


Abstract
Tezosentan (Ro 61-0612) [5-isopropyl-pyridine-2-sulfonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2-1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide] is a new endothelin (ET) receptor antagonist specifically designed for parenteral use. Tezosentan competitively antagonizes the specific binding of 125I-labeled ET-1 and of the selective ETB receptor ligands 125I-labeled ET-3 and125I-labeled sarafotoxin S6c on cells and tissues carrying ETA and ETB receptors, with inhibitory constants in the nanomolar range, and has high water solubility. Tezosentan exhibits high functional inhibitory potency for inhibiting contraction induced by ET-1 on isolated rat aorta (ETAreceptors; pA2 = 9.5) and by sarafotoxin S6c on rat trachea (ETB receptors; pA2 = 7.7). In vivo, tezosentan inhibits the pressor effect of big ET-1 in pithed rats and increases ET-1 plasma concentrations in conscious rats in a dose-dependent fashion. In spontaneously hypertensive rats, i.v. injection of tezosentan has acute hemodynamic effects and decreases blood pressure. Tezosentan is also able to prevent the acute renal failure that complicates rhabdomyolysis in a rat model of myoglobinuric nephropathy. Finally, tezosentan exhibits an apparent elimination half-life of less than 1 h in rabbits and primates and of 2 h in rats. In conclusion, tezosentan, a potent mixed ET receptor antagonist with a short half-life, may offer a novel medical approach for the i.v. treatment of acute pathological conditions.
Endothelin (ET)-1 plays a major pathogenic role in acute pathological conditions such as acute heart failure and renal failure (Kaddoura and Poole-Wilson, 1996; Rabelink et al., 1996). ET antagonists show efficacy in animal models of radiocontrast renal injury, myoglobinuric nephropathy, and ischemic renal failure. Acute oral administration of bosentan, an orally active antagonist of both ETAand ETB receptors, is efficacious for decreasing mean arterial blood pressure in a rat model of heart failure secondary to myocardial infarction (Teerlink et al., 1994), and parenteral administration of bosentan is clinically efficacious in patients with severe decompensated heart failure (Kiowski et al., 1995). ET receptor antagonists therefore have a therapeutic potential in the acute treatment of emergency indications. However, the presently available ET receptor antagonists have not been optimized for this type of hemodynamic disturbances. Most have been selected for oral activity and have a prolonged half-life that does not allow them to reach a rapid plateau of efficacy. Tezosentan (Ro 61-0612) [5-isopropyl-pyridine-2-sulfonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2-1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide sodium salt, 1:2] (Fig. 1), a follow-up compound of bosentan (Clozel et al., 1994), is a new water-soluble ET receptor antagonist optimized to get high potency on both ETA and ETB receptors as well as high water solubility. Tezosentan is a weak diacid with pKa values of 4.4 and 4.1, corresponding to its isopropylpyridylsulfonamido and tetrazole functional groups, respectively. Its solubility reaches 14% at physiological pH values. It has a short half-life in various animal species.

Chemical structure of tezosentan disodium salt, C27H25N9Na2O6S (mw 649.6).
In the present report, we describe the general pharmacology of tezosentan, its pharmacokinetics in animals, and its profile in pathological models.
Materials and Methods
Cell Culture
Recombinant baculovirus-infected insect cells [Spodoptera frugiperda (Sf9)] and Chinese hamster ovary (CHO) cells expressing human ETA receptors were grown as previously described (Clozel et al., 1994; Breu et al., 1996). Sf9 cells were grown for 4 to 5 days in IPL-41 medium (Gibco BRL, Basel, Switzerland) supplemented with lipids and 1.5% FCS and subsequently infected with recombinant baculovirus at a multiplicity of infection of 1. Three days after infection, cells were harvested by centrifugation and frozen. CHO cells were grown in α-minimal essential medium supplemented with 0.1 μM methotrexate, 5% dialyzed FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Rat aortic endothelial cells were obtained by an explant technique modified from Cole et al. (1986). They were grown in RPMI 1640 supplemented with 20% FCS, 10 mM HEPES, 300 μg/ml endothelial cell growth supplement, 1 mM sodium pyruvate, and antibiotics (Clozel et al., 1993b).
Preparation of Membranes
Microsomal membranes were prepared from Sf9 cells in culture and from two tissues: human placenta and porcine trachea. Baculovirus-infected insect cells expressing recombinant human ETA receptors were broken by three freeze/thawing cycles in hypotonic 5 mM Tris buffer, pH 7.4, containing 1 mM MgCl2, resuspended in the same buffer with 250 mM sucrose, and stored in aliquots at −80°C. Membranes from human placenta and porcine trachea were prepared as described earlier (Fischli et al., 1989). Briefly, the tissues were homogenized in 5 mM Tris buffer, pH 7.4, containing 1 mM MgCl2 and 250 mM sucrose with a Polytron (Kinematica Ltd., Littau, Switzerland) and subsequently with a Potter homogenizer (Vetter Ltd., Ammerbuch, Germany). After centrifugation at 3000g for 15 min at 4°C, the supernatant was centrifuged again at 72,000g for 40 min. The resulting pellet was finally suspended in 2.5 ml of 75 mM Tris buffer, pH 7.4, containing 25 mM MgCl2 and 250 mM sucrose and stored frozen at −80°C. Protein content was determined according to the method of Lowry using BSA as a standard.
Binding Assays on Membranes
Suspensions of microsomal membranes were defrosted and centrifuged at 25,000g for 10 min. The pellet was resuspended at 22°C in 50 mM Tris buffer, pH 7.4, containing 25 mM MnCl2, 1 mM EDTA, and 0.5% (w/v) BSA. Then, 50 μl of this suspension containing 5 μg of recombinant Sf9 cells, 35 μg of placenta, or 30 μg of protein (porcine trachea) was used in a 250-μl assay containing the same buffer with 32 pM125I-labeled tracer (ET-1 for recombinant Sf9 cells and placenta and sarafotoxin S6c for porcine trachea) and increasing amounts of unlabeled tezosentan. After a 2-h incubation at 22°C, bound and free ligands were separated by filtration. Each assay was performed three times in triplicate, and nonspecific binding was assessed in the presence of 100 nM unlabeled ET-1 or sarafotoxin S6c.
Binding Assays on Attached Cells
Binding experiments were performed as previously described (Clozel et al., 1989). Briefly, recombinant CHO cells expressing ETA receptors and rat endothelial cells (∼105 cells/16-mm-diameter well) were washed three times and incubated at room temperature with 500 μl of binding medium (Dulbecco’s modified Eagle’s medium supplemented with 2 mg/ml BSA and 25 mM HEPES, pH 7.4) containing125I-labeled ET-1 (for CHO cells expressing ETA receptors) or125I-labeled ET-3 (for rat endothelial cells; ∼60,000 cpm; final concentration 36 pM) and various concentrations of tezosentan. After 2 h, the cells were extensively washed with binding medium and solubilized in 1% SDS with 0.5 M sodium hydroxide and 0.01 M EDTA at 37°C, and the radioactivity of bound125I-labeled ET-1 or125I-labeled ET-3 was measured (total binding). Nonspecific binding was determined simultaneously in the presence of 100 nM unlabeled ET-1 or 1 μM unlabeled ET-3, respectively. Maximal specific binding was calculated as total binding minus nonspecific binding. Specific binding represented 80 to 90% of total binding. The inhibitory constant of tezosentan (Ki) was calculated according to Cheng and Prusoff (1973). The Hill coefficient was calculated according to Weiland and Molinoff (1981).
Specificity Assay
The specificity of tezosentan as an ET receptor antagonist was assessed by measuring its ability to compete with various neurotransmitters, neuropeptides, growth factors, eicosanoids, and ions in 30 different ligand binding assays. Tezosentan was tested at 1 μM in duplicate (Cerep, Celle l’Evescault, France).
In Vitro Functional Inhibitory Potency
Isolated Rat Aortic Rings.
Male 14- to 16-week-old Wistar-Kyoto rats were anesthetized with sodium thiobutabarbital (Inactin, 100 mg/kg i.p.), and the thoracic aorta was removed and cut into 5-mm rings. The endothelium was removed by gentle rubbing of the intimal surface, and each ring was suspended in a 10-ml isolated organ chamber containing gassed 95% O2/5% CO2 and warmed (37°C) Krebs-Henseleit solution of the following composition: 115 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.5 mM KHPO4, 25 mM NaHCO3, 2.5 mM CaCl2, and 10 mM glucose. Isometric force was recorded. The rings were stretched to a resting force of 3g. After a 60-min equilibration period, the rings were contracted using norepinephrine (10−7 M). Endothelium denudation was assessed by the absence of relaxation to acetylcholine (10−5M). The rings were then washed and stretched if necessary until a stable baseline force was obtained. The rings were incubated with various concentrations (10−9 to 10−8 M) of tezosentan. After 20 min, cumulative doses of ET-1 were added, and the interval between doses was determined by the time required for the force generated to reach a plateau.
Isolated Rat Tracheal Rings.
Male 14- to 16-week-old Wistar-Kyoto rats were anesthetized with 100 mg/kg i.p. inactin, and the trachea was removed and cut into 5-mm rings. The epithelium was removed by gentle rubbing of the luminal surface, and each ring was suspended in a 10-ml isolated organ chamber containing gassed and warmed Krebs-Henseleit solution as described above. The rings were stretched to a resting force of 2g. After a 60-min equilibration period, the rings were contracted using potassium chloride (50 mM). The rings were then washed and stretched if necessary until a stable baseline force was obtained. After a 20-min incubation with tezosentan (10−7 to 10−6 M), cumulative doses of sarafotoxin S6c were added. The interval between doses was determined by the time required for the force generated to reach a plateau.
Analysis and Calculations.
The maximum force was defined as the force generated with the highest concentration yielding a maximal effect, and from this the ET-1 or sarafotoxin S6c concentration yielding a half-maximal effect (EC50) was calculated. Contractile responses are expressed as absolute tension (aorta), percentage of contraction to potassium chloride (trachea), and as percentage of the maximal response. The pA2value (negative logarithm of the molar concentration of antagonist that causes a 2-fold parallel shift to the right of the agonist concentration-response curve) as an index of functional inhibitory potency was determined for each individual curve by the equation pA2 = log(concentration ratio − 1) − log[B], where concentration ratio is the ratio of EC50 values with or without antagonist, and [B] is the concentration of antagonist. Regression analysis of the plot log(concentration ratio − 1) against log[B] (Schild plot) allowed us to confirm the competitive nature of the antagonist by assessing its slope (Arunlakshana and Schild, 1997).
In Vivo Functional Inhibitory Potency
Inhibition of Pressor Effect of Big ET-1.
Male Wistar rats (340–360 g) were anesthetized with sodium hexobarbital (Evipan, 150 mg/kg i.p.). After tracheal intubation, the rats were pithed with a steel rod and artificially ventilated with room air using a rodent ventilator (model 683; Harvard Apparatus, South Natick, MA) at a tidal volume of 2 ml and a rate of 65 strokes/min. The animals were kept warm at 38°C. The femoral artery and vein were cannulated for blood pressure measurement and i.v. injection of drugs, respectively. After stabilization of blood pressure, various doses of tezosentan or saline (1 ml/kg) were injected. At 5 min later, the first dose of big ET-1 was injected i.v. in saline containing 0.1% BSA (0.5 ml/kg). Increasing doses were injected in a cumulative manner, with each dose being given after stabilization of the effect of the previous dose on blood pressure.
Effects on ET-1 Levels in Conscious Wistar Rats.
Male Wistar rats were anesthetized with Evipan (230 mg/kg i.p.) and instrumented with catheters in the left carotid artery and jugular vein for drug injection and blood sampling, respectively. After complete recovery, saline or tezosentan (1 or 10 mg/kg i.v. bolus) was injected in the conscious rats. Samples for measurement of plasma concentration of ET-1 were withdrawn before and at various times after drug injection. Blood was drawn into heparinized tubes and chilled immediately. Plasma was separated by centrifugation and stored at −20°C until use. Plasma immunoreactive ET-1 concentration was measured by radioimmunoassay as previously described (Löffler and Maire, 1994). Briefly, duplicates of 400 μl of plasma were extracted on Sep-Pak Vac C18 cartridges (Waters Associates) after previous conditioning with 2 ml of methanol, followed by 2 ml of 0.2 M phosphate-citric acid, pH 7. Cartridges were eluted with 2 ml of methanol/water (90:10, v/v). The eluates were dried and reconstituted in assay buffer [20 mM borate-HCl, 0.1% (w/v) BSA, 0.1% (w/v) NaN3, pH 7.4]. Radioimmunoassay was then performed using a rabbit anti-ET-1 antiserum (RAS-6901; Peninsula Laboratories, Merseyside, UK). Cross-reactivity with ET-3 and big ET-1 was 14 and 6%, respectively. Free and bound tracers were separated by adsorption at 25°C for 15 min on 250-μl Amerlex-M magnetobeads (Amersham, Zurich, Switzerland) supplemented with 0.1% (w/v) Tween 20.
Comparison with ETA-Selective Compounds in Spontaneously Hypertensive Rats (SHR)
To evaluate the maximal efficacy of the combined ETA/ETB antagonist tezosentan compared with that of ETA-selective antagonists and understand more of the contribution of ETA and ETB receptors in the control of blood pressure in hypertension, dose-response curves of the mixed antagonists tezosentan and bosentan and of the ETA-selective antagonists BQ-123 (a cyclic pentapeptide; Ihara et al., 1992) and BMS182′874 (a nonpeptide small-molecular weight antagonist; Webb et al., 1995) were performed in anesthetized SHR. Male SHR were anesthetized with 100 mg/kg i.p. inactin. The femoral artery (for blood pressure monitoring) and vein (for injection) were cannulated. After stabilization of blood pressure, increasing doses of tezosentan, bosentan, BQ-123, or BMS182′874 were injected, with each dose being given after stabilization of the effect of the previous dose on blood pressure, until a plateau of blood pressure was obtained. Drugs were compared for their maximal efficacy at decreasing blood pressure.
Effects of Tezosentan in Myoglobinuric Acute Renal Failure in Rats
Rhabdomyolysis or other causes of massive myoglobin release are often complicated by acute renal failure. It was recently shown that ET-1 plays a major role in this complication (Karam et al., 1995). A pseudocrush syndrome was simulated by injection of i.m. glycerol as described previously (Karam et al., 1995). A control group did not receive glycerol and was used as a reference. Tezosentan or bosentan for comparison or saline as control was injected as two bolus i.v. doses of 10 mg/kg 1 h and 20 min before glycerol. Rats were allowed to recover for 2 h and then were placed in individual metabolic cages for 48 h. Blood samples withdrawn from a catheter placed in the abdominal aorta and urine free of food and feces were collected at 24 and 48 h. Plasma and urinary creatinine levels were measured with a centrifugal analyzer (Roche-Cobas Fara II; F. Hoffmann-LaRoche Ltd., Basel, Switzerland). Renal function was assessed by calculating creatinine clearance at 24 and 48 h after glycerol administration.
Single-Dose Pharmacokinetics
Single-dose pharmacokinetic studies with tezosentan were performed in rats, rabbits, and cynomolgus and rhesus monkeys after i.v. bolus doses of 5 to 10 mg/kg. Tezosentan was quantified in plasma using HPLC assay after protein precipitation with methanol. Analysis was performed on silica gel plates using ethyl acetate/methanol/water/diethylamine as eluent, followed by postchromatographic fluorescence enhancement by immersion in Triton X-100 and scanning of the plates with a densitometer in fluorescence mode. Quantification was based on external standards using peak heights. The quantification limit was 0.1 μg/ml with 0.1 ml of plasma.
Expression of Results
Results are expressed as mean ± S.E. ANOVA for repeated measures and Dunnett’s test were used to assess the effect of tezosentan on dose-response curves of big ET-1, on ET-1 plasma concentrations, and on creatinine clearance in renal failure rats. The comparison of the different drugs for their effects on blood pressure in SHR were assessed using Student’s t test. A value ofP < .05 was considered significant.
Drugs
The 125I-labeled ET-1 and ET-3, and [125I-His]sarafotoxin S6c were obtained from Anawa Trading SA (Wangen, Switzerland). ET-1, big ET-1, and sarafotoxin S6c were obtained from Peninsula Laboratories. They were dissolved in methanol/water (50:50) for in vitro studies or saline plus 0.1% BSA for in vivo studies. Dilutions were always performed in solutions containing 0.1% BSA. Tezosentan (Actelion Ltd.) was synthesized at F. Hoffmann-La Roche Ltd. and was dissolved in water immediately before use. Norepinephrine hydrochloride and potassium chloride were obtained from Fluka Chemical (Buchs, Switzerland), and acetylcholine hydrochloride was obtained from Sigma Chemical Co. (St. Louis, MO). Culture reagents were from Gibco Laboratories (Paisley, Scotland).
Results
Binding Affinity and Specificity.
Affinity of tezosentan for the ET receptors was assessed in different cells and tissues (Table1). Tezosentan inhibited the specific125I-labeled ET-1 binding to ETA receptors with an inhibitory potency (Ki) of 0.3 nM on CHO cells and of 18 nM on membranes of baculovirus-infected insect cells (Table 1). Similarly, tezosentan inhibited the specific binding of125I-labeled ET-1, ET-3, or sarafotoxin S6c to ETB receptors with an inhibitory affinity of 10 to 21 nM (Table 1). Tezosentan up to a concentration of 1 μM did not exhibit any binding inhibitory activity in 27 radioligand binding assays different from ET binding. On H1 central, 5-hydroxytryptamine2A, and vasopressin V1 receptors, tezosentan (1 μM) induced a weak inhibition of less than 20%.
Binding inhibitory potency (Ki) and Hill coefficient of tezosentan on the two ET receptor subtypes
In Vitro Functional Inhibition.
In isolated endothelium-denuded rat aortic rings (ETAreceptors) and in epithelium-denuded rat tracheal rings (ETB receptors), tezosentan produced concentration-dependent, parallel shifts to the right of the ET-1 and sarafotoxin S6c concentration-response curves, respectively (Fig.2), with no agonistic effect and without any significant change in the maximal responses. On aortic rings, maximal contraction to ET-1 was 4.6 ± 0.3, 5.6 ± 0.4, 4.8 ± 0.2, and 5.4 ± 0.2g in the absence and in presence of 10−9, 3 × 10−9, and 10−8M tezosentan, respectively. On tracheal rings, maximal contraction to sarafotoxin S6c was 121 ± 6, 117 ± 16, 83 ± 9, and 123 ± 6% of potassium chloride contraction in the absence and in presence of 10−7, 3 × 10−7, and 10−6M tezosentan, respectively. Schild analysis yielded a pA2 value of 9.5 ± 0.3 (slope = 1.2 ± 0.2, n = 19) on rat aortic rings (ETA receptors) and of 7.7 ± 0.2 (slope = 1.1 ± 0.1, n = 15) on rat tracheal rings (ETB receptors). Both slopes did not significantly differ from unity, suggesting that tezosentan behaves as a competitive antagonist on both ETA and ETB receptors.
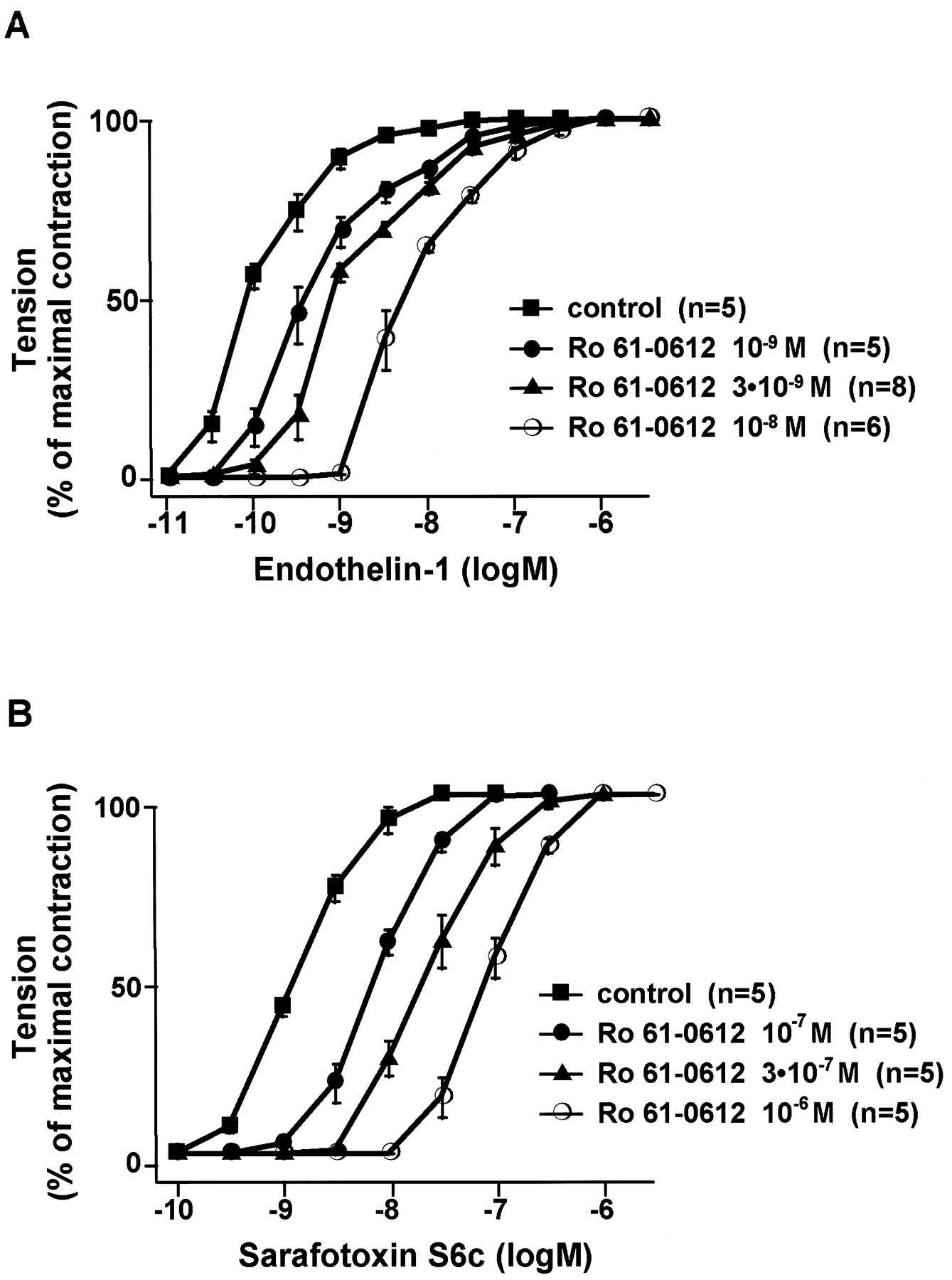
A, concentration-response curve of ET-1 on isolated rat aortic rings without endothelium in presence or in absence of tezosentan. B, concentration-response curve of sarafotoxin S6c on isolated rat tracheal rings without epithelium in presence or absence of tezosentan.
In Vivo Inhibition of Big ET-1 Effects.
In pithed Wistar rats, tezosentan dose-dependently inhibited the pressor effect of big ET-1 (P < .001 at all doses; Fig.3). At the lowest dose tested of 1 mg/kg, tezosentan inhibited the pressor effect of the various doses of big ET-1 by 50 to 80%. Tezosentan had no effect by itself on blood pressure in these pithed rats.

Effect of tezosentan on the pressor effect of big ET-1 in pithed rats. ***P < .001 compared with control.
Effects of Tezosentan on ET-1 Levels.
Tezosentan dose-dependently increased ET-1 plasma concentrations after a bolus administration in conscious rats (Fig.4). The increase was 2.6-fold at a dose of 1 mg/kg and 8.6-fold at a dose of 10 mg/kg (P < .001 at both doses).

Effect of tezosentan on ET-1 plasma concentration in conscious Wistar rats. ***P < .001 compared with control (saline).
Comparison with ETA-Selective Compounds.
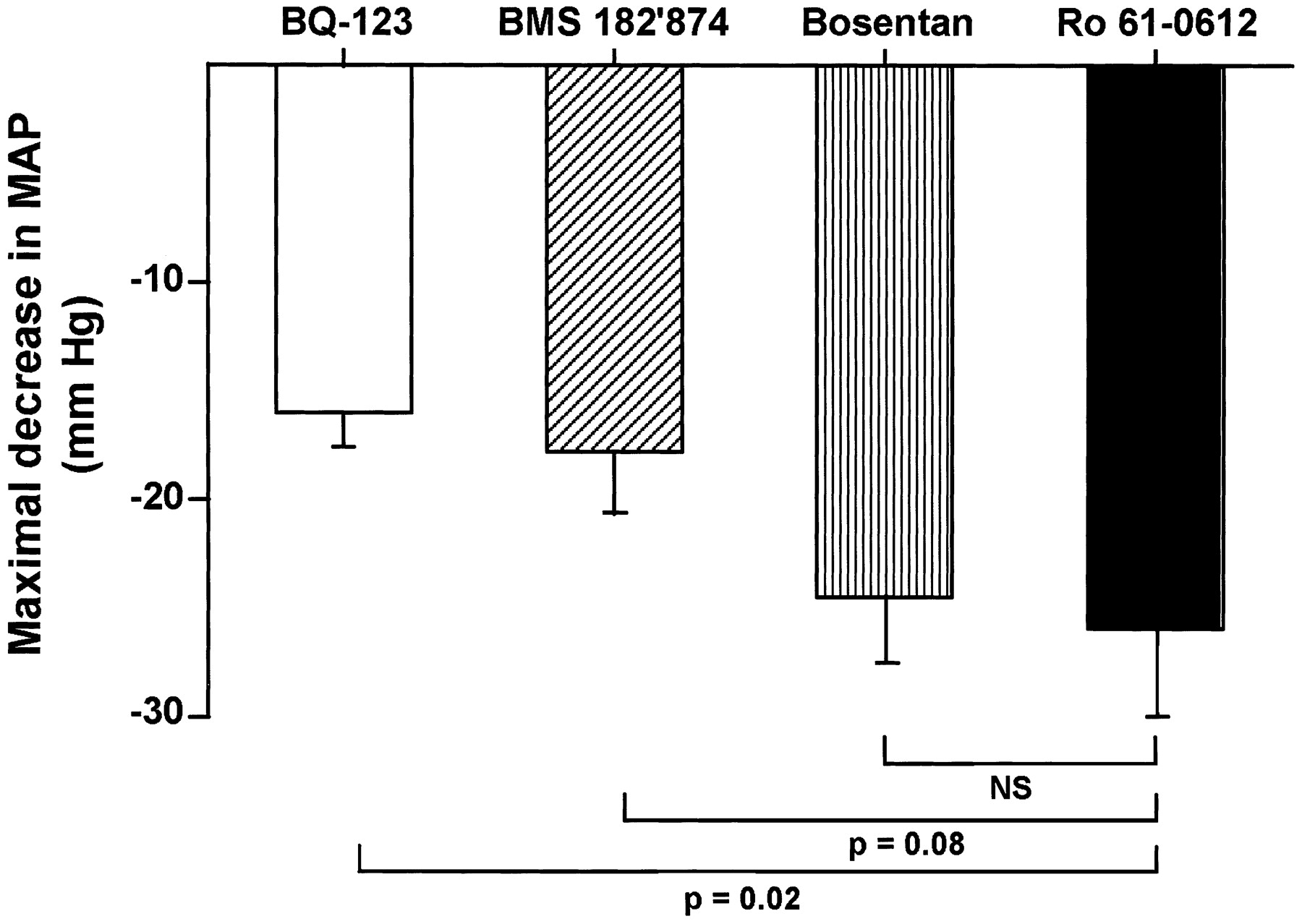
All compounds induced dose-dependent decreases in blood pressure in anesthetized SHR. Maximal efficacy was reached at a dose of 10 mg/kg for all four compounds. At maximal efficacy, the blood pressure lowering induced by tezosentan was 25 mm Hg, similar to that of bosentan (Fig. 5). In comparison to tezosentan, the maximal effect of BMS182,874 and BQ-123 was less (P = .08 and .02, respectively). BQ-123 and BMS182,874 decreased blood pressure by a maximum of 17 mm Hg.

Maximal effect of tezosentan, bosentan, BQ-123, and BMS 182′874 (all at 10 mg/kg i.v.) on blood pressure in anesthetized spontaneously hypertensive rats.
Effects of Tezosentan in Myoglobinuric Acute Renal Failure in Rats.
After 24 h, glycerol injection alone induced a 43% decrease in creatinine clearance compared with the control group that did not receive glycerol. At 24 and 48 h, both bosentan and tezosentan almost completely prevented the decrease in creatinine clearance (Fig. 6).

Effects of tezosentan and bosentan (10 mg/kg i.v. 1 h and 20 min before glycerol injection) on creatinine clearance in a rat model of crush syndrome (glycerol-induced rhabdomyolysis). *P < .05, **P < .01 versus untreated glycerol group.
Pharmacokinetics.
The main pharmacokinetic characteristics of tezosentan are shown in Table 2. Tezosentan exhibited relatively low systemic clearance in rats but not in rabbits and primates, where systemic plasma clearance was high and approximated liver blood flow. The volume of distribution corresponded to the distribution volume of serum albumin in both primate species and to the extracellular space in rats and rabbits. The apparent half-life for the elimination of tezosentan was shorter than 1 h in rabbits and in both primate species but was longer in rats.
Pharmacokinetic variables of tezosentan after i.v. dosing in different species
Discussion
Tezosentan is a potent, specific, and highly water-soluble ET receptor antagonist. It binds with a high affinity to human ETA receptors. The lower inhibitory potency observed for the human ETA receptors expressed in baculovirus-infected insect cells compared with CHO cells is likely to be due to a different glycosylation pattern, a modified receptor conformation due to the marked overexpression, or an abnormal G protein coupling of the receptor in this expression system. The high-affinity binding of tezosentan on ETA receptors is also confirmed in functional assays. Tezosentan also has a high affinity for ETB receptors and is about 30-fold more potent on ETA receptors than on ETBreceptors. It is a competitive antagonist, and therefore its inhibitory activity will depend on the agonist concentration. It is a very specific antagonist for ET receptors.
The in vivo pharmacological properties of tezosentan have been evaluated by its inhibition of the pressor effect of big ET-1 and by the increase in ET-1 concentrations in rats. As opposed to ET-1, which causes both a depressor and a pressor effect when injected i.v., big ET-1 induces almost exclusively a pressor effect. We have suggested that i.v. injection of big ET-1 may represent a much more physiological tool for pharmacological evaluation of antagonists than i.v. injection of mature ET-1 (Clozel et al., 1993a). Indeed, big ET-1 is the inactive precursor of ET-1 and can exert its pressor effect only after enzymatic processing to ET-1 in the vascular wall (Corder and Vane, 1995;Teerlink et al., 1995). Therefore, big ET-1 injection mimics much better than ET-1 injection the abluminal release of ET-1 from endothelial cells toward smooth muscle cells and the tissular processing of ET-1. The increase in ET-1 that follows the administration of ET receptor antagonists is most likely due to the prevention of binding of ET-1 to its receptors, particularly to the ETB receptors (Löffler et al., 1993). For both tests, tezosentan behaved as a potent antagonist. Indeed, a dose of 1 mg/kg inhibited by more than 50% the pressor effect of big ET-1 and increased by 3-fold ET-1 levels in plasma.
In SHR, the blood pressure-lowering effect of tezosentan tended to be superior at its maximal efficacy to that of two ETA-selective antagonists. Both ETA and ETB receptors may mediate the pathophysiological role of ET-1 (Seo et al., 1994; Seo and Lüscher, 1995; McCulloch et al., 1996), in particular its pressor effect. There appears to exist cross-talk between ETA and ETB receptors, allowing one receptor to compensate for the other if only one receptor is blocked, suggesting that combined blockade may be necessary in certain situations (Clozel and Gray, 1995; Fukuroda et al., 1996;Vitola et al., 1996; Mickley et al., 1997). Therefore, mixed ETA/ETB receptor antagonists may have specific therapeutic advantages over receptor-selective blockers, as, for example, in experimental hypertension. However, further studies will be needed to evaluate and compare the chronic efficacy of combined and selective receptor antagonists and their efficacy in various pathological models other than hypertension.
Tezosentan was very effective in a rat model of acute renal failure. ET antagonists have been shown to prevent the vasoconstriction and the renal failure that follow acute renal ischemia in rats (Shibouta et al., 1990; Gellai et al., 1994; Kusumoto et al., 1994; Hunley and Kon, 1997; Birck et al., 1998). We showed previously that ET is also a major mediator of ischemia, tubular necrosis, and renal failure in myoglobinuric nephropathy (Karam et al., 1995), as confirmed recently (Shimizu et al., 1998). Tezosentan showed an efficacy similar to that of bosentan in acute renal failure complicating rhabdomyolysis.
In conclusion, tezosentan is a novel mixed ET receptor antagonist that represents an important new research tool for blocking both ET receptors. Tezosentan is optimally suited for i.v. use because of its solubility and potency. Its short half-life should allow a plateau of effect to be reached rapidly and the dosage to be tuned easily. If the present preclinical data are confirmed in humans, it has the potential of being an innovative drug in a new therapeutic field for treating acute hemodynamic conditions.
Acknowledgments
We thank Martine Hug, Hans Gloor, Benoı̂t Lack, Brigitte Butscha, and Rolf Osterwalder for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Martine Clozel, Actelion Ltd., Innovation Center, Gewerbestrasse 16, CH-4123 Allschwil, Switzerland. E-mail: martine.clozel{at}actelion.com
- Abbreviations:
- ET
- endothelin
- CHO
- Chinese hamster ovary
- SHR
- spontaneously hypertensive rats
- Received October 5, 1998.
- Accepted April 26, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}