Article Text

Abstract
In normal conditions, human gut mucosa is infiltrated with a large number of mononuclear cells. This is a reflection of the fact that human intestine is continuously subjected to a massive stimulation by luminal antigens. This state of “physiological” inflammation is a tightly controlled phenomenon, as several mucosal cells interact to generate and maintain an appropriate local immune response. Changes in cell type number and/or function, including the release of soluble mediators, have been associated with the development of chronic inflammatory diseases, such as Crohn's disease (CD) and ulcerative colitis (UC), the two major forms of inflammatory bowel disease. Evidence also indicates that the type of inflammatory response occurring in the intestine of patients with CD differs from that in UC, and this probably reflects distinct pathways of immune activation. In CD mucosa, a Th1 response with high IL-12 and IFNγ production prevails, while in UC a humoral immunity appears to be predominant. Despite this, CD and UC share downstream inflammatory events, characterised by high levels of inflammatory cytokines, free radicals, matrix-degrading enzymes and growth factors.
- immunoregulation
- inflammatory bowel disease
- Crohn's disease
- ulcerative colitis
- APC, antigen presenting cell
- CD, Crohn's disease
- IBD, inflammatory bowel disease
- IFN, interferon
- Ig, immunoglobulin
- IL, interleukin
- KGF, keratinocyte growth factor
- LPL, lamina propria lymphocyte
- LPMC, lamina propria mononuclear cell
- MMP, metalloproteinase
- NF, nuclear factor
- PP, Peyer's patches
- PWM, pokeweed mitogen
- SEB, staphylococcal enterotoxin B
- TGF, tumour growth factor
- TIMP, tissue inhibitor of metalloproteinase
- T-LPL, lamina propria T lymphocyte
- TNBS, trinitrobenzene sulphonic acid
- UC, ulcerative colitis
Statistics from Altmetric.com
- APC, antigen presenting cell
- CD, Crohn's disease
- IBD, inflammatory bowel disease
- IFN, interferon
- Ig, immunoglobulin
- IL, interleukin
- KGF, keratinocyte growth factor
- LPL, lamina propria lymphocyte
- LPMC, lamina propria mononuclear cell
- MMP, metalloproteinase
- NF, nuclear factor
- PP, Peyer's patches
- PWM, pokeweed mitogen
- SEB, staphylococcal enterotoxin B
- TGF, tumour growth factor
- TIMP, tissue inhibitor of metalloproteinase
- T-LPL, lamina propria T lymphocyte
- TNBS, trinitrobenzene sulphonic acid
- UC, ulcerative colitis
A continuous, low grade or “physiological” inflammation is a characteristic feature of the gastrointestinal tract. This is a reflection of the fact that the intestine has a large surface area (400 m2), which is continuously exposed to dietary antigens and microorganisms.1 T cells normally constitute one-third of the cells in the intestinal lamina propria, and the phenotypic distribution of CD4+ and CD8+ T cells is similar to that of peripheral blood lymphocytes, with a preponderance of CD4+ and αβTcR+ cells.1 The majority of CD4+ T-lamina propria lymphocytes (LPL) are L selectin negative, and 66–96% of them express the CD45RO isoform of the CD45 molecule associated with memory-like T cells.1 As result of their state of activation, T-LPLs produce high levels of cytokines. Freshly isolated human T-LPL contain a very high frequency of cytokine-secreting cells compared to autologous blood T cells, and the response is dominated by interferon (IFN)-γ. There are about 10-fold fewer cells spontaneously secreting interleukin 4 (IL-4), whereas IL-5-secreting cells are uncommon.2 Stimulation of T-LPL via the alternative pathways of activation (for example CD2 and CD28) elicits a strong cytokine response, which is again dominated by IFN-γ_.2 The reason why T-LPL should respond to cell activation with an enhanced synthesis of Th1 cytokines remains unclear, given that Th1-inducing cytokines (for example IL-12, IL-18, IFN-α) are barely detectable in the intestinal lamina propria of normal subjects.3 The explanation more likely is that this type of response is due to T cells derived from the Peyer' s patches (PP), where bacterial antigens, transported promiscuously across the epithelium by M cells into the dome region, stimulate IL-12 production and Th1 cell polarisation.4 Indeed, the majority of T-LPLs expresses α4β7, an integrin that interacting with the gut specific vascular addressin MadCAM-1, allows the PP derived T cells to migrate across the endothelium into the lamina propria.5 Despite the fact that T-LPLs are persistently stimulated, the gut has evolved effector mechanisms, which specifically prevent excessive inflammation and tissue injury. First, the reactivity of T-LPL to TCR/CD3 engagement is reduced when compared with peripheral blood T lymphocytes. This state of hyporesponsiveness has been associated with the local production of suppressive molecules (for example IL-10, TGFβ1, prostaglandin2), insufficient delivery of co-stimulatory signals, and synthesis of low molecular weight non-protein mediators with oxidative capacities.6 Second, it is known that T-LPLs undergo immunologically induced programmed cell death (apoptosis) and are thus eliminated before they can mediate a deleterious immune response. T-LPLs express the apoptotic molecule FAS and a subpopulation of them is FAS ligand positive. They exhibit an increased level of apoptosis in comparison to PBL, and respond to CD2 stimulation with enhanced FAS dependent apoptosis.7, 8 Finally, there is evidence that intestinal cells produce high levels of TGF-β1 and IL-10, which are able to counter-regulate detrimental immune responses in the mucosal environment. This effect is at least in part dependent on the ability of TGF-β1 and IL-10 to suppress the expression of co-stimulatory molecules on resident antigen presenting cells9, 10 (fig 1).
Crohn's disease (CD) and ulcerative colitis (UC), the major forms of inflammatory bowel disease (IBD) in humans, are distinguished by a complex chronic inflammatory process. Growing evidence indicates the role of a genetically determined disregulation of the host immune response towards the resident bacterial flora in the pathogenesis of human IBD.11
In human IBD tissue, CD4+ T cells represent the vast majority of activated mononuclear cells infiltrating the gut. A large proportion of T cells bear the phenotypic characteristics of circulating naive lymphocytes, and they are recruited from the blood into the intestinal mucosa, probably as a result of an enhanced expression of adhesion molecules and chemokines in the inflamed gut of IBD patients.11 Evidences indicate that CD4+ T cells play a key role in the pathogenesis of tissue damage in IBD, especially in CD.11–13 CD is indeed associated with non-caseating granuloma, the hallmark of cell mediated immunity. The role of T cells in the pathogenesis of IBD is supported by clinical and pathophysiological observations, showing that stable remission is observed in CD patients who develop symptomatic HIV infection or undergo bone marrow transplantation.12–14
The factors/mechanisms leading to this uncontrolled T cell activation are not fully clarified. Loss of tolerance towards the resident bacterial flora appears an important factor and it is believed that counter regulatory molecules (TGFβ1, IL-10) involved in maintaining tolerance towards the resident flora may play a role in regulating mucosal T cell activation (fig 2).15, 16 Consistently, it has recently been shown that TGFβ1 activity is defective in IBD mucosa because of enhanced production of Smad7, an inhibitor of TGFβ1 signal transduction.17
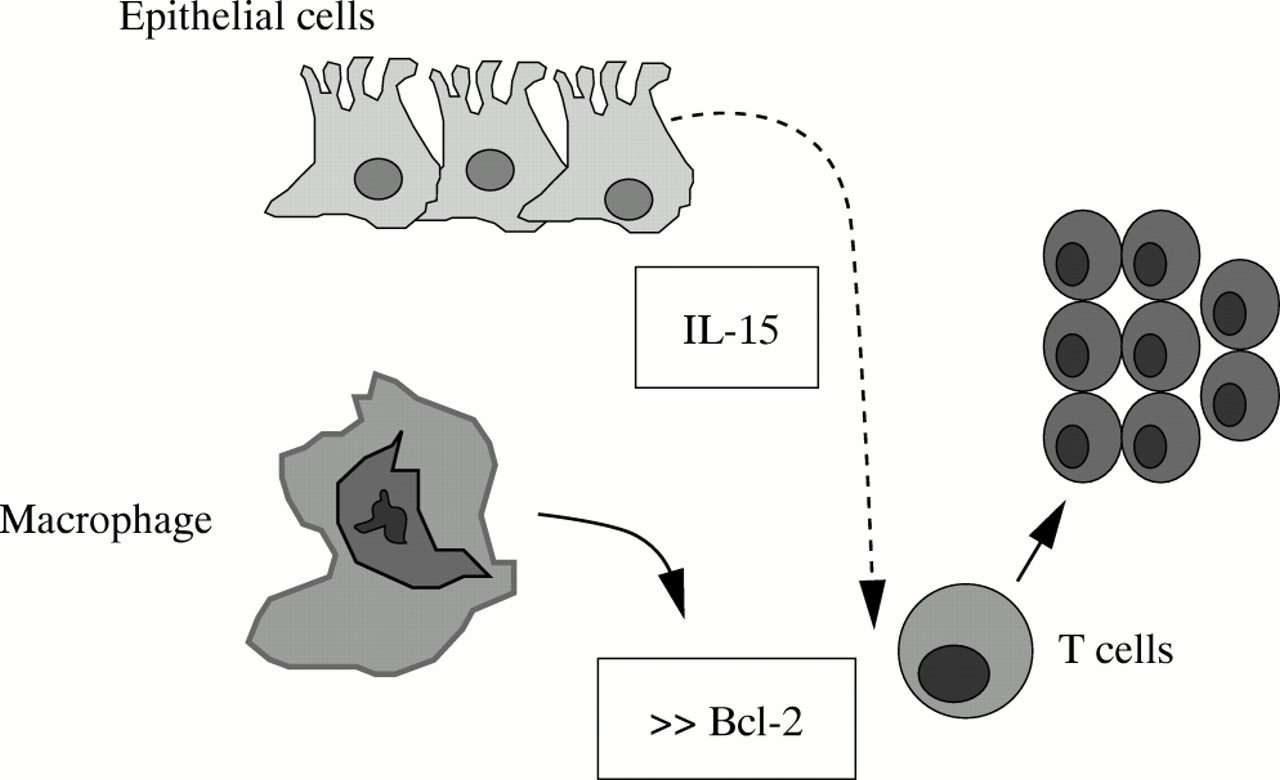
Furthermore, it has been widely shown that CD mucosal T cells display a defective apoptosis to several stimuli, associated with an increased ratio between the antiapoptotic protein Bcl-2 and proapoptotic molecule Bax.18–20 There is also evidence that activation of the transcription factor STAT3 by IL-6/IL-6R interaction contributes to rescue resident T cells from apoptosis, extending the life span and eventually favouring T cell accumulation.20, 21 Consistent with these observations, administration of a neutralising antibody against IL-6 receptor enhances apoptosis of intestinal lamina propria T cells and suppresses established colitis in various animal models of CD.20–22 It is likely, however, that the resistance of CD intestinal T cells to apoptosis may depend on other factors released within the inflamed tissue. Indeed, in the inflamed intestine of these patients there is enhanced production of IL-15, a cytokine which, by signalling through the common γ chain cytokine receptor, enhances expression of Bcl-2 (fig 3).23
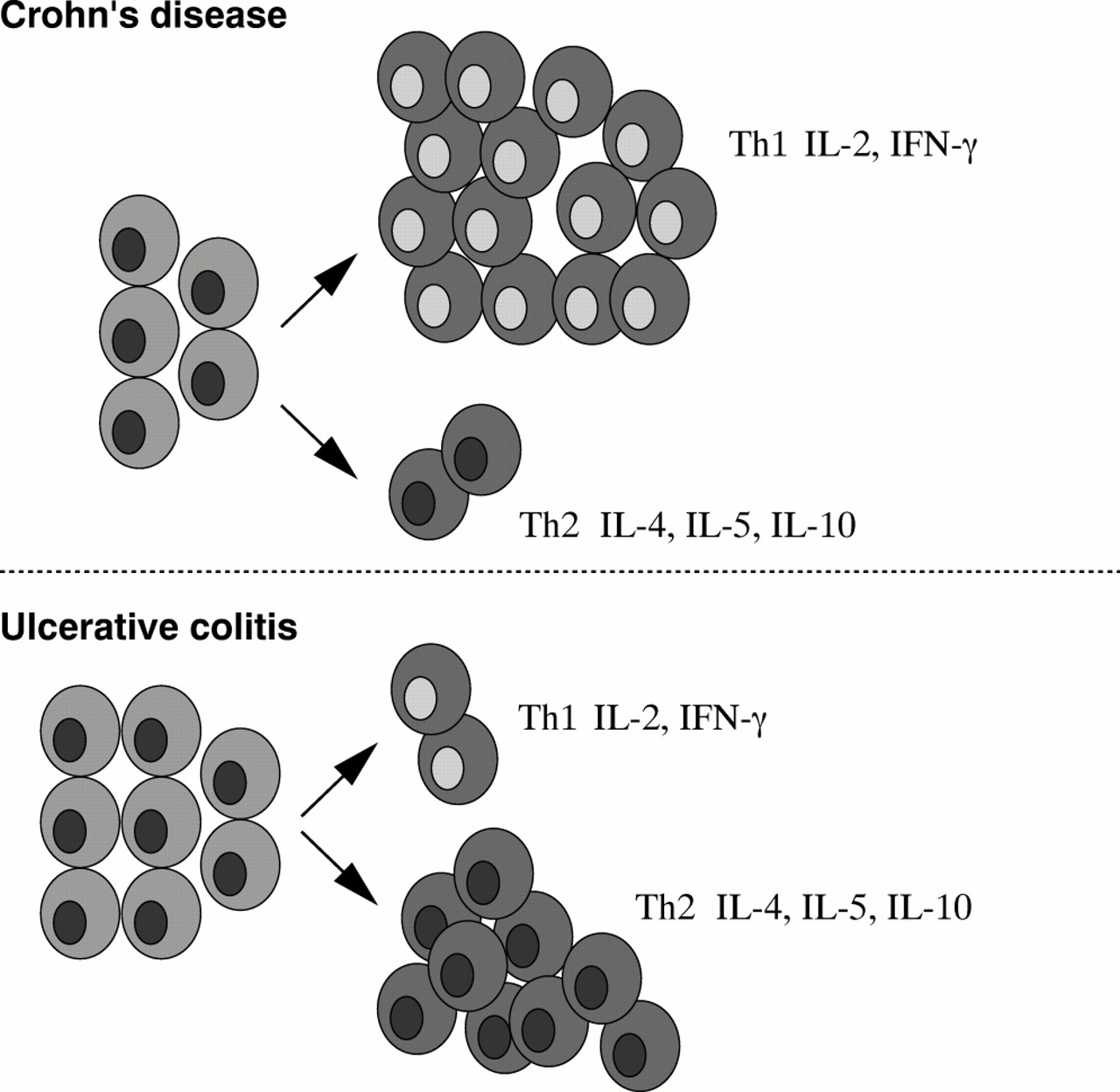
As a result of their state of activation, T cells synthesise large amounts of cytokines, which directly or indirectly contribute to the inflammatory process. It has been shown that in CD the cytokine profile is polarised towards the Th1 type, and that Th1 cells (for example, IFNγ producing cells) are essential mediators of the tissue damaging intestinal inflammation (fig 2). Lamina propria T lymphocytes (T-LPLs) isolated from the inflamed intestinal mucosa of CD patients contain increased amounts of IFNγ mRNA and spontaneously release higher levels of IFNγ than controls in vitro.2, 24
There is only one study showing that IL-4 transcripts are elevated in the early lesions of recurrent CD.25 This observation was not confirmed and may well reflect a transient non-specific response, as the biopsies were performed in patients with relatively non-specific gross changes (that is, the edge of apthoid ulcers).
Some immunological phenomena observed in CD inflamed tissue can be related to the effects of Th1 cytokines. These include increased epithelial expression of MHC class II antigens and CD68, enhancement of ICAM-1 expression on both endothelium and mononuclear cells, and the presence of multinucleated giant cells. IFNγ can also facilitate the activation of resident macrophages and the release of proinflammatory cytokines, such as IL-1, IL-6, and TNFα, which in turn maintain and increase the local inflammatory response.12
The characteristic cytokine products of Th1 and Th2 cells are mutually inhibitory for the differentiation and effector functions of the reciprocal phenotype. The differentiation of Th2 cytokine producing cells (for example, IL-4, IL-5, and IL-13 secreting cells) is inhibited by IFNγ, whereas activation of Th1 cells (for example, IFNγ secreting cells) is blocked by IL-10.26 Furthermore, other factors, such as the nature of the antigen and environmental stimuli to which the cells are exposed, influence the differentiation of Th1 or Th2 cell subtype. Intracellular bacteria, which activate macrophages, promote Th1 cell differentiation by stimulating the synthesis of IL-12, the major Th1 inducing cytokine. In contrast, the differentiation of Th2 cells requires IL-4, produced by basophils and mast cells, usually in response to allergens and extracellular microbes.26 This is consistent with the demonstration that, in CD tissue, IFNγ production is associated with expression of IL-12.27 The functionally active IL-12 heterodimer is released by lamina propria mononuclear cells (LPMC) isolated from inflamed mucosal areas of CD patients; its neutralisation by a specific antibody results in a dramatic decrease in the number of IFNγ producing cells.27, 28 CD T-LPL also exhibit high levels of the IL-12Rβ2 chain, the signalling component of the IL-12 receptor, and active STAT4, a transcription factor essential in the IL-12 dependent Th1 polarisation, in comparison to those from UC patients and normal controls.29 Despite the fact that IL-12 has the ability to sustain Th1 immunity, it is likely that the IL-12 induced Th1 cell polarisation may be expanded by other cytokines produced within the CD mucosa, such as IL-7, IL-15, and IL-18 (fig 4).30–33
Several studies in animal models of IBD support the role of the IL-12/STAT-4 signalling pathway in Th1 cell mediated injury in the gut. Indeed, STAT4 deficient T cells manifest impaired IFNγ production in response to IL-12, and are unable to promote the development of colitis when transferred in immunodeficient mice.34 On the other hand, it has been reported that overexpression of STAT-4 proteins in transgenic mice results in the induction of colitis, which is characterised by the presence of a diffuse infiltration of IFNγ and TNFα secreting cells in the intestinal wall.35 Finally, studies conducted in murine 2,4,6-trinitrobenzene sulphonic acid (TNBS) induced colitis (which mimics some characteristics of CD), showed that neutralising IL-12 antibodies prevent the onset of colitis when given at the time of TNBS administration, or treat the mucosal inflammation if given after colitis is established.21, 36
The type of pathological changes (for example, epithelial damage), the occurrence of multiple (auto)antibodies, and data from several immunopathological studies suggest that the mucosal immune response in UC differs from that occurring in CD. In UC, an enhanced humoral immunity appears to predominate. Mucosal plasma cells isolated from the intestine of UC patients produce high levels of immunoglobulins, particularly IgG1.37 In addition, a large variety of autoantibodies (anticolon and antineutrophil antibodies) may be detected in the serum of these patients.38, 39 These autoantibodies are not tissue specific and seldom correlate with clinical variables of the disease, suggesting that they have no pathogenic role. For some of these autoantibodies, the potential target has been identified. For example, the putative antigen of anticolon antibodies is a protein of 40 kilodalton size that is exclusively recognised by IgG eluted from the inflamed colon of UC patients.39 This autoantigen appears to be fraction 5 of the tropomyosin family of cytoskeletal proteins and is codeposited with IgG1 and complement on UC colonocytes.40, 41 Antibodies directed against this protein also identify epitopes expressed in human biliary epithelium, eye, skin, and joints, locations compatible with the extraintestinal manifestations of UC.42 No study has, however, shown that these autoantibodies can cause intestinal damage. The profile of regulatory T cell cytokines produced in UC is consistent with the findings mentioned above. Indeed, expression of IFNγ is lower than that in CD and not different from that in normal controls.2 UC T lymphocytes release high levels of IL-5 after stimulation via the CD3/CD28 pathway, and this cytokine may be relevant in helping antibody response.2 However, despite enhanced IL-5, the same cells still produce 50 times as much IFNγ. The idea that a Th2 type response is prevalent in UC remains unfounded (fig 5). In agreement with the profile of cytokines released by T-LPL, the production of IL-12 is rare or undetectable in the intestine of UC patients.27 The reasons why this occurs remain unclear, given that in UC mucosa there is a massive infiltration of activated macrophages.
In both CD and UC, an increased mucosal synthesis of proinflammatory cytokines, including IL-1β, IL-6, IL-8, IL-16, and TNFα characterises the excessive local immune response.11, 43, 44 The elements that play a key role in control of synthesis of these inflammatory cytokines remain unclear. Important candidates may be transcription factors that bind to the gene promoter regions. Among these, nuclear factor κB (NFκB) appears to be a regulator of the inducible expression of many cytokine genes (for example, IL-1, IL-6, IL-8, TNFα) in lymphocytes, monocytes, and epithelial cells in the gut, and its activity has been reported to be enhanced in the mucosa of patients with active IBD.45 Furthermore, a wide variety of inflammatory cytokines, including IL-1, IL-18, and TNF-α, extensively upregulate NFκB. This could thus provide a positive feedback mechanism, which is able to perpetuate the inflammatory process in the gut. Furthermore, bacterial product signalling through members of a family of transmembrane receptors, which are designed Toll like receptors (TLRs), may activate NFκB. TLR4 is abundantly expressed by epithelial and lamina propria mononuclear cells in IBD mucosa.46 As TLR4 serves as a major transducing subunit of the LPS receptor complex, it is conceivable that increased LPS recognition and signalling may contribute to the enhanced NFκB activity and subsequent synthesis of inflammatory cytokines in IBD.
Among inflammatory cytokines TNFα has a broad spectrum of biological effects of importance in IBD.11, 43 It can activate resident macrophages, promote the release of other proinflammatory mediators (including nitric oxide, prostacyclin, and platelet activating factor), and induce expression of adhesion molecules on vascular endothelium, favouring the influx of new inflammatory cells into the mucosa. TNFα can also contribute to the intestinal damage by directly altering the integrity of epithelial membranes. The contribution of inflammatory cytokines to the pathogenesis of IBD is also a result of their ability to activate mechanisms which eventually cause important morphological changes. IL-1β and TNFα stimulate gut fibroblasts to synthesise matrix metalloproteinases (MMPs), a family of zinc containing neutral endopeptidases which slowly replace the matrix.47
In the normal human gut, lamina propria stromal cells secrete low amounts of MMPs, as well as tissue inhibitors of MMPs (TIMPs) which prevent excess enzyme activity, together with inhibitors such as α2 macroglobulin.47 IL-1β and TNFα rapidly upregulate MMP production without altering TIMP production.47 Studies using the ex vivo model of T cell mediated gut inflammation show the importance of this pathway. Indeed, the T-LPL cell response in the fetal gut explants induced by pokeweed mitogen (PWM) or anti-CD3 and IL-12 is strongly Th1 biased, and within four days there is severe tissue injury, with destruction of the mucosa.48, 49 Furthermore, these organ culture supernatants contain increased concentrations of MMP-1 (collagenase) and MMP-3 (stromelysin 1). Importantly, tissue damage, without altering T cell activation, is prevented by the addition of a stromelysin 1 inhibitor to the fetal gut organ culture.48, 49 In addition, a p55 TNFα receptor human IgG fusion protein may prevent the mucosal degradation and inhibit stromelysin 1 production.49, 50 Stromelysin 1 has a broad substrate specificity, being capable of degrading proteoglycans, laminin, fibronectin, collagen core protein, and non-helical cross linked regions of type IV collagen. Stromelysin 1 has therefore the potential to destroy the structure of the intestinal lamina propria, thereby removing the scaffolding on which the epithelium lies. As enterocytes adhere to basement membrane through extracellular matrix receptors, modification and loss of the basement membrane may result in decreased adhesiveness and epithelial cell shedding.47 Consistently, abundant stromelysin 1 has been found in the mucosa of patients with IBD, particularly near ulcers.51–53
Another pathway by which cytokines could indirectly produce changes in the gut is through their effects on stromal cell production of epithelial growth factors, such as members of the fibroblast growth factor family. Keratinocyte growth factor (KGF) is one of these molecules; it is produced by stromal cells, mainly in response to TNFα.54 It is over expressed in IBD patients, and could to be responsible for the crypt cell hyperplasia observed in these conditions. Indeed, in the fetal gut model, KGF by itself enhances epithelial cell proliferation, whereas a neutralising KGF antibody partially inhibits the crypt cell hyperplasia resulting from activation of lamina propria Vβ3+ T cells by staphylococcal enterotoxin B (SEB).54 In this model, SEB induced crypt hyperplasia is also associated with an enhanced expression of TGFα, a member of the epidermal growth factor family, which is produced by epithelial cells and is able to modulate epithelial cell function.54 Addition of anti-TGFα prevents the SEB stimulated crypt hyperplasia. Interestingly, stimulation of explants with recombinant KGF enhances TGFα induction, suggesting molecular cross talk between KGF and TGFα in T cell mediated crypt cell hyperplasia.54

CD4+ T cell subsets and their products in the human intestinal mucosa.
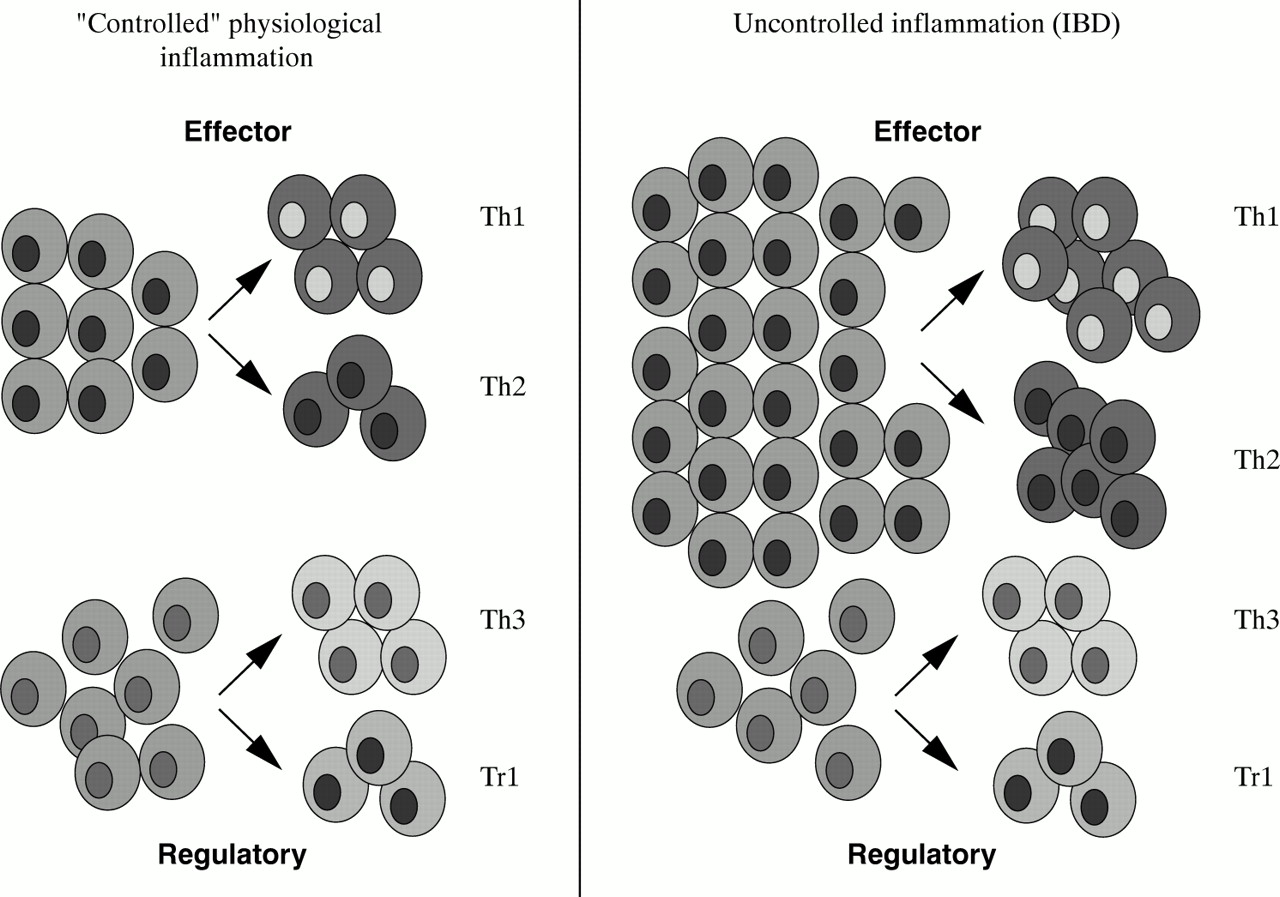
CD4+ T cell subsets in human intestinal mucosa. During uncontrolled inflammation the number of CD4+ effector T cells is increased while the proportion of CD4+ regulatory T cells tend to remain relatively stable.
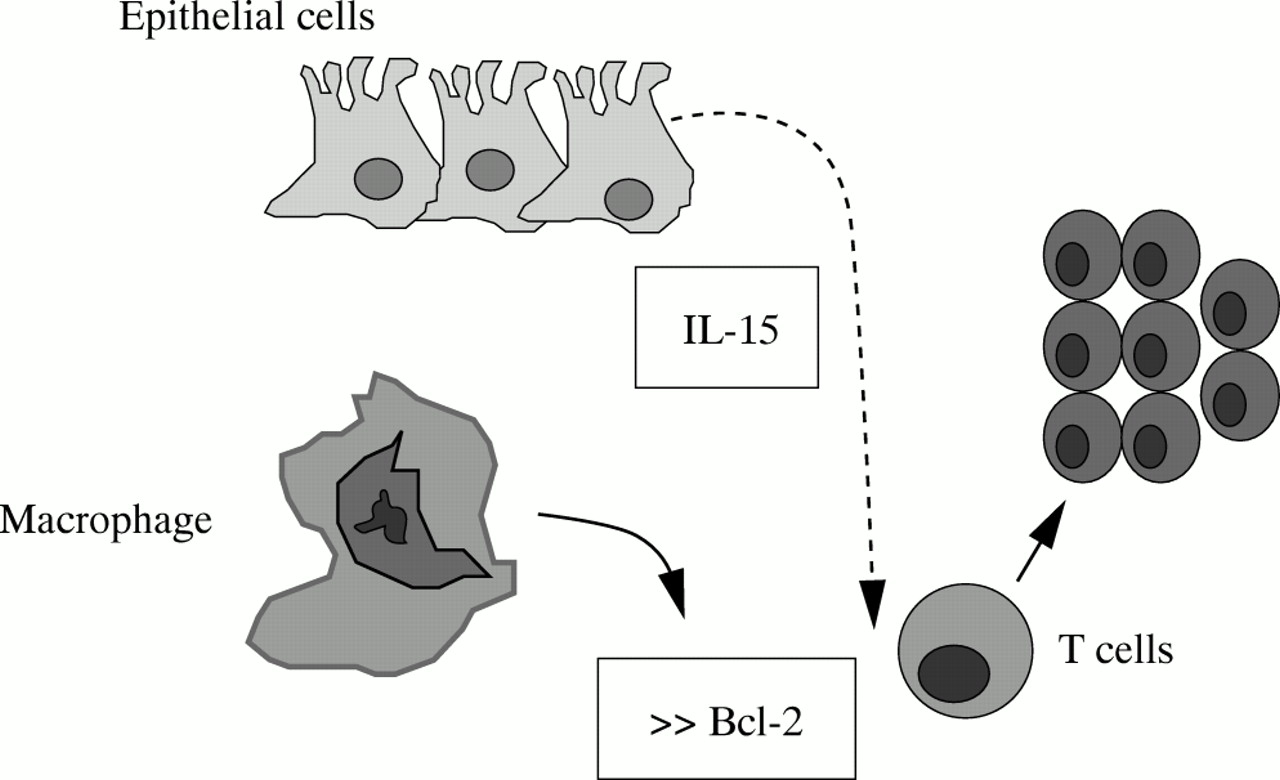
IL-15 enhances T-LPL cell resistance to apoptosis by stimulating the release of Bcl-2.
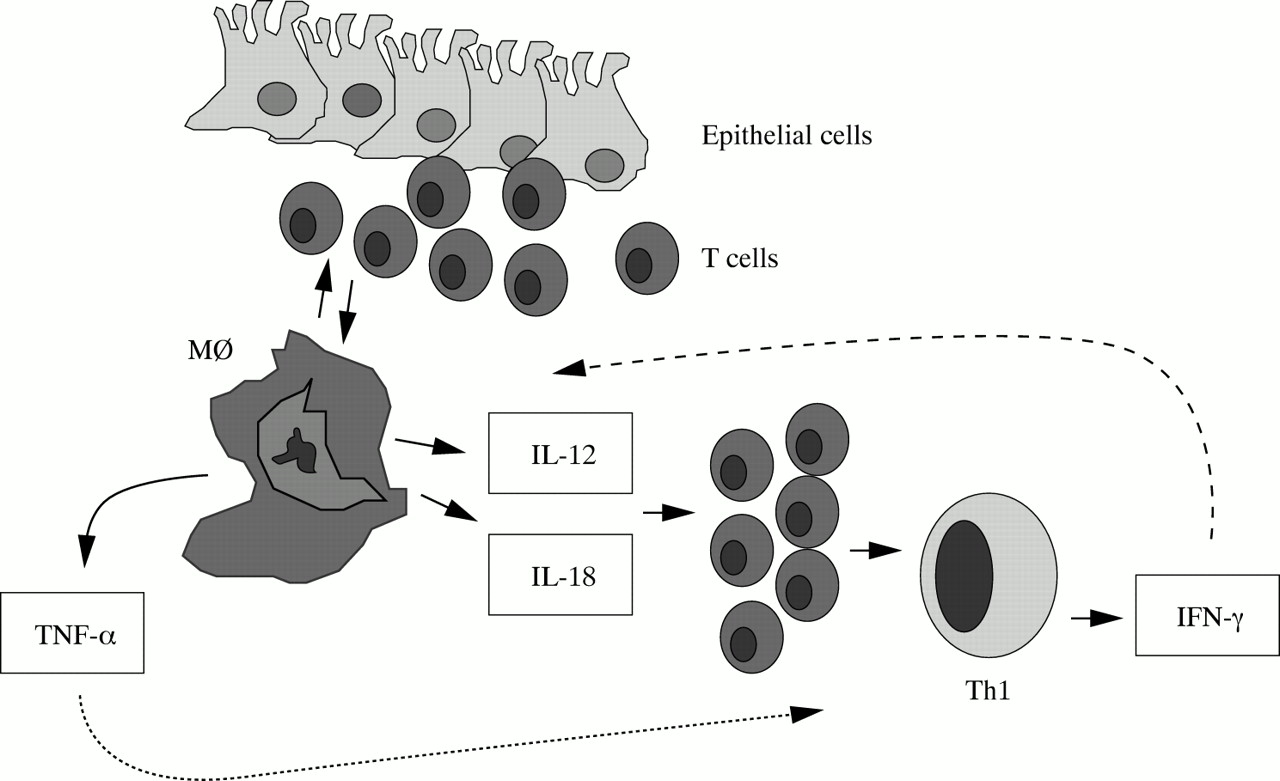
Driving a Th1 response in Crohn's disease.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Th1 and Th2 profiles in inflammatory bowel disease: a dogma to be disputed.