Article Text

Abstract
Objective Cardiovascular events remain the leading cause of death in rheumatoid arthritis (RA). To study the role of cytokines in these observations, the effects of tumour necrosis factor α (TNFα) and interleukin (IL)-17, a classical and a new key player in RA, were assessed in endothelial cell (EC) dysfunction.
Methods Primary human EC were treated with IL-17 alone or combined with TNFα. mRNA expression was quantified by qRT PCR and Affymetrix microarrays. The role of IL-17 was studied using functional assays of platelet aggregation, EC migration and invasion.
Results IL-17 alone induced 248 pro-inflammatory genes and 9803, when combined with TNFα. IL-17 plus TNFα induced synergistically chemokine genes such as CCL5, IL-8 and cytokine genes such as IL-6. In contrast, IL-17 decreased genes involved in the regulation of inflammation such as IL-33. IL-17 induced EC migration and invasion in synergy with TNFα. Such invasion was inhibited with an antiCXCR4 antibody, indicating the contribution of the stromal cell-derived factor-1/C-X-C chemokine receptor type 4 axis. Supernatants of IL-17-treated EC induced strong platelet aggregation. IL-17 inhibited endothelial CD39/ATPDase expression, an inhibitor of platelet activation. Finally, IL-17 enhanced genes critical for coagulation such as tissue factor and decreased thrombomodulin, leading to a pro-thrombotic state.
Conclusion These results indicate that IL-17 specifically when combined with TNFα has major pro-coagulant and pro-thrombotic effects on vessels.
Statistics from Altmetric.com
Patients with rheumatoid arthritis (RA) and systemic lupus erythematosus have a striking increase in the incidence of cardiovascular disease (CVD).1 2 While this is secondary to premature atherosclerosis, Framingham risk factors do not account for this increase.3 Pro-inflammatory mediators are strongly linked to endothelial cell (EC) activation and dysfunction, now considered as an early step for atherosclerosis preceding atherosclerotic plaque formation.4 Endothelial dysfunction combines reduced vasodilatation to a pro-inflammatory and pro-thrombotic state.
EC lining all blood vessels play an important role during systemic inflammation. Direct EC exposure to inflammatory mediators leads to changes in gene expression for cytokines, adhesion molecules, and coagulation factors. Conversely, control of inflammation with tumour necrosis factor α (TNFα) inhibitors has a beneficial effect on CVD incidence.5
In addition to TNFα, interleukin (IL)-17 is another cytokine involved in RA pathogenesis. This pro-inflammatory cytokine produced by the Th17 T cell subset has pleiotropic activities, including the induction of other inflammatory cytokines (eg, IL-6, TNFα, IL-1β), chemokines (CCL20 and IL-8), adhesion molecules (eg, ICAM-1) and pro-inflammatory mediators (prostaglandin E2, nitric oxide and cyclooxygenase-2) in synoviocytes, a cell critical for the local pathogenesis of RA.6,–,8
While the role of IL-17 in atherosclerosis is now admitted, its global effect on endothelial cells has not yet been fully described.9 The aim of this study was to describe the effects of IL-17 alone and combined with TNFα on several vascular functions. Human umbilical vein EC (HUVEC) were selected because they reflect large vessel biology and furthermore keep their coagulation phenotype in culture. Microarray analysis was performed to define the IL-17-induced expression profile with an extensive statistical analysis. Focusing on the cooperation between IL-17 and TNFα, some of these results were compared to those we have obtained with synoviocytes.10 Functional assays were performed, focusing on proliferation, migration, invasion and coagulation.
Materials and methods
Cells
HUVEC were collected from umbilical cords by collagenase perfusion of umbilical veins. Ten umbilical cords were used with informed and written consent obtained from the mother. The cells were maintained in endothelial cell basal medium (EBM2) with 12 ng/ml bovine brain extract, (1 mg/ml) hydrocortisone, 10 ng/ml epidermal growth factor, 50 ng/ml gentamycin, 50 ng/ml amphotericin-B, 5% fetal calf serum (FCS)S (all from LONZA, Cologne, Germany). EC were used at passage 3–5.11 After 12 h in 1% FCS, 90% confluent EC were treated or not with TNFα (1 ng/ml), or IL-17 (100 ng/ml) alone or in combination, for 12 h for mRNA studies. To assess interaction between both cytokines, TNFα was added 2 h after pretreatment with IL-17 as described previously.12 All experiments were performed at least four times, and two microarrays were performed per condition.
Cytokines, ELISA
Human recombinant TNFα and IL-17A were purchased from R&D systems (San Diego, California, USA). IL-6, IL-8 and CCL20 levels were quantified in supernatants by ELISA (eBioscience; Diaclone; R&D Systems, San Diego, California, Minneapolis, Minnesota, USA, respectively).
mRNA microarray hybridisation and analysis
Total RNA was isolated using RNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA was obtained from cells 12 h after treatment with IL-17 and/or TNFα. 2 µg of RNA were analysed using HG-U133+2 arrays (Affymetrix, Santa Clara, California, USA), according to the manufacturer's instructions.
RNA integrity number was assessed using RNA 6000 nanochips and the Agilent 2100 Bioanalyser (Agilent Technologies, Santa Clara, California, USA). Total RNA was used to prepare double-stranded cDNA containing the T7 promoter sequence. cRNA was synthesised and labeled with biotinylated ribonucleotide (GeneChip IVT Labeling kit; Affymetrix). The fragmented cRNA was hybridised on HG-U133A+2 oligonucleotide arrays (54 234 probe sets). The arrays were washed and stained using the fluidic station FS450 (Affymetrix) (protocol EukGE-WS2v4). The arrays were scanned with the Agilent G2500A Gene Array Scanner. The NETAFFX web site (www.affymetrix.com) was used to select candidate genes.
Microarray data preprocessing and validation
Statistical analysis was performed using the Affymetrix Data Mining Tool Software (version MAS 5.0). Gene products that showed a signal intensity of 30 or greater were considered as significantly expressed and genes that showed a change of twofold or greater compared with the control situation were considered as significantly regulated. The NETAFFX web site (www.affymetrix.com) was used to select candidate genes.
For all statistical analysis, we used open source software available from BioConductor for the R statistics environment (BioConductor, www.bioconductor.org). Raw data were preprocessed using a combination of the Robust multi-array average and the MAS5 methodologies as implemented in the Bioconductor 2.0 environment (Wilson, bioinformatics, 2005). The preprocessing consisted of three main steps. First, probe-specific background correction was performed to compensate for non-specific binding using perfect match distribution rather than perfect match – mismatch values. Second, probe level data were normalised using the MAS5 approach. Third, the median polished procedure was used to summarise the log-transformed probe-level data.
Using the local-pooled-error test for identifying significant differentially expressed genes in microarray experiments, the sensitivity of detecting subtle expression changes can be dramatically increased and differential gene expression patterns can be identified with both small false-positive and small false-negative error rates.
All Affymetrix identifications on the applied gene chip that showed at least one P detection call in the two compared samples in each of the duplicated experimental sets served as reference. Two ways were used for independent confirmation of the results: in silico analysis using published results, and experimental validation by real-time reverse transcriptase-PCR (RT-PCR). The validation by real-time RT-PCR was tested on four target genes (IL-6, IL-8, E selectin and ICAM).Due to the allometric relation between PCR- and microarray-derived data, the correlation was calculated after log transformation. See supplemental data for details. To examine the molecular function and genetic networks, microarray data were analysed using David database (http://david.abcc.ncifcrf.gov/) and ingenuity pathways analysis tool (IPA version 8.7, Ingenuity Systems Inc., Redwood City, California, USA; http://www.ingenuity.com), a web-based software application which enables identification of biological mechanisms, pathways, and functions most relevant to experimental datasets or differentially expressed genes. Networks of the IL-17+TNFα gene were constructed using ingenuity pathway analysis (IPA). Genes found in the IPA knowledge database are labeled ‘focus’ genes. Based on the focus genes, IPA generated a set of molecular networks with a cutoff of 40 genes for each network based on interactions between uploaded genes and all other genes/proteins stored in the knowledge base.
Quantitative real-time reverse transcriptase-PCR
The procedures and conditions for real-time RT-PCR using the QuantiTect SYBR Green PCR kit (Roche) were the same as described previously.10 Briefly, total RNA was denatured by incubating for 5 min at 65°C with 4 µM of oligo(dT) primer and then reverse transcribed by using in final concentration of 0.5 mM dNTP, 40 µl, RNase OUT, 0.01 M dithiothreitol and 10 U/µ of ThermoScript reverse transcriptase. Reverse transcription was performed by incubation at 50°C for 60 min followed by 85°C for 5 min. The obtained cDNA was diluted 1 in 10 with distilled water and 10 µl used for amplification. Primers (see list of primers in supplemental data) were designed using the primer express software package (Applied Biosystems, Foster City, California, USA) and obtained from LC search Biotech (Ebersberg, Germany) or Eurogentec (Liege, Belgium). Gene expression was normalised with respect to the endogenous housekeeping control gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Relative expression differences of respective genes were calculated using the comparative threshold cycle method as described by the manufacturer. mRNA expression of target genes was normalised with GAPDH mRNA expression and data are expressed as the fold induction compared to untreated controls. Data are expressed as the mean±SEM. Statistical significance of changes was determined by the student's t test. Differences resulting in p values < 0.05 were considered statistically significant. All data are the result of at least three separate experiments.
Functional assays
In vitro cell growth assay
To evaluate the effect of IL-17 on proliferation, HUVEC were suspended in EBM with 2% FCS and were plated at 1.5×105 cells in 10 cm culture dish coated with type II collagen (Becton Dickinson, Bedford, Massachusetts, USA). After 24 h, medium was then replaced with EBM with or without cytokines. Cell number at day 5 after carboxyfluorescein diacetate, succinimidyl ester incorporation was obtained by flow cytometry.
Migration assay
Migration of HUVEC through a gradient of vascular endothelial growth factor was evaluated by using a modified Boyden chamber assay, as described.13 Cells were cultured in EBM with 2% FCS for 8 h and plated at 10×104 cells/cm2 onto a polycarbonate filter with 8 µm pores (Kurabo Industries Ltd., Osaka, Japan) coated with 10 µg/ml fibronectin (Sigma).Cytokines (TNFα 1 ng/ml, IL-17 100 ng/ml, and their combination) in EBM with 1% FCS were applied in the lower compartment of the chamber, and cells suspended in EBM with 1% FCS were cultured for 4 h at 37°C. The filters were fixed and stained with Hoechst, and the number of migrating cells was quantified by counting cells in five randomly selected high power microscopic fields (HPF) (x200) in each well. For the inhibitory assay, a neutralising antihuman IL-17 mAb (20 µg/ml) was added to the lower and upper compartments of the chamber. Each experiment was performed with three different cell lines.
Invasion assay
The effect of IL-17A on the invasiveness of HUVEC was examined using the BD Bio-Coat Matrigel invasion assay system (BD Biosciences, Bedford, Massachusetts, USA) according to the manufacturer's instructions. Each chamber contains a thin layer of growth factor reduced Matrigel basement matrix that acts as a reconstituted basement membrane in vitro. The layer prevents non-invasive cells from migrating through the membrane. Cells that migrate beneath the chamber are able to invade through the Matrigel matrix and the eight micron membrane pores.
HUVEC (5×104 cells) were suspended in medium containing 2% FCS and seeded into the Matrigel precoated transwell chambers. The transwell chambers were then placed into 24-well plates, to medium with or without cytokines. After 16 h the upper surface of the transwell chambers was wiped with a cotton swab. Transwell filter was fixed in 1% gluteraldehyde for 15 min. Following fixation, the filters were stained using Hoechst solution before washing. Representative fields were captured digitally and cells in five random high powered fields for each well were counted in order to assess the average number of migrating cells. Each condition was assessed in triplicate. To assess the role of CXCR4 in invasiveness induced by IL-17, the invasion assay was performed through a SDF1 gradient (5 ng/ml) (ALMAC, Craigavon, UK). Monoclonal antibody directed toward CXCR4 was added to confirm its involvement in the invasiveness (R&D Systems, Minneapolis, Minnesota, USA).
Platelet aggregation
Platelet aggregation was studied in Platelet Rich Plasma (PRP) containing 250×106 platelets/ml using the Born's method with a Lumi-aggregometer (ChronoLog Corporation, Lille, France). PRP was preincubated at 37°C in an aggregometer cuvette. Platelet activation was started by addition of supernatants of EC stimulated or not by cytokines. Addition of adenosine diphosphate served as positive control and platelet aggregation was monitored and quantified by the increase of light transmission. Platelet-poor plasma as negative control performed with EC incubated with ADP, a mediator of platelet aggregation that is not hydrolysed.14
Results
Effects of IL-17A on major genes of inflammation
Confluent EC were cultured with IL-17A alone and in combination with TNFα for expression of target genes (IL-6, IL-8/CXCL8) known to be induced in synoviocytes by IL-17A. Expression was assessed at 12 h. Time and dose course experiment assessing the effect of IL-17 on IL-6 and IL-8 mRNA expression as a positive control, are shown as supplemental material (figure S1–5).
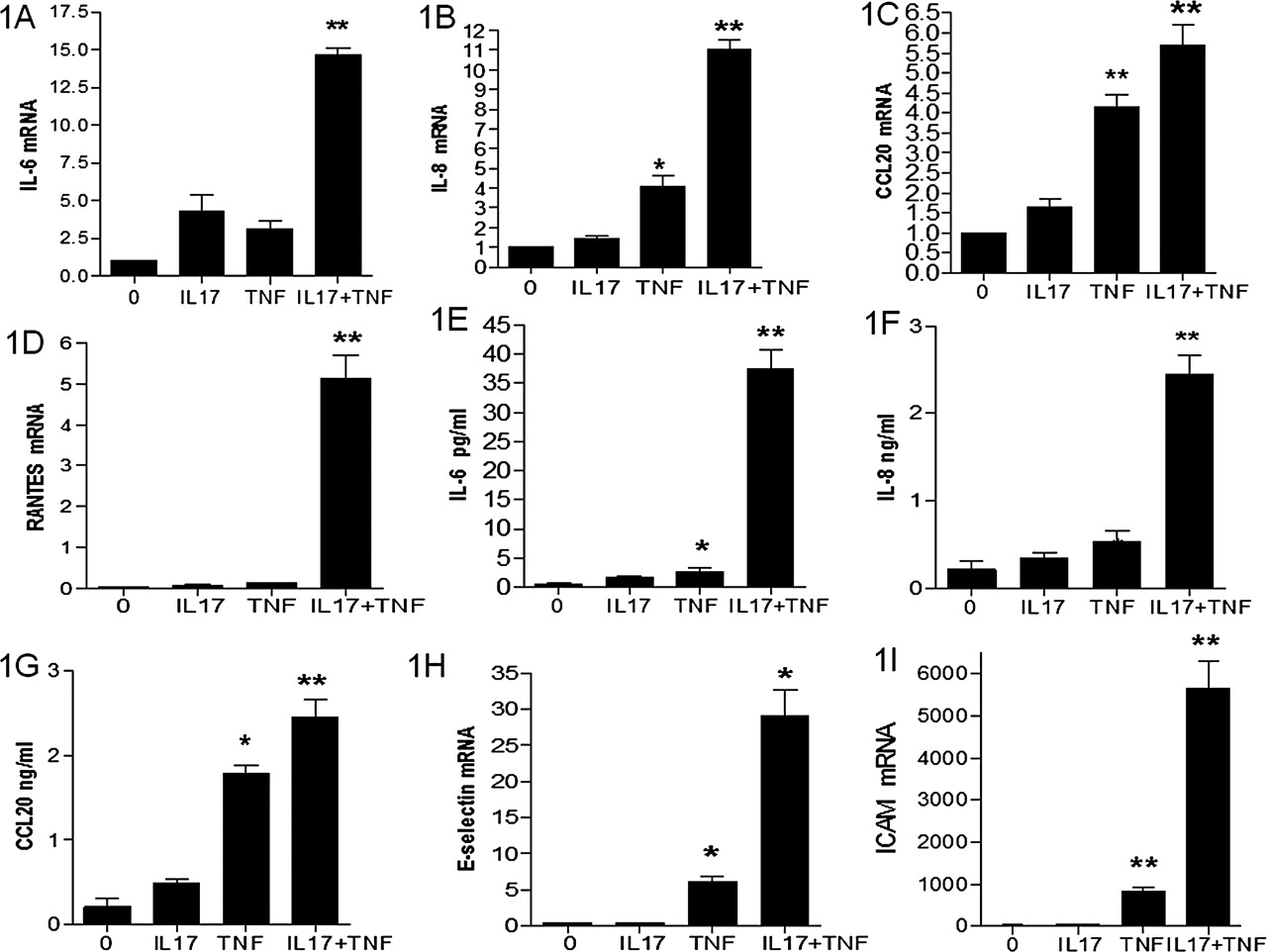
Although IL-17A had a limited effect alone, it potently induced IL-6 mRNA expression in the presence of TNFα (15-fold increase compared with the basal level; figure 1A). At 12 h, IL-17A induced a 10-fold increase in IL-8 mRNA expression when combined to TNFα (figure 1B). While IL-17 has no effect on CCL20 expression, combination with TNFα had the same effect as TNFα alone (figure 1C). This is in contrast to the results in synoviocytes where a synergistic effect was observed.10 IL-17A and TNFα alone had no effect on RANTES mRNA expression but their combination induced a massive expression (figure 1D). This is also in contrast to the inhibition of IL-17A on TNFα induced RANTES expression in synoviocytes.10

Effect of IL-17A, alone or in combination with TNFα, on mRNA and protein levels of pro-inflammatory mediators. EC were stimulated for 12 h with IL-17A, alone or in combination with TNFα. IL-6 (A), IL-8 (B), CCL20 (C), and RANTES (D) mRNA levels were quantified by real-time RT-PCR. Values (mean±SEM of four independent experiments) were normalised with GAPDH mRNA expression and are expressed as fold induction compared with the untreated condition. *p<0.05; **p<0.001 by two-tailed Mann–Whitney test. EC Supernatants were collected 48 h after stimulation with IL-17, TNFα, or their combination, using supernatants of untreated EC as a negative control. Concentrations of IL-6 (E), IL-8 (F), and CCL20 (G) were tested by ELISA. Results are expressed as mean±SEM of five experiments. Adhesion molecules intercellular adhesion molecule 1 and E selectin mRNA expression in EC were assessed at 12 h (H, I).
Similar effects were obtained at the protein level. Although IL-17A alone had no effect, it potently induced IL-6 production in the presence of TNFα (38-fold increase compared with basal level; figure 1E). The same results were obtained for IL-8 production (figure 1F). While IL-17 has no effect on CCL20 production, combination of IL-17 with TNFα had only a modest effect compared to TNFα alone (figure 1G).
EC-specific genes were used to confirm the specific vascular activation. IL-17A alone had no effect while a clear synergistic effect for E selectin (figure 1H), and ICAM (figure 1I) was observed in combination with TNFα.
Transcript profiling data analysis and validation
Microarrays were used to obtain a broad view of gene expression in EC stimulated by IL-17 and TNFα. The numbers of genes that were either down- or up-regulated at least twofold and had a raw intensity value of at least 1000 in the experimental sample in each of the two independent experiments are presented in a Venn diagram (figure 2). Two hundred and forty-five genes were modified by IL-17 alone, 1036 by TNFα alone and 10873 by their combination (figure 2A). These raw findings were used to select new genes involved in inflammation and vascular biology.

(A) Microarray-based analysis of IL-17A induced effects alone or in combination with TNFα. EC were stimulated with IL-17A alone or in combination with TNFα for 12 h. Gene products that showed a signal intensity of 30 or greater were considered as expressed and genes with a twofold or greater change were considered as regulated (see Materials and Methods). Among the genes selected for this analysis, 245 were regulated by IL-17A; 199 were exclusively regulated by IL-17A (81.6%), 8 were also regulated by TNFα (3.2%); 1036 genes were regulated by TNFα and 10873 genes were regulated by the combination of both cytokines with 9803 genes specifically modified by the combination. A Venn diagram summarises these findings. (B) The network derived from the genes synergistically induced by combination of IL-17 with TNFα derived from ingenuity pathway analysis (IPA) software is presented. Edges (gene relationships) are displayed with labels that describe the nature of the relationship between nodes (genes). The network clearly showed central connection represented by the pro-inflammatory cytokines and the connection with F3 or tissue factor and direct relation between tissue factor and inflammatory cytokines, and indirect relation between lipid proteins and inflammation (HDL and IL-1).
Pathway analysis of microarray analysed data
Among the genes induced by IL-17 and TNFα, 1046 were induced more than five times, so these genes were selected for IPA. Of the 1046 differentially expressed genes, 768 genes were determined to be network eligible. The top few genes most dramatically up-regulated with a fold change greater than 10 (p<0.05) are listed in table 1.From IPA, 25 networks were identified with 21 networks having 10 or more focus genes among the differentially expressed genes. The top gene networks identified were Network 1 (score 47, 28 focus molecules): cell-to-cell signalling and interaction, immune cell trafficking and cell movement; network 2 (score 37, 24 focus molecules figure 2B) cell haematological system development and function, including genes as tissue factor,) and network 4 was identified as cellular development, cell death, and cellular growth and proliferation. Table S1 summarises the list of the 15 networks and the list of the associated genes (see supplemental material).
List of genes induced by IL-17 combined with TNF-α
Some of these genes were validated by qRT-PCR, (figure 3A–I). As suggested by the IPA analysis, our study focused on biological functions such as cell proliferation, cell movement and migration, cell invasion and coagulation.
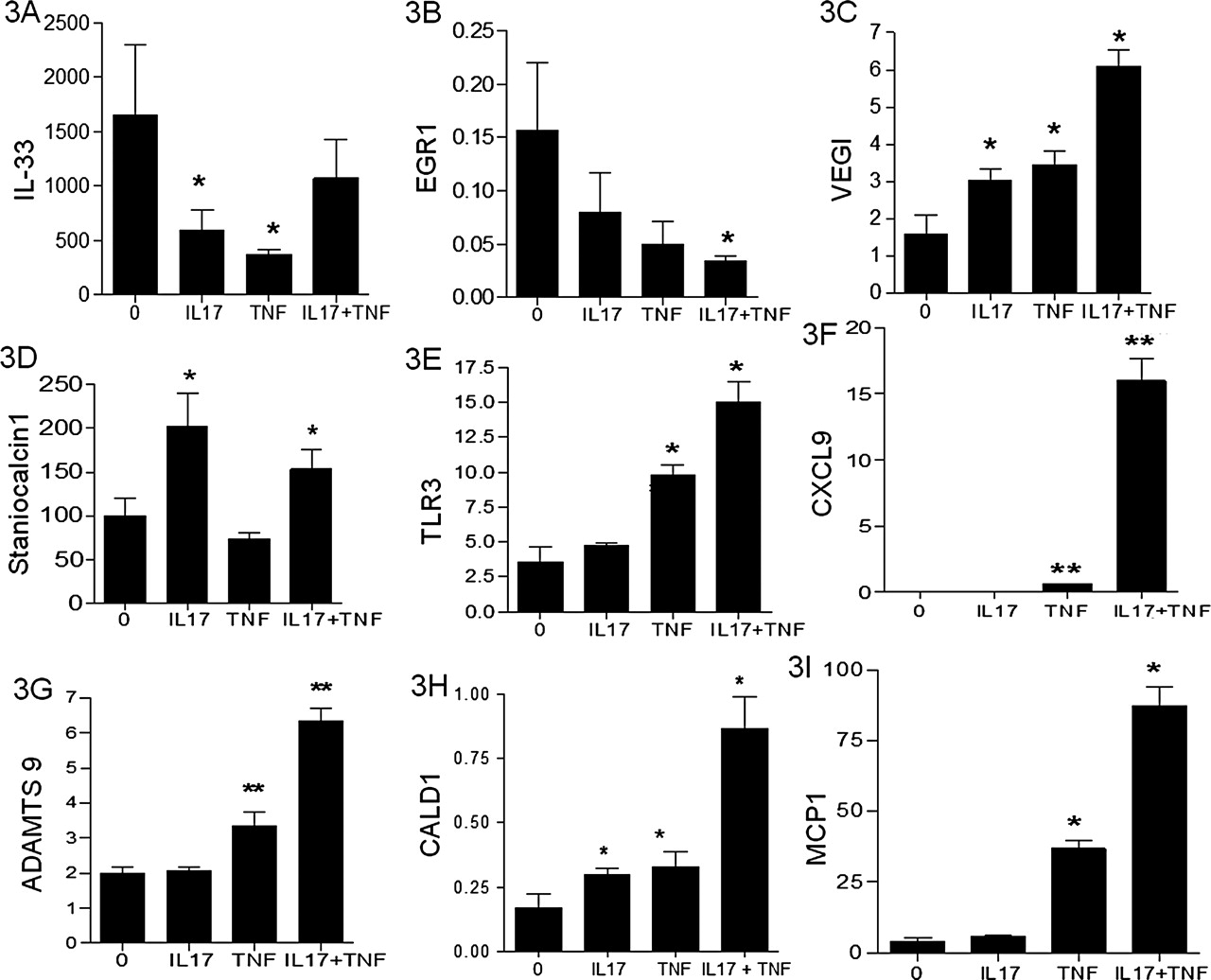
Validation of target gene mRNA expression by real-time RT-PCR. Ten genes selected from microarrays were validated by real-time RT-PCR: IL-33 (A), EGR1 (B), VEGI (C), Staniocalcin (D), TLR3 (E), CXCL9 (F), ADAMTS9 (G), CALD1 (H) and MCP1 (I). The values were normalised with GAPDH mRNA expression and expressed as fold induction compared with the untreated condition. Values represent the mean±SEM of four independent experiments. *p<0.05, **p<0.001 by the two-tailed Mann–Whitney test.
Identification of new target genes in EC
Analyses of a few selected new genes that were regulated by IL-17, TNFα or their combination are described in figure 3. Some of the genes regulated by IL-17, known to play a role in both RA and atherosclerosis are presented here. For instance, IL-17 decreased the expression of IL-33 transcript as for TNFα, but this decrease was not modified with their combination (figure 3A). IL-17 reduced the expression of cytokine receptors such as EGR1 which is involved in cell proliferation but also in atherosclerosis (figure 3B).15 IL-17 induced VEGI (figure 3C), which inhibits EC growth and EC progenitor differentiation and is considered as an antiangiogenic cytokine.16
Staniocalcine was increased by IL-17 but not by TNFα (figure 3D). This gene is involved in cell proliferation and atherosclerosis.17 TLR3 was induced by the combination of IL-17 with TNFα (figure 3E). CXCL9 was induced with a synergistic effect by IL-17 combined with TNFα (figure 3F). ADAMTS 9 (figure 3G) and CALD1 (figure 3H) are genes involved in tumour angiogenesis, which were also increased by the combination.18 Similar effects were seen for MCP1, a chemokine involved in vascular diseases.19
Using these microarrays, we have also identified genes involved in EC function leading to a pro-inflammatory and pro-atherosclerotic state. In order to confirm these results, time course experiments were performed for some of these genes, and results for IL-33, tissue factor, TLR3, and CD39 are shown as supplemental material (figure S6–9). These results were then extended using functional assays.
IL-17 effects on angiogenic related functions
Such analysis suggested a role of IL-17 in EC growth. Using bFGF and VEGF as positive controls, IL-17 alone had no effect on EC proliferation (figure 4A and B). However, the combination of IL-17 with VEGF or FGF increased EC numbers in a dose dependent fashion. In addition, combination of IL-17 and TNFα had an additive effect on VEGF mRNA expression with a 5 fold increase compared to baseline, with a massive effect on VEGF production (figure 4C and D)

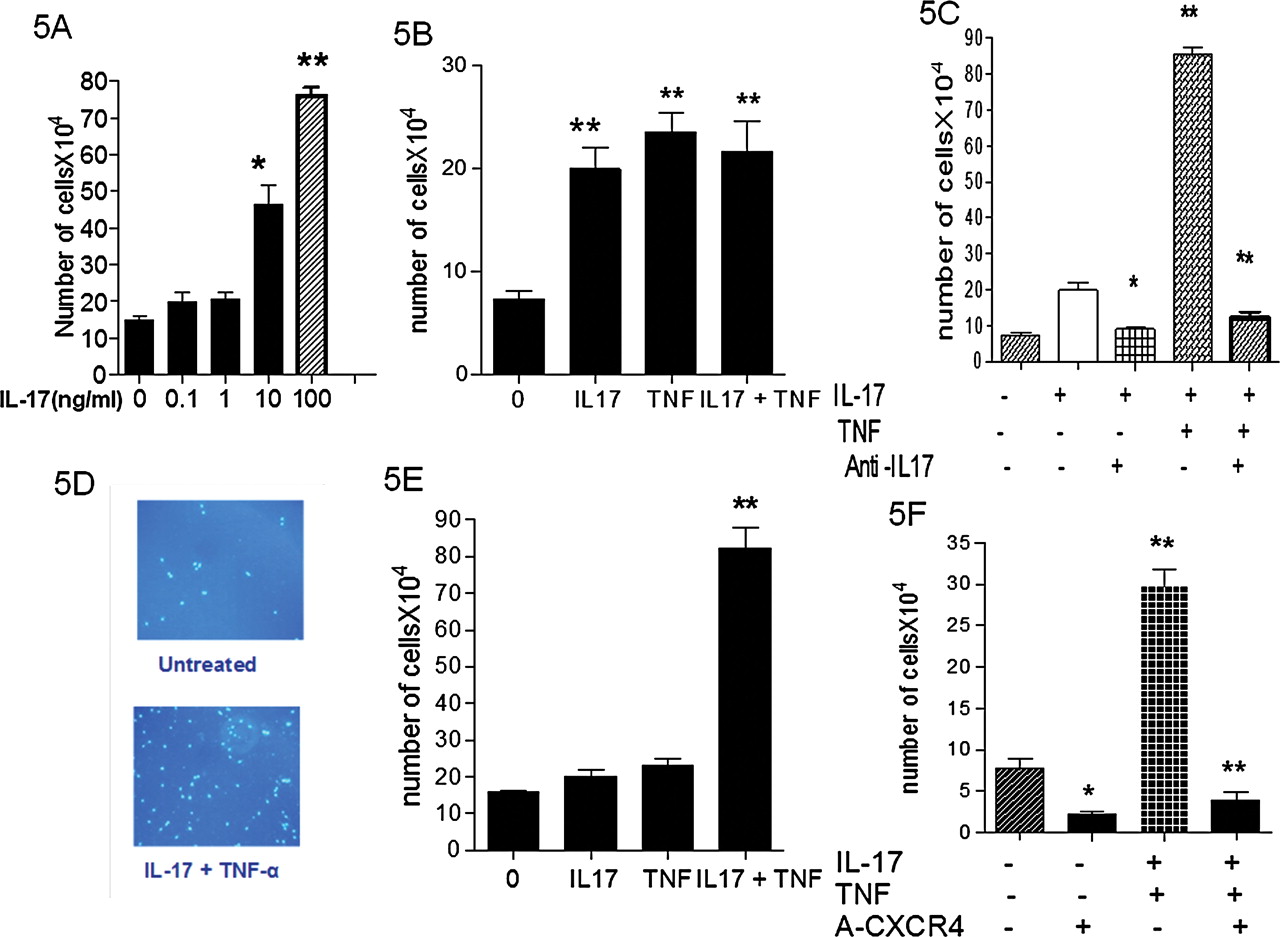
IL-17 effects on EC angiogenic related functions: EC were plated at 2×105 cells in 10 cm culture dish. After 24 h, cells were treated by IL-17 (0.1–100 ng/ml) and bFGF (10 ng/ml, A) or VEGF (10 ng/ml, B) and medium was changed each day. The cell number was measured at day 5 by CFSE using flow cytometry. The results represent the mean±SEM, n=4; control versus IL-17 and bFGF or VEGF: *p<0.05; **p<0.001. EC were stimulated with IL-17 and TNFα. VEGF mRNA expression was determined by real-time RT-PCR after 12 h (C). Data were taken from at least three experiments performed with each sample obtained from three different umbilical vein cords. Levels of VEGF in supernatants were determined by ELISA after 48 h (D). Data are expressed as mean concentration±SEM; *p<0.05; **p<0.001 by Mann–Whitney test.
EC migration in Boyden chambers was increased by IL-17 in a dose dependent fashion (figure 5A). Such IL-17-induced EC migration was similar to that induced by bFGF (data not shown) or TNFα (figure 5B). Combination of both cytokines had no further effect. The specificity of this response was determined using a neutralising antiIL-17 mAb (20 µg/ml) with VEGF as a chemoattractant, which gave a full inhibition (figure 5C).

Migration and invasion of EC following stimulation with IL-17 and TNFα (A) ECs were allowed to grow over 3 days with IL-17 (0–100 ng/ml). Bars represent mean number of migrated cells±SEM per 5 HPF (x400). These result are representative of three independent experiments (VEGF 10 ng/ml was used as chemoattractant, *p<0.05, **p<0.001). (B) The number of migrated cells following stimulation with IL-17 and TNFα was assessed at 24 h. (C) The specificity of this chemotactic response was determined using a neutralising mAb to human IL-17 (10 µg/ml). Representative pictures show EC invasion following IL-17 and TNFα combination stimulation (D). 8 µm membrane Matrigel invasion chambers containing EC were used with serum free EC media and IL-17A, TNFα, and their combination in the lower wells. At 16 h, migrating cells attached to the lower membrane were fixed (1% gluteraldehyde) and stained (Hoechst, original magnification x20). (F) SDF1 as chemoattractant and an antiCXCR4 (10 µg/ml) monoclonal antibody were used to confirm the contribution of the CXCR4/SDF1 axis EC invasion induced by IL-17.
The effect of IL-17 on EC invasiveness was examined using transwell Matrigel invasion chambers. IL-17 and TNFα acted in synergy to promote EC invasiveness from a control level of 18±4 to 74±18 cells/HPF through a SDF1 gradient (p<0.005) (figure 5D and E).
SDF1 has recently been described to be involved in synoviocyte migration and SDF1 production is regulated by IL-17.20 EC invasiveness induced by IL-17 and TNFα was strongly reduced with an antiCXCR4 monoclonal antibody, blocking the receptor of SDF1 expressed by EC. The number of cells clearly decreased after exposure to a monoclonal antibody directed to CXCR4 (70×104/HPF–6×104/HPF, p=0.003; figure 5F).
Activation of thrombosis in IL-17A stimulated EC
Thrombosis is the result of platelet aggregation and pro-coagulation activation in EC. To study the role of IL-17 in platelet aggregation, a functional assay with PRP was performed. Supernatants of EC treated with IL-17 and TNFα were added to PRP and ADP, acting as an agonist of platelet aggregation. Aggregation was monitored by changes in light transmittance in a lumi-aggregometer. IL-17 alone enhanced the level of platelet aggregation from 20% to 45–50% (p=0. 001; figure 6A). Similar effect was seen with TNFα combined to IL-17.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-17 promotes thrombosis in EC: EC were incubated with IL-17α with or without TNFα for 12 h. ADP and ATP (100 µmol each) were added for 20 min; supernatants were tested for aggregation (n=5). Bars show mean±SEM of percentage of aggregated platelets triggered by culture supernatants. *p<0.05, **p<0.01 (A). EC were incubated with IL-17 with or without TNFα for 12 h. Total mRNA was isolated, and analysed for tissue factor (B), thrombomodulin (C) and CD39 (D). Data represent the mean±SEM from three independent experiments. *p<0.05 **p<0.001.
Based on our microarray data on coagulation, IL-17 and TNFα induced synergistically the expression of tissue factor (F3, 151 fold), a key player to initiate the coagulation cascade (figure 6B). Furthermore, IL-17 and TNFα induced plasminogen activator mRNA expression by six fold with an additive effect (data not shown).
Conversely, IL-17 in combination with TNFα inhibited by 10-fold the expression of thrombomodulin, an inhibitor of coagulation, thus increasing the EC pro-thrombotic phenotype (figure 6C). Microarrays identified CD39, an inhibitor of platelet aggregation, among the genes inhibited by IL-17. The preserved function of CD39/ATPDase is crucial in the inhibition of platelet activation by keeping adenosine nucleotide levels low to inhibit platelet aggregation.21 After 12 h of incubation with IL-17, TNFα and their combination, CD39mRNA expression was divided by 3.7 (figure 6D). Thus, IL-17A was able to induce an imbalance between a pro-thrombotic and anticoagulant phenotype.
Discussion
The role of TNFα, then that of IL-17 has been well described in the local manifestations of RA.22 23 Here we show that the same cytokines specifically when combined affect EC biology inducing an inflammatory, pro-coagulant and pro-thrombotic state. We have used in these studies HUVEC because they keep their properties for a long time and are good tools to assess anticoagulant properties of endothelium. They are usually a good model for invasion and migration assessment. In addition, most of our studies are based on mRNA levels, which imply large quantities of cells, difficult to obtain with small vessel-derived EC. Nevertheless, these studies need to be performed with EC from arterial vessels to better reproduce the clinical situation of atherosclerosis.
Regarding inflammation, IL-17 alone had limited effects to promote inflammatory gene expression, but it acted in concert with TNFα to induce the expression of characteristic genes such as IL-6 and IL-8. When EC and synoviocyte gene expression was compared, the pattern was rather similar but with interesting differences. For instance, IL-17 acted here synergistically with TNFα to promote the expression of RANTES in EC while IL-17 decreased that expression induced by TNFα in RA synoviocytes.10
Similarly, IL-17 had limited effects on CCL20 mRNA expression in EC compared to TNFα, and their combination did not change the effect of TNFα alone. Conversely, in RA synoviocytes, a clear synergistic effect was seen with the combination of IL-17 and TNFα.10 These results are in line with differences between the local and systemic mechanisms in RA pathogenesis. Among the mechanisms involved in the cooperation between TNFα and IL-17, IL-17 can promote the expression of TNF type II receptor and the use of TNFR II mAb was able to inhibit the effect of the combination of TNFα and IL-17.10
Regarding protective genes toward atherosclerosis, IL-17 reduced the expression of cytokine genes such as IL-33 which is mostly expressed in EC and with an antiatherosclerotic effect.24 Vessel formation and repair are clearly affected by inflammation, IL-17 induced genes involved in angiogenesis, and affecting EC proliferation. For instance IL-17 inhibited the expression of EGR1, involved in EC proliferation, and enhanced the expression of VEGI, an inhibitor of angiogenesis. These genes are crucial for neoangiogenesis, suggesting an inhibitory effect of IL-17 on new vessel formation.16 However, these results are not in line with those of the functional assay where IL-17 was found to induce growth factor-induced EC growth. Here again, regulation at the local and systemic levels might be different. IL-17 could at the same time induce angiogenesis to promote local inflammation including migration of inflammatory cells and inhibit blood vessel repair. Similar conflicting results have been described in the context of tumor growth. Murine fibrosarcoma cells transfected with IL-17 demonstrated increased growth compared with control tumors.25 Moreover, IL-17 increased blood vessel development in rat cornea and vascularity in arthritis models (24). In our studies, IL-17 did not induce angiogenesis alone, but it could do so by promoting the mitogenic activity of VEGF and bFGF, as previously shown for human lung microvascular EC.26
Angiogenesis is linked to new blood vessel formation with invasiveness properties. The cooperation of IL-17 with TNFα appears crucial for EC invasiveness using SDF1 as chemoattractant with a clear synergistic effect. The involvement of SDF1 in migration was confirmed by the antiCXCR4 blockade. This clear inhibitory effect indicated that increased CXCR4 expression at the surface of EC results from the combined effects of the two cytokines.
EC are characterised by a precise balance between antithrombotic and pro-coagulant activities. As expected, IL-17 alone and massively in combination with TNFα, increased the expression of coagulation genes such as tissue factor. This was already demonstrated for other cytokines such as IL-1β and TNFα.27 In addition, IL-17 may affect the antithrombotic properties of EC. IL-17 and TNFα had an additive effect to reduce CD39mRNA level.28 In addition, IL-17 decreased anticoagulant factors such as thrombomodulin which is expressed at the luminal surface of resting EC, acting as a physiologic anticoagulant.29 30 Regarding platelet aggregation, IL-17 alone had an enhancing effect but not modified by the addition of TNFα.
Combined together, our results indicate a clear involvement of IL-17 in EC activation, contributing to endothelial dysfunction and these effects were in general massively increased in combination with TNFα. Results obtained in mouse experiments are in line with these findings. LDLR/IL-6 double knockout (KO) mice, which exhibit a decrease of IL-17 levels, were found to have a reduction in atherosclerotic lesion development, suggesting a potential role for IL-17 and Th17 cells in the promotion of atherogenesis.31 In another study using the apoE/IL18 double KO mice, the exacerbated atherosclerotic lesion formation correlated with increased Th17 cells in blood.32 Using bone marrow chimera studies and transplanted irradiated LDLR-deficient recipient mice with IL-17R-deficient bone marrow, Western type diet-induced atherosclerotic lesions were reduced.33 In addition, IL-17A-blocking antibody injected in apoE–/– mice reduced atherosclerotic lesion development and plaque vulnerability, cellular infiltration, and tissue activation in apoE-deficient mice.34 At this stage, limited results are available on the direct effect of IL-17 on EC function and especially its antithrombotic properties but its role remains controversial in vascular remodeling.35 IL-17 KO mice, when on high fat diet displayed significantly diminished aortic lesion size and macrophage accumulation.36 Expanding these results to the human situation, a high peak of IL-17 plasma level was found in patients with unstable angina, if followed by myocardial infarction.37 Most importantly, concomitant presence of IL-17 and interferon γ in clinical specimens of coronary atherosclerosis has been observed.38 The cooperation of IL-17 with other cytokines remains crucial to understand their systemic effect because it could explain that a vascular risk remains increased despite therapy with biologics blocking only TNFα.39 Finally, our study is in line with a recent report showing a correlation between IL-17 plasma levels in RA patients and endothelial dysfunction.40
Conclusion
IL-17 is becoming an emerging therapeutic target in RA, a disease with systemic inflammation leading to an increase of cardiovascular morbidity. Although the role of IL-17 in atherosclerosis remains to be confirmed, these results indicate its involvement in EC dysfunction, an early step of atheroma. IL-17 was able to induce the expression of pro-inflammatory genes, to activate coagulation and vascular remodeling by enhancing EC migration and invasiveness. These effects were commonly increased in the presence of TNFα. Accordingly IL-17 blockade could lead to a decrease in vascular risk in RA and in other chronic inflammatory disorders.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding This work was supported by the Hospices Civils de Lyon, the Région Rhone-Alpes and a Mérieux research grant. Arnaud Hot was supported by a grant from the French society of internal medicine, SNFMI.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Miscellaneous