal inhabitants of the small intestine (Booijink et al., 2010; Zoetendal et al., 2012). Characteristics of the isolates are included in Tables 0003 and S2.
Characteristics of the isolates are included in Table S2. A graphic representation of the data included in this Table and Table S2 is added as Fig. S3.
All isolates were obtained from a 79-year-old male ileostomist (subject 1; Table 0001).
Isolated obtained from MS agar.
AFLP analysis unlike Rep-PCR genomic fingerprinting identified a single Streptococcus isolate as a separate genetic lineage.
Isolates obtained from VSA.
N.D. not determined because AFLP analysis and/or rep-PCR genomic fingerprinting did not reveal discriminative lineages.
Characteristics of the isolates are included in Table S2. A graphic representation of the data included in this Table and Table S2 is added as Fig. S3.
All isolates were obtained from a 79-year-old male ileostomist (subject 1; Table 0001).
Isolated obtained from MS agar.
AFLP analysis unlike Rep-PCR genomic fingerprinting identified a single Streptococcus isolate as a separate genetic lineage.
Isolates obtained from VSA.
N.D. not determined because AFLP analysis and/or rep-PCR genomic fingerprinting did not reveal discriminative lineages.
Based on the near-full-length 16S rRNA gene sequences, isolates identified as Streptococcus were divided in three phylotypes, and Veillonella was represented by a single phylotype at a sequence identity threshold of 97%. The 16S rRNA gene sequences within each Streptococcus phylotype showed > 99% sequence identity. The least abundant phylotype, consisting of three isolates, showed highest similarity to species in the SILVA database belonging to S. parasanguinis (> 99%; S. mitis species group), while the second phylotype consisting of 81 isolates showed highest similarity to S. equinus (> 98.5%) and S. lutetiensis (> 99.7%; S. bovis species group). The last phylotype represented by 76 isolates showed highest similarity to S. salivarius subsp. salivarius (> 98.7%) and S. vestibularis (> 99.3%; S. salivarius group; Fig. 0002). These identifications were confirmed by MALDI-TOF MS analysis of randomly picked isolates from each of the Streptococcus phylotypes (data not shown). Considering the high sequence similarity with multiple species for isolates from one phylotype, in the remainder of the paper, the phylotypes are indicated with the Streptococcus species group names as they are also used in the Bergey's Manual of Systematic Bacteriology (De Vos et al., 2009).
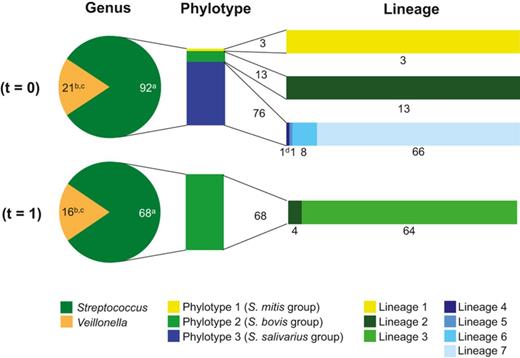
Relative contribution of Streptococcus and Veillonella isolates obtained from ileostoma effluent (t = 0 and 1) on genus, phylotype, and lineage level. The bar plots next to the pie charts represent the division of the Streptococcus isolates in phylotypes and genetic lineages. The numbering of the Streptococcus phylotypes and lineages are based on the groupings in Table 0003. Characteristics of the isolates are included in Tables 0003 and S2. A graphic representation of the complete data is included as Fig. S3: aisolated obtained from MS agar; bisolates obtained from VSA; cVeillonella lineage groupings are not determined because AFLP analysis and/or rep-PCR genomic fingerprinting did not reveal discriminative lineages; dAFLP analysis unlike Rep-PCR genomic fingerprinting identified a single Streptococcus isolate as a separate genetic lineage. According to rep-PCR genomic fingerprinting, this isolate belongs to Streptococcus lineage 7.
Isolates from the single Veillonella phylotype showed highest similarity to V. parvula, V. dispar, and V. atypica on the basis of the 16S rRNA gene sequence classification. This indicates that Veillonella 16S rRNA gene sequences do not allow discrimination of these Veillonella spp. Therefore, MALDI-TOF MS analysis was employed to identify an isolate of this group as V. parvula, and in combination with consistent phylogenetic fingerprinting obtained for all isolates of this group, we decided to use this species identification for the Veillonella isolates.
Strain diversity of ileostoma Streptococcus populations exceeds the phylotype level
Amplified fragment length polymorphism analysis and Rep-PCR genetic fingerprinting were employed to discriminate different bacterial lineages within a 16S rRNA gene phylotype group. Both techniques generated consistent results in terms of subtyping of the 152 bacterial isolates obtained from ileostoma effluent at t = 0 (Fig. S2, Table S2). It should be noted, however, that AFLP fingerprinting discriminated isolates within the S. salivarius species group into two distinct lineages (lineages 4 and 7), while these isolates were grouped together by Rep-PCR genetic fingerprinting (Fig. S2), illustrating the higher resolution of AFLP as a fingerprinting technique. Because of its higher throughput, Rep-PCR fingerprinting was used to classify the 120 isolates from ileostoma effluent at t = 1, identifying seven genetic lineages within the three Streptococcus phylotypes (Figs 0002 and S3), whereas no separate genetic lineages were identified for the Veillonella phylotype as indicated above. Notably, seven distinct genetic lineages could be identified among the two Enterococcus phylotypes (Tables 0003 and S2; Fig. S3).
These findings illustrate the high degree of bacterial richness for the small intestinal ecosystem and confirm that 16S rRNA gene–based approaches underestimate the true diversity of microbial ecosystems that extends to subphylotype levels.
Temporal dynamics of ileostoma Streptococcus populations
To determine whether the occurrence of identified groupings on phylotype and subphylotype levels varies in time, the dynamics of the ileostoma effluent populations were assessed by comparing two samples collected 1 year apart from the same individual.
Isolates from the same Veillonella phylotype were obtained from ileostoma effluent at both time points. Intriguingly, only one of the three Streptococcus phylotypes that were cultivated from ileostoma effluent at t = 0 was recovered from the second ileostoma effluent sample obtained a year later (Fig. 0002). Detailed analysis revealed that of the seven Streptococcus lineages that were cultivated from ileostoma effluent, five Streptococcus lineages were exclusively cultivated from ileostoma effluent at t = 0, while one Streptococcus lineage was only recovered from ileostoma effluent at t = 1. Additionally, one Streptococcus lineage was cultivated from ileostoma effluent at both time points (Fig. 0002). These findings suggest that while the same species are detected, the small intestinal Streptococcus populations display population dynamics at the level of the genetic lineages within the phylotypes.
Carbohydrate fermentative capabilities differ between Streptococcus lineages
To assess whether genetic differences among the Streptococcus lineages are also reflected in their phenotypic characteristics, the carbohydrate fermentation capabilities of one or two (if available) randomly picked bacterial isolates representing each of the six Streptococcus genetic lineages from ileostoma effluent at t = 0 (Fig. 0002) were tested using the API 50 CH system (Table S1). Fermentation profiles between duplicate tests for the same isolate were concordant, albeit that in some cases, a clear positive or negative result was obtained for one duplicate, while the second duplicate showed a weak reaction. Furthermore, fermentation patterns for isolates from the same lineage only showed minor differences (Table S1).
All of the tested Streptococcus isolates were able to ferment the monosaccharides galactose, glucose, and fructose as well as the disaccharides maltose, lactose, and saccharose (sucrose). The three different Streptococcus phylotypes and most of the Streptococcus lineages could be discriminated based on their disparate capacity to ferment arabinose, N-acetylglucosamine, amygdalin, arbutin, esculin, salicin, cellobiose, melibiose, trehalose, raffinose, amidon (starch), glycogen, and gentiobiose (Table S1). Members of Streptococcus lineages 4 and 7 as well as lineages 5 and 6 could, however, not be distinguished based on their fermentation profiles (Table S1). For the isolates belonging to lineages 4 and 7, this appears to reflect their close relatedness as was also concluded from the failure to distinguish these lineages by rep-PCR genetic fingerprinting. However, the fermentation capabilities of the strains tested here did differ from phenotypic characteristics for closely related streptococci, including S. parasanguinis, S. equinus, S. lutetiensis, S. salivarius, and S. vestibularis (De Vos et al., 2009). This illustrates the difference in metabolic capabilities exhibited by individual strains of a species, as is also apparent from the distinct fermentation profiles obtained for the four S. salivarius strains in this study (Table S1).
Isolates were further cultured in basal medium (MSbasal medium) supplemented with different sugars as sole carbohydrate and energy source to test exemplary whether the isolates could also utilize the substrates as a carbon source for growth (Table 0004). Growth was observed for the same substrates that were fermented in the API 50 CH assay, except for isolates from Streptococcus lineages, which were able to ferment N-acetyl-glucosamine (lineages 4 and 7), d-raffinose (lineages 5 and 6), and glycogen (lineage 2) according to the API50 assay, but were not able to use this substrate as a carbon source for growth (Table 0004).
OD600 measurements of Streptococcus cultures in MSbasal medium supplemented with sugars after 18-h incubation at 37 °C
Black: positive growth; white: regarded as no growth.
The numbering of the phylotypes and lineages are based on the order of groupings in Table 0003 and (clockwise) groupings in Fig. S3.
AFLP analysis unlike rep-PCR genomic fingerprinting identified a single Streptococcus isolate as a separate genetic lineage. According to rep-PCR genomic fingerprinting, this isolate belongs to Streptococcus lineage 7.
OD600 measurements of Streptococcus cultures in MSbasal medium supplemented with sugars after 18-h incubation at 37 °C
Black: positive growth; white: regarded as no growth.
The numbering of the phylotypes and lineages are based on the order of groupings in Table 0003 and (clockwise) groupings in Fig. S3.
AFLP analysis unlike rep-PCR genomic fingerprinting identified a single Streptococcus isolate as a separate genetic lineage. According to rep-PCR genomic fingerprinting, this isolate belongs to Streptococcus lineage 7.
Overall, these results demonstrate that the phenotypic diversity of the Streptococcus isolates is in good agreement with the determined phylotype grouping and, to some extent, is reflecting the grouping on the subphylotype level.
Discussion
Culture-(in)dependent analysis of small intestinal Streptococcus and Veillonella populations
The current study employed cultivation and polyphasic molecular typing to provide increased insights into the richness and dynamics of the small intestinal Streptococcus and Veillonella populations. Ileostoma effluent samples were used as a representation of the luminal content of the human small intestinal ecosystem. A recent study, however, postulated that oxygen penetration disrupts the ileostoma microbiota and therefore does not represent that of the terminal ileum in healthy subjects (Hartman et al., 2009). Although the influence of oxygen cannot be ruled out, investigations in our laboratory revealed that the ileostoma effluent microbiota contain a high relative abundance of strict anaerobes (Booijink et al., 2010). Moreover, the ileostoma effluent microbiota resemble that encountered in the proximal part of the small intestine of individuals with an normal intestinal tract that includes a colon (van den Bogert et al., 2011b; Zoetendal et al., 2012). Streptococcus and Veillonella populations were detected in all ileostoma effluent samples and showed fluctuations in relative abundance in a 72-h time frame, which are most likely due to the subject's diet composition. These results are in good agreement with a previous study performed in our laboratory (Booijink et al., 2010). Although the relative abundances of Streptococcus and Veillonella populations varied per subject and time of sampling, two groups of Streptococcus (S. salivarius and S. mitis group) and Veillonella spp. (affiliated with V. atypica, V. rogosae, V. parvula, and/or V. dispar) were consistently dominant in all ileostoma effluent samples. Therefore, we decided to proceed with deciphering the diversity of the Streptococcus and Veillonella populations at the genetic lineage level. To facilitate in-depth analysis of isolates from ileostoma effluent, we focused on samples collected from a single male ileostomist (subject 1), rather than samples collected from several subjects. Samples were collected with a long time interval (1 year apart) to expand the collection of distinct bacterial genetic lineages relative to the diversity that may be expected from ileostoma effluent collected over a relative short time frame (Zoetendal et al., 2012). Selective cultivation conditions enabled the targeted isolation of these Streptococcus and Veillonella spp., although isolates within the genera Enterococcus, Lactobacillus, and Bacteroides were also obtained. Although the latter genera were also detected in other ileostoma samples from subject 1, their relative abundance was generally low (< 0.7%; B. van den Bogert et al., unpublished results). The number of obtained Enterococcus isolates might be explained by a cultivation bias resulting from the use of selective media that preferentially allow growth of specific microbial groups and are known to provide a distorted view of bacterial abundances in the original samples (Dunbar et al., 1997; Muniesa et al., 2005). Of each of the streptococcal species groups identified by pyrosequencing, representative bacterial isolates, including streptococci affiliated with S. equinus and S. lutetiensis belonging to the S. bovis species group, were cultured from ileostoma effluent. Unambiguous affiliation of the Veillonella isolates to specific species, that is, V. atypica, V. rogosae, V. parvula, and/or V. dispar, was not possible based on 16S rRNA gene sequences, which is in agreement with what has previously been described for species within this genus (De Vos et al., 2009). Nonetheless, MALDI-TOF MS analysis identified the Veillonella phylotype as V. parvula. The fact that isolates from Streptococcus, Veillonella, and three other genera were obtained shows that these populations were alive at the time of sampling and may be part of the active small intestinal microbiota.
The Streptococcus isolates obtained from ileostoma effluent collected at two time points could be clustered into multiple species groups, which could be further subdivided into strains belonging to distinct genetic lineages on basis of genetic fingerprinting. Although not the prime subject of this study, also the Enterococcus isolates were found to display a substantial level of phylogenetic richness, indicating that these groups of small intestinal cocci encompass a high degree of genetic diversity. The two ileostoma samples that were taken 1 year apart from the same individual revealed distinct Streptococcus lineages, while only representative strains of one of the Streptococcus lineages were cultivated at both time points. This indicates that the fluctuations in relative abundance seen at the genus and species level are confirmed and expanded at the genetic lineage level. Moreover, while representative strains of the same species are frequently detected in multiple samples from one or several individuals, the occurrence of the corresponding genetic lineages may be quite different. Genetic fingerprinting methods have been widely applied to discriminate strains from Streptococcus spp. such as S. pneumoniae (Dunne et al., 2011) and S. pyogenes (Enright et al., 2001), and were used to assess the oral Streptococcus diversity at the strain level, which showed that the oral cavity of most subjects harbored multiple genotypes of S. mutans (Cheon et al., 2011; Zhou et al., 2011) and S. oralis (Do et al., 2009). Similarly, Enterococcus faecium strains from different sources were differentiated (Homan et al., 2002). Furthermore, the temporal fluctuations of strain abundances of Lactobacillus and Bifidobacterium spp. in fecal samples were analyzed previously (McCartney et al., 1996), revealing similar results as described here. However, to the best of our knowledge, the (small) intestinal Streptococcus richness has not yet been assessed to the subphylotype level. We hypothesize that this is important for the functioning of the ecosystem, because the substrate conversion capacities were different among the Streptococcus lineages. Furthermore, these observations underpin the limitation of species-level identifications of intestinal bacteria on basis of 16S rRNA gene sequences alone, when it comes to the prediction of function of the microbiota. This is especially true for the novel high-throughput sequencing technologies that provide only partial 16S rRNA gene sequences.
Possible interactions of small intestinal bacterial populations with the human host
Notably, streptococci and Veillonella are also abundant in other sections of the upper GI tract (Pei et al., 2004; Aas et al., 2005; Bik et al., 2006; Andersson et al., 2008) and likely originate from their abundant populations in the oral cavity (Walter & Ley, 2011). Molecular typing of oral Streptococcus and Veillonella strains, isolated from the ileostomist studied here (subject 1), identified a Veillonella phylotype and three S. salivarius lineages that, remarkably, group together with those that were cultivated from ileostoma effluent (B. van den Bogert et al., unpublished results). Although speculative, this first comparison between oral microbiota and small intestinal microbiota suggests that the oral microbiota may serve as an inoculum for the upper GI tract. Although this suggests that these populations are allochthonous to the small intestine (Walter & Ley, 2011), the considerable common abundance of the streptococci and their high activity in efficient uptake and fermentation of the available (diet-derived) carbohydrates (Zoetendal et al., 2012) suggest that Streptococcus populations play a prominent role in the primary carbohydrate metabolism occurring in the small intestinal ecosystem.
Because the number of isolates obtained from the ileostoma effluent samples is limited, it cannot be ruled out that lineages that were recovered only from one of the two ileostomy samples would in fact be shared lineages when larger numbers of isolates were to be analyzed. However, some of these lineages were represented by a relatively large number of isolates (e.g. Streptococcus lineages 3 and 7), indicating that even if these lineages were present at both time points, their relative abundance would have differed considerably.
Preliminary investigations in our laboratory identified unique genetic markers for several of the different Streptococcus lineages. Remarkably, these genetic markers were detected in small intestinal samples of other human individuals, suggesting that these lineages are common commensals of the small intestinal microbiota (van den Bogert et al., unpublished results). It is well known that dietary changes lead to an alteration in intestinal microbial composition, which has a profound influence on responses of the host immune system [for a recent review see (Maslowski & Mackay, 2011)]. Furthermore, recent studies revealed that substantially different mucosal responses and immunoregulatory cascades can be modulated by closely related species (van Baarlen et al., 2011), different bacterial strains (Meijerink et al., 2012), and even different preparations of the same strain (van Baarlen et al., 2009). Based on these findings, in combination with the discriminating fermentation and growth patterns among the Streptococcus strains and lineages described here, it is tempting to speculate that there is a potential for directed modulation of mucosal immune responses by dietary modulation of the endogenous Streptococcus populations. To this end, elucidating the role of the small intestinal microbiota, especially of the abundant and diverse Streptococcus population, is a task for the future.
General conclusion
The work presented here demonstrates high intragenus and intraspecies genetic diversity of the small intestinal microbiota, focusing on populations of Streptococcus and Veillonella. It is of particular interest to assess whether the small intestinal Streptococcus and Veillonella isolates have the potential for metabolic interaction similar to what is observed in the oral cavity (Egland et al., 2004). In addition, our findings show that the Streptococcus population in the small intestine has a high phenotypic variability, which may be a dominant driver of the high population dynamics of the small intestinal streptococci in response to varying nutrient availability that is caused by variable food intake. These dynamic streptococcal populations may profoundly influence local host–microorganism interactions, thereby modulating small intestinal physiology and immune system functions.
Acknowledgements
This project was partially supported by the Netherlands Bioinformatics Centre (NBIC). We appreciate the help of Freddy Troost in obtaining approval for this study by the Maastricht Medical Ethics Committee (METC). We thank Milkha Leimena for assistance with collecting ileostoma effluent samples and Kun Xie for assistance with cultivation of isolates and sequencing of 16S rRNA genes, as well as Hauke Smidt and Detmer Sipkema for critical reading of the manuscript. We are very grateful for the participation of all ileostomy subjects who provided samples. We thank Hermie Harmsen of the University Medical Centre Groningen (UMCG; Groningen, The Netherlands) for facilitating the MALDI-TOF MS analyses.
References
Supporting Information
Data S1. Materials and methods.

{kind=link}
{kind=link}