Article Text

Abstract
Background and aims: Oesophageal adenocarcinoma frequently develops on a background of metaplastic Barrett’s epithelium. The development of malignancy is accompanied by genetic alterations, which may be promising biomarkers of disease progression.
Methods: A case control study was conducted nested within a large unselected population based cohort of Barrett’s patients. Incident oesophageal malignancies and high grade dysplasias were identified. For each case up to five controls were matched on age, sex, and year of diagnosis. Biopsies from the time of diagnosis of Barrett’s epithelium were stained immunohistochemically for TP53, cyclin D1, cyclooxygenase 2 (COX-2), and β-catenin proteins.
Results: Twenty nine incident oesophageal malignancies and six cases of high grade dysplasia were identified. The odds of diffuse or intense TP53 staining were substantially elevated in biopsies from patients who developed oesophageal adenocarcinoma compared with controls (odds ratio (OR) 11.7 (95% confidence interval (CI) 1.93, 71.4)). This difference was also present when all cases were considered (OR 8.42 (95% CI 2.37, 30.0). Despite the association with TP53 staining, only 32.4% of cases had an initial biopsy showing diffuse/intense TP53 staining. There were no significant associations between cyclin D1, COX-2, or β-catenin staining and case control status. The OR for positive staining for both TP53 and COX-2 was markedly increased in cases compared with controls (OR 27.3 (95% CI 2.89, 257.0)) although only 15% of cases had positive staining for both markers.
Conclusions: Immunohistochemical detection of TP53 expression is a biomarker of malignant progression in Barrett’s oesophagus but sensitivity is too low to act as a criterion to inform endoscopic surveillance strategies. Additional biomarkers are required which when combined with TP53 will identify, with adequate sensitivity and specificity, Barrett’s patients who are at risk of developing cancer.
- OA, oesophageal adenocarcinoma
- BO, Barrett’s oesophagus
- SIM, specialised intestinal metaplasia
- LOH, loss of heterozygosity
- COX-2, cyclooxygenase 2
- H&E, haematoxylin and eosin
- NIBR, Northern Ireland Barrett’s Oesophagus Register
- TBS, Tris buffered saline
- OR, odds ratio
- Barrett’s oesophagus
- cyclin D1
- TP53
- oesophageal adenocarcinoma
- biomarkers
Statistics from Altmetric.com
- OA, oesophageal adenocarcinoma
- BO, Barrett’s oesophagus
- SIM, specialised intestinal metaplasia
- LOH, loss of heterozygosity
- COX-2, cyclooxygenase 2
- H&E, haematoxylin and eosin
- NIBR, Northern Ireland Barrett’s Oesophagus Register
- TBS, Tris buffered saline
- OR, odds ratio
Oesophageal adenocarcinoma (OA) is commonly associated with a precancerous condition termed Barrett’s oesophagus (BO) in which the normal squamous epithelium of the distal oesophagus is replaced by a specialised intestinal metaplasia (SIM) characterised by the presence of goblet cells.1 BO patients have an elevated risk of adenocarcinoma,1,2 a malignancy for which five year survival following diagnosis is generally less than 15%.3,4 Consequently, interest has focused on surveillance by upper gastrointestinal endoscopy in an attempt to provide earlier detection and more effective treatment. Several studies have shown that patients diagnosed with OA within a BO surveillance programme have earlier stage disease and longer survival times than patients diagnosed outside such programmes5–,7 but these studies may be subject to length and lead time bias. Randomised controlled trial evidence of the effectiveness of surveillance programmes in reducing mortality in BO patients is not available. The relatively low incidence of OA among BO patients,8,9 competing mortalities, and loss to follow up in surveillance programmes10 have led to questions concerning their effectiveness,11,12 particularly in light of competing economic pressures on healthcare systems.13,14
Surveillance of BO is commonly undertaken both in the USA and Europe.15–,17 Identification of BO patients at highest risk of development of adenocarcinoma using biomarkers may permit current surveillance strategies to be more individually tailored and, presumably, more cost effective. A number of genetic alterations have been associated with the metaplastic and dysplastic changes that represent early stages in the natural history of OA.1,18 In particular, aneuploidy, loss of heterozygosity (LOH) on chromosome 9p21 and 17p, hypermethylation and mutation of CDKN2 (p16), TP53 mutations, overexpression of cyclin D1 and cyclooxygenase 2 (COX-2), and nuclear accumulation of β-catenin have all been reported in BO tissue.2,19,20
While there are many cross sectional studies of genetic alterations in relation to the development of OA2 there are fewer longitudinal studies of BO patients that have examined these alterations in biopsy specimens in advance of the later stages of the disease.1,2 The former would be termed phase 1 and 2 studies and the latter as phase 3 and 4 in the recently proposed classification system by the USA National Cancer Institute’s Early Detection Research Network.21 In this context, Reid and colleagues22 have reviewed biomarkers commonly applied to BO and OA, notably dysplasia, ploidy, TP53, cyclin D1, and p16 status. In a case control analysis nested within a cohort of 350 Barrett’s patients in Leeds, UK, cyclin D1, and to a lesser extent TP53, immunostaining in biopsies of BO patients was associated with an increased risk of progression to adenocarcinoma.23 In a prospective study of 269 patients in Seattle, USA, 17p LOH was also strongly associated with progression in BO patients.24 However, subjects in both of these studies were drawn from selected populations of BO patients with the attendant risks of selection bias.
In this study, we examined whether altered expression of four candidate biomarkers—namely, cyclin D1, TP53, COX-2, and β-catenin—was associated with a risk of oesophageal malignancy using a case control design nested within a large unselected population based cohort of BO patients. TP53 and cyclin D1 were selected because of their promise in earlier longitudinal studies22–,24 while COX-2 was studied because of the documented increase in expression during progression from BO to dysplasia and OA.25–,27 Inclusion of β-catenin was based on earlier observations that nuclear accumulation of this protein was common in OA28 and that promoter hypermethylation of the APC gene was frequent in BO patients, suggesting a potential mechanism for dysregulation of the WNT APC β-catenin pathway and the observed nuclear translocation of β-catenin.2,29 Increased nuclear β-catenin could also be a consequence of reduced expression of E-cadherin which has been reported to occur in Barrett’s metaplasia.30
MATERIALS AND METHODS
Patient selection
A nested case control study was conducted within a population based cohort of BO patients, the Northern Ireland Barrett’s Oesophagus Register (NIBR).8,31 The NIBR comprises every adult identified within Northern Ireland (NI, population 1.7 million) between January 1993 and December 1999 as having oesophageal columnar epithelium. The register was constructed by examining pathology reports relating to all oesophageal biopsies taken in NI during this period. BO was defined histologically as the presence of SIM (including the presence of goblet cells), irrespective of whether Barrett’s mucosa was reported visually by the endoscopist. Biopsies stated (by the endoscopist) as having been taken at the oesophagogastric junction were excluded. Infrequent provision of data on BO segment length precluded its incorporation into the definition but these data were recorded where available.
Individual patients were identified within the dataset and were followed up for death and oesophageal malignancy until the end of 2000 by matching with death files from the NI Registrar General’s Office and the NI Cancer Registry database of incident cancers. Cases were defined as patients within NIBR who developed OA, or undifferentiated/unspecified oesophageal carcinoma during the follow up period and at least a minimum of six months from their first biopsy showing BO as in our previous study.23 Squamous cell tumours of the oesophagus were not included. Patients undergoing treatment (oesophagectomy or ablative treatment) for high grade dysplasia within the same period were also identified and included as cases for certain analyses. For each of the incident cases, five control patients who did not develop oesophageal carcinoma during the follow up period were selected from the register. Controls had to have biopsies that showed evidence of SIM and by preference those with long segment columnar lined epithelium were selected. Controls were also matched with cases on age (within five years), sex, and year of diagnosis; controls had a period of follow up of at least as long as that of their respective case.
Ethics approval for the study was granted by the Research Ethics Committee of Queen’s University, Belfast.
Histology
The original oesophageal biopsies on which the classification of BO was made were obtained for each of the cases and controls from the four pathology laboratories in the province (Belfast Link Laboratories, Antrim Hospital, Craigavon Area Hospital, and Altnagelvin Hospital). New sections were prepared from each histology block. One section of each biopsy was stained with haematoxylin and eosin (H&E) and examined for confirmation of SIM, as defined by the presence of goblet cells. Only biopsies containing goblet cells were used for immunohistochemistry. H&E sections were scored by consensus by two histopathologists (MS and DMcM) for the presence and grade of dysplasia according to a modification of the Vienna classification: 0 = no dysplasia, 1 = indefinite for dysplasia, 2 = low grade dysplasia, 3 = high grade dysplasia, and 4 = intramucosal adenocarcinoma. Inflammation was also graded, where 0 = no inflammation, 1 = mild inflammation, 2 = moderate inflammation, and 3 = severe inflammation.
The remaining sections were shipped to Leeds for immunohistochemical staining for cyclin D1, TP53, COX-2, and β-catenin. Investigators in Leeds were blinded as to the case and control status of the slides.
Immunohistochemistry
The protocol for immunohistochemistry was similar for each of the four markers examined. Initially, sections were dewaxed in xylene before rehydration through alcohol to water. Endogenous peroxidase activity was then blocked by immersing the slides in methanol containing 2% hydrogen peroxide for 30 minutes. After washing in water, slides were rinsed in Tris buffered saline (TBS) (50 mM Tris, pH 7.6) prior to antigen retrieval by pressure cooking (see specific conditions below). Staining was performed on a Sequenza Work Station (ThermoShandon, Runcorn UK). Non-specific binding was blocked using a casein solution (SP-5020 Vector Labs) as per the manufacturer’s instructions followed by treatment with the Avidin-Biotin Blocking kit (SP-2001; Vector Labs., Peterborough, UK). Sections were then incubated in primary antibody (see specific conditions below) diluted in TBS with 0.01% Tween 20.
After incubation with primary antibody, slides were washed twice with TBS prior to incubation with the biotinylated secondary antibody for 30 minutes at room temperature. Slides were washed twice with TBS to remove secondary antibody and stained with ABC solution for 30 minutes (Vector Stain Elite ABC kits, PK-6102; Vector Labs). Slides were removed from the Sequenza Work Station and staining was developed by addition of a working concentration of 0.025% w/v diaminobenzidine substrate in TBS. Slides were rinsed in TBS, placed into a copper sulphate solution for five minutes to enhance staining, washed in water, lightly counterstained with haematoxylin, and then dehydrated, cleared, and mounted in DPX for evaluation.
Matched case and control sections were run together, coded, in a single batch to minimise any risk of bias in immunostaining between the two groups. The same positive and negative control sections for each biomarker were also included with each batch of immunostaining on the test sections to monitor any batch-to-batch variations in staining. The controls comprised tissue sections from the same tissue blocks throughout, obtained from histologically normal or carcinoma lung tissue, where the negative or positive staining for the various antigens was validated in preliminary experiments; optimal conditions for signal to noise ratio of immunostaining were also determined on these sections.
Specific antibodies and conditions for each staining process were:
TP53 (Novocastra NCL-L-TP53-D07; mouse monoclonal IgG2b raised against a recombinant human wild-type p53 protein): 90 seconds antigen retrieval; 1:100 dilution primary antibody, one hour at 25°C; 1:200 dilution secondary antibody.
Cyclin D1 (Novocastra NCL-L-CYCLIND1-GM; mouse monoclonal IgG2a raised against a prokaryotic fusion protein corresponding to the human cyclin D1 molecule): 60 seconds antigen retrieval; 1:50 dilution primary antibody, one hour at 25°C; 1:100 dilution secondary antibody.
β-catenin (Transduction Laboratories, BD Pharmingen, G10153; mouse monoclonal IgG1, generated against the C terminal of mouse β-catenin): 90 seconds antigen retrieval; 1:240 dilution primary antibody, one hour at 25°C; 1:100 dilution secondary antibody.
COX-2 (Cayman Chemicals, Alexis Biochemicals, 160112; mouse monoclonal raised against a synthetic peptide corresponding to COX-2): 90 seconds antigen retrieval; 1:250 dilution primary antibody, overnight at 4°C; 1:100 dilution secondary antibody.
Evaluation of immunohistochemical staining
The stained sections were independently scored for specific staining within columnar mucosa by the two histopathologists blinded to the case control status of the sections. Nuclear staining was evaluated for TP53, cyclin D1, and β-catenin using a modification of a method previously applied to a series of gastro-oesophageal tumours where TP53 mutation status was known.32 With regard to β-catenin, only nuclear positivity was assessed.28 Biopsies were assigned a score of 0–3 based on the following criteria: 0 = no positive cells or only occasional scattered positive cells (no staining); 1 = <10% of epithelial cells positive (focal staining); 2 = 10–50% positive (diffuse staining); and 3 = >50% of epithelial cells positive or confluent groups of positively stained glands (intense staining). COX-2 sections were examined for the presence of cytoplasmic staining and were also scored 0–3 by assessing both staining intensity and the proportion of positive epithelial cells (stromal cells were not considered) in a modification of a scoring system previously described33: 0 = no positive cells or occasional scattered weakly positive cells; 1 = <10% of epithelial cells with weak to moderate positive staining; 2 = 10–50% weakly or moderately positive or <10% strongly positive; and 3 = >50% moderately to strongly positive. Scores for sections containing more than one tissue fragment were defined according to the highest scoring fragment. The location of staining (that is, in crypts, on the surface, or both) was also recorded for each marker.
Statistical methods
All statistical analyses were performed using STATA version 8. The kappa statistic was used to assess agreement between the histopathologists assessment of immunohistochemical staining. Analyses were initially performed with cases defined as patients who developed high grade dysplasia, undifferentiated/unspecified carcinoma, and definite OA. Subsequent analyses excluded firstly all high grade dysplasia patients and then patients with undifferentiated/unspecified carcinoma. Matched controls for these patients were also excluded. Conditional logistic regression models were developed with case control status as the dependent variable and staining for the molecular markers as explanatory variables. No staining (grade 0) was used as the reference category and the diffuse (grade 2) and intense (grade 3) categories were combined because of small numbers in each of these categories. The analyses were repeated with grades 0 and 1 combined as the reference category to permit the additional comparison of weak (0, 1 grade) versus strong (2, 3 grade) staining. The potential confounding effect of the conditions in which the biopsies were stored on the results of immunohistochemical staining was addressed by including pathology laboratory as an explanatory variable in all statistical models.
RESULTS
Between January 1993 and December 1999, pathologists reported on 15 670 oesophageal biopsy specimens from patients who underwent upper gastrointestinal endoscopy within hospitals in NI. All of these reports were examined and 4955 biopsies (oesophagogastric biopsies excluded) from 2969 patients (1701; 57.3% male) showed oesophageal columnar epithelium. In 1670 (56.2%) of these patients, SIM was reported to be present in at least one oesophageal biopsy. Mean period of follow up in the whole cohort was 3.7 years (range 0–8.0); it was 2.3 years (range 0.5–7.4) in the cases and 3.9 years (range 1.1–7.8) in controls.
Twenty two patients developed OA a minimum of six months after the date of their first biopsy showing BO; one patient was diagnosed with an oesophageal adenosquamous carcinoma (treated as OA in the analyses) and a further six patients developed undifferentiated/unspecified carcinomas of the oesophagus, giving a total of 29 incident oesophageal malignancies (22; 76%, in men) within the cohort. Review of pathology specimens did not result in further classification of the six undifferentiated/unspecified carcinomas mentioned above. An additional four patients underwent oesophagectomy for high grade dysplasia within their segment of BO and two further patients had ablative treatment for BO by laser. Review of the pathology specimens of these patients confirmed high grade dysplasia without malignancy and they were included in the study to yield a total of 35 cases.
All but one of the original pathology specimens relating to the 35 cases were obtained; the missing biopsy was from a patient who subsequently developed OA. Twelve selected control specimens (6.9%) could not be obtained but specimens were available for a minimum of three controls per case. Insufficient material in some biopsy specimens meant that COX-2 staining was not performed on one further control and β-catenin staining was not performed on one further case and two further controls.
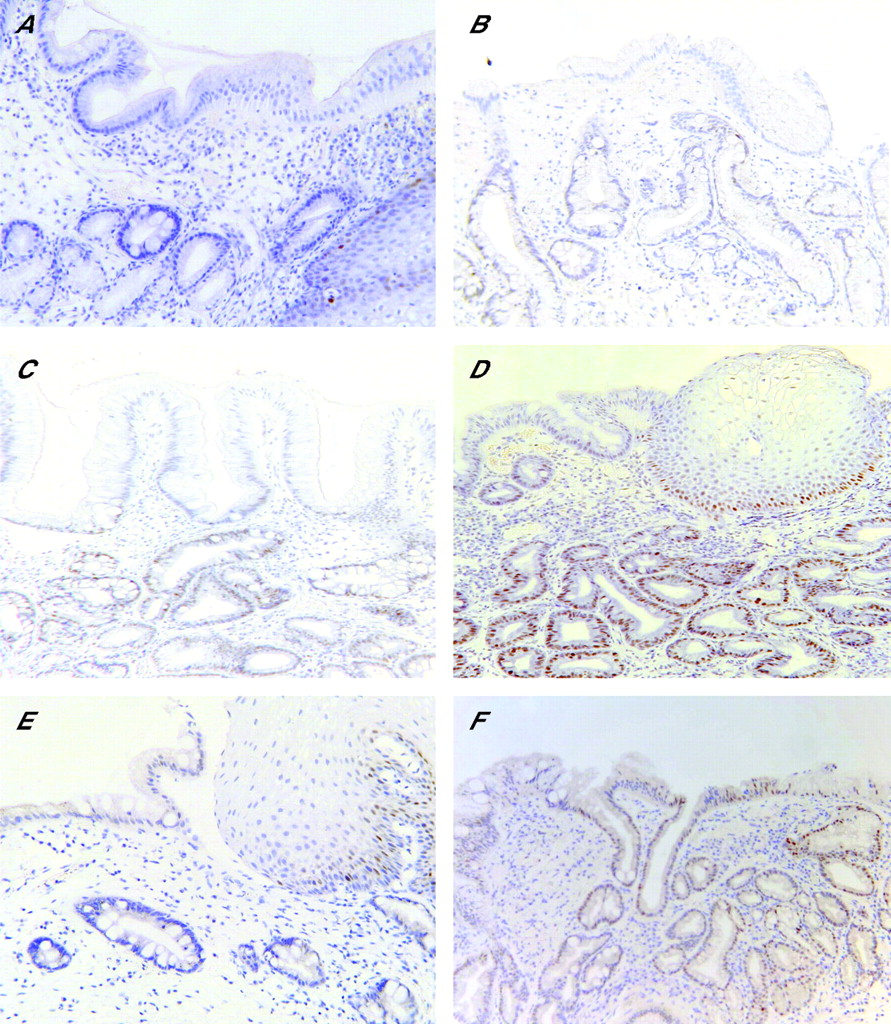
SIM was confirmed to be present in at least one biopsy fragment from every available case and control specimen. Cyclin D1, TP53, COX-2, and β-catenin staining scores ascribed independently by the histopathologists agreed in all but 4.6%, 8.6%, 9.6%, and 7.1% of the specimens, respectively (kappa statistics 0.92, 0.86, 0.83 and 0.85, respectively), and in all cases the scores differed by only one category. The lowest category was used in the analyses when the categories ascribed by the histopathologists differed. Examples of the staining for TP53 and cyclin D1 are presented in fig 1⇓. Staining in the columnar epithelium only was the subject of this study; some positive staining in the basal cell layer of the normal squamous epithelium was visible.
{kind=link}
Immunohistochemical staining for TP53 and cyclin D1. (A–D) Examples of TP53 staining graded as 0 (none), + (focal), ++ (diffuse), and +++ (intense), respectively. (E, F) Examples of staining for cyclin D1, classified as 0 (none) and ++ (diffuse), respectively. As described in the materials and methods, only staining in the columnar epithelium was considered in relation to cancer risk; low levels of TP53 and cyclin D1 staining in the basal layer of the normal squamous epithelium in (A) and (E), respectively, was expected and serves to illustrate that the immunostaining reaction was working in these sections. Sections processed in the absence of primary antibody showed staining of similar background to that classified here as “0” or no staining. Magnification ×200.
Comparison of cases and controls on matching criteria showed that males represented 77.1% of cases (27 males, eight females) and controls (135 males, 40 females) and that age did not significantly differ between the groups (68.0 (10.6) years in cases and 67.1 (10.5) years in controls; t = 0.48, p = 0.95). In addition, the number of biopsy fragments did not differ significantly between slides prepared from cases and controls (4.50 (2.3) and 4.39 (2.1), respectively; t = 0.28, p = 0.5). The characteristics of oesophageal biopsies are shown in table 1⇓. Only four specimens showed evidence of dysplasia; one showed high grade dysplasia and this patient developed adenocarcinoma within a year. None of the specimens from cases showed low grade dysplasia. Modest or severe inflammation was more common in control specimens (35%) than case specimens (26.5%) but the difference was not statistically significant (χ2 5.60, df 3, p = 0.13).
Characteristics of initial oesophageal biopsies
The results of staining for cyclin D1, TP53, COX-2, and β-catenin are shown in table 1⇑ and the results of the paired (conditional) logistic regression analyses are shown in table 2⇓. Cyclin D1 staining was observed in >10% of cells (grade 2 and 3, diffuse or intense) in 39.3% of controls and in 29.4% of cases. Staining was mainly restricted to the crypts but was also seen on the surface of the epithelium in cases where strong staining occurred (see fig 1⇑). Immunohistochemistry for COX-2 revealed the two strongest categories of staining to be present in 17.3% of controls and in 26.4% of cases. The pattern of COX-2 staining was similar to cyclin D1 and in the case of both markers there was no difference in the patterns between cases and controls. In some biopsies, staining for COX-2 was negative in the mucosa but strongly positive in inflammatory debris and granulation tissue on the surface of the oesophageal biopsy.
Odds of cases compared with controls showing evidence of staining for markers in the initial oesophageal biopsies
Diffuse or intense TP53 staining was observed in 11.6% of controls and in 32.3% of cases. In just over half (52%) of samples, TP53 staining was seen in both the crypts and surface, and in the other samples staining was limited to the crypts; there was no difference between cases and controls in the distribution of staining. The majority of sections were negative for strong (grades 2 or 3) nuclear β-catenin staining. In almost all samples (96%) in both cases and controls, β-catenin staining was confined to the crypts.
The odds ratio for biopsies from patients who developed incident OA showing diffuse or intense TP53 staining was substantially elevated compared with controls (odds ratio (OR) 11.7 (95% confidence interval (CI) 1.93, 71.4)). This elevated OR was also seen when all oesophageal malignancies or all cases, including high grade dysplasias, were included in the analysis (table 2⇑). No increased risk was associated with focal TP53 staining, defined as <10% cells positive for this marker. When diffuse or intense staining was compared with none or focal staining, the OR (95% CI) for patients who developed definite OA showing TP53 staining was 9.28 (1.78, 48.3).
The odds of positive staining for cyclin D1 was not raised in cases compared with controls, either for focal staining or diffuse/intense staining. This was true when all oesophageal malignancies and high grade dysplasias were considered (OR for diffuse/intense staining v no staining 0.93 (95% CI 0.21, 4.22)) or when the analysis was restricted to those with definite OA (OR 0.81 (95% CI 0.14, 4.58)) (table 2⇑). Exclusion of only high grade dysplasia cases from the analysis did not significantly alter the results. When the two lowest categories of staining were used as the reference and compared with diffuse/intense staining, there was still no significant difference between cases and controls (OR 0.66 (95% CI 0.22, 1.99)).
Moderate elevation in the odds of positive staining for COX-2 or β-catenin was seen in cases (especially when restricted to definite OA) but statistical significance was not reached (table 2⇑).
Despite the high odds ratio for TP53 staining in cases compared with controls, only 11 of 34 patients (32.4%) who developed oesophageal malignancy or high grade dysplasia had an initial biopsy showing diffuse/intense TP53 staining. This percentage was similar when considering all patients who developed oesophageal malignancy (10 of 28; 25.7%) or only those who developed OA (eight of 22 patients (36.3%)).
The effect of combining biomarkers on the proportion of cases that were biomarker positive (grade 2/3 staining) and the odds of biomarker positivity in cases compared with controls are shown in table 3⇓. Staining positively for either TP53 or one of the other biomarkers increased the proportion of cases who were classified as “biomarker positive” but reduced the strength of the association between biomarker positivity and risk of malignancy. On the other hand, biopsies from cases were much more likely to stain positively for both TP53 and COX-2 (OR 27.3 (95% CI 2.89, 257.0)) but only 15% of cases were positive for both biomarkers.
Effect of the combination of biomarkers on the proportion of cases that were biomarker positive and the odds of biomarker positivity in cases compared with controls
DISCUSSION
Four potential biomarkers for predicting progression of BO to OA were examined in a nested case control study within a population based cohort of BO patients. All patients with SIM of the oesophagus diagnosed within NI during the study period were included and population based registers utilised to identify deaths and incident oesophageal malignancies within this group. Patients leaving NI may have been lost to follow up but emigration is unusual among the age group studied. The rate of oesophageal malignancy among patients in the NIBR is 0.4% per year for patients with SIM.8 It is important to note that this is a population based register of all Barrett’s patients in a specific community and not a surveillance cohort. As such, questions concerning the stage of diagnosed cancers and clinical effectiveness of the register are not relevant.
Particular strengths of the study were the relatively high number of cases in comparison with previous longitudinal studies of BO and OA, the unselected population, the fact that the analysis was applied to biopsies collected under the varied protocols that typify much of this field of clinical practice, and the use of immunostaining that is feasible within a routine setting using archival biopsies. Thus the study design approximates to what would be encountered in translating research findings into clinical practice; in this sense the results have relevance to a broad range of clinical settings rather than only to high intensity surveillance in specialised facilities.
There are some weaknesses in design, in particular the relatively short follow up (mean follow up 3.7 years) which means it is highly unlikely that all oesophageal malignancies that will occur within the cohort of BO patients have occurred during follow up. In addition, oesophageal biopsy protocols were not systematic. While this means the results may be more readily generalised, this does present potential problems. Firstly, individuals within the cohort could be misclassified with respect to the presence of SIM. However, all patients with columnar oesophageal mucosa were followed up, irrespective of whether SIM was detected or not, and SIM was retrospectively confirmed to be present in at least one biopsy fragment from every available case and control specimen in the nested case control study. Secondly, differences in biopsy protocol could affect the number of biopsies and hence the chances of identifying positive immunostaining. However, there were no significant differences in numbers of biopsies on the slides between cases and controls, making a systematic bias unlikely. Thirdly, it is possible that some of the biopsies evaluated may have been cardiac rather than oesophageal SIM. It is uncertain what effect this may have on the biomarker-disease associations. However, the likelihood of including cardiac SIM was minimised by excluding biopsies taken from the oesophagogastric junction. Also, this issue reflects a common problem in the clinical setting and the findings may therefore be more relevant to practice than studies employing very strict biopsy protocols that are unlikely to be routinely applied.
Although all of the selected biomarkers are involved in pathways previously implicated in the pathogenesis of BO and OA, only staining for TP53 protein was significantly associated with the risk of malignant progression. The original biopsy specimens from patients who developed OA, undifferentiated/unspecified carcinoma, or high grade dysplasia were eight times more likely to show diffuse/intense staining for TP53 than were specimens from individuals who did not develop these outcomes. When the more specific outcome of histologically confirmed OA was employed, biopsies from cases were 11 times more likely than controls to show TP53 staining. The OR for diffuse/intense COX-2 staining was also elevated, especially in confirmed adenocarcinomas (threefold increase), although this result was not statistically significant. Staining for cyclin D1 was not associated with case control status and nuclear β-catenin staining was uncommon in cases and controls.
Few other longitudinal studies have examined the relationship between TP53 protein immunostaining in BO and the risk of OA. Three studies have shown that detection of TP53 in the presence of low grade dysplasia is a risk factor for progression to high grade dysplasia or cancer.34–,36 We previously observed a threefold increased risk, albeit not statistically significant, of progression to adenocarcinoma in BO patients staining positively for TP53.23 Combining data for that study and the current one results in a statistically significant pooled OR of 4.97 (95% CI 1.73, 14.3). Using a molecular approach, Reid and colleagues24 showed that BO patients with 17p LOH (at the TP53 locus) had a 16-fold higher risk of progression to cancer than those without this abnormality. Neither of the latter studies were undertaken from a population perspective but both are consistent with an elevated risk of malignant progression in BO patients with TP53 abnormalities. The current study indicates that TP53 overexpression without dysplasia is predictive of cancer risk as only one of the 11 patients whose initial biopsies were TP53 positive, and who developed malignancy, also had dysplastic changes (high grade dysplasia).
One important question with regard to TP53 immunostaining is exactly what does the measure indicate? It is frequently interpreted as a surrogate for TP53 mutation.22 However, TP53 immunostaining does not always correlate with mutation.37 For example, mutations resulting in deletion or truncation of the protein will not be detected by immunostaining. Direct comparisons of TP53 mutation and immunohistochemistry in OA are rare and therefore the magnitude of discrepancy between the two in oesophageal tissue is uncertain. The few published studies involve small numbers of cases, do not confirm mutation screening methods by DNA sequencing, consider only exons 5–8, use different TP53 antibodies, and define widely different cut off points for positive staining.38–,42 Nevertheless, the data do indicate that in a proportion of cases TP53 staining will not reflect mutation. However, the presence of TP53 protein can be considered as a biomarker in its own right because other mechanisms leading to overexpression, including accumulation of wild-type TP53 in response to alterations in other genes43 or DNA damage,44 may in themselves represent an increased risk for disease progression. Thus we would argue that positive TP53 immunostaining should not be interpreted merely as a surrogate for TP53 mutation but as a more general marker of an epithelium that is susceptible to disease progression. In this context it is of interest that DNA damage is elevated in Barrett’s epithelium compared with squamous epithelium.45 This could result in inactivation of TP53, impaired DNA repair, and clonal expansion46,47 of cells predisposed to further genetic damage.45,48
TP53 immunostaining alone could not be employed as a risk stratification tool on which to base entry of BO patients into a surveillance programme because the biopsies of two thirds of the patients who progressed to malignancy or high grade dysplasia did not stain positively for this marker. This contrasts with studies of tumour samples from OA patients where the majority (>80%) have TP53 mutations.2 The discrepancy may be because TP53 mutations occurred after the time of biopsy collection in some patients or alternatively TP53 independent pathways could be important in the early stages of a subset of OA. It is possible that some of the control subjects staining positively (diffuse/intense) for TP53 might have progressed to low grade dysplasia during follow up. However, as this study was not a surveillance cohort, we only had follow up biopsies on nine control patients; only one of these showed low grade dysplasia making it unlikely that many progressed to dysplasia. As discussed above, TP53 status may be misclassified in some patients, either due to sampling limitations combined with heterogeneity of TP53 alterations in the Barrett’s epithelium46 or due to the nature of the immunohistochemical assay employed.
Absence of a relationship between cyclin D1 staining and risk of malignancy is at odds with our previous finding in a cohort of BO patients from Leeds, UK.23 When the data from the NI and Leeds studies were combined, the OR for cyclin D1 staining was not significant (OR 2.21 (95% CI 0.75, 6.31)). There are several differences between the two studies that may account for these discrepant results. The current study was substantially larger and was population based rather than drawing patients from one centre, which may be associated with some unidentified bias in recruitment. Patients were approximately 10 years older in NI and the mean follow up was slightly shorter; the period of time that a BO patient had experienced undiagnosed SIM (prior to entry in to the cohort) and the time between BO diagnosis and cancer occurrence could possibly affect the nested case control analysis. For example, if cyclin D1 overexpression in the early stages following development of SIM identifies high risk BO patients, but overexpression eventually occurs in an increasing proportion of all BO patients, then the discrimination between cases and controls will tend to diminish with duration of presence of SIM. In the current study, the older age of patients and the short follow up are consistent with a more “advanced” group of BO patients in the NI cohort. These questions are difficult to resolve because the time of onset of SIM in BO patients is unknown. No other longitudinal studies have examined the relationship between malignant progression in BO and cyclin D1 overexpression but given the contrasting results to date further studies are warranted.
COX-2 overexpression was weakly positively related to cancer risk. Previously this relationship has been the subject of contradictory reports.25,26,49,50 However, Abdalla and colleagues50 reported that, independent of case control status, COX-2 expression increased during follow up of Barrett’s patients and gastrin was shown to be a key factor in influencing expression. Of interest in our study was the observation that patients who were both TP53 and COX-2 positive were at very high risk of developing malignancy but these patients comprised only 15% of patients who developed OA. The mechanistic role of COX-2 in BO and OA therefore requires further study while an evaluation of COX-2 should be conducted in larger prospective studies in combination with other functionally related biomarkers.
β-Catenin is a key component of cell adhesion and cell signalling pathways. However, current evidence suggests that abnormalities in β-catenin may not play an important role in the early stages of most OA.2 In this study, we specifically scored nuclear accumulation of β-catenin, as an indicator of alterations of the Wnt signalling pathway, but found nuclear staining to be uncommon in BO. This observation is consistent with another study where increased expression of nuclear β-catenin occurred only in dysplastic epithelium.28
In conclusion, it is clear that immunohistochemical detection of TP53 is a biomarker of malignant progression in BO but alone possesses too low a sensitivity for cancer risk for it to be applied as a criterion for endoscopic surveillance. Detection of TP53 mutations by direct sequencing or other screening techniques such as SSCP would potentially offer better sensitivity and specificity compared with immunohistochemistry, but such techniques are not widely available in NHS laboratories and moreover would be difficult to adapt to analysis of small routinely fixed and processed endoscopic biopsies. In addition, the predictive value of TP53 immunostaining in the absence of genetic alterations should be considered. Combination of TP53 status with dysplasia would not appear to improve on its utility, as so few patients had dysplasia. Furthermore, combining TP53 status with the other markers examined also appears to be of limited value; although patients who were both TP53 and COX-2 positive were at very high risk, these patients comprised only 15% of patients who subsequently developed cancer. Identification of complementary biomarkers with adequate sensitivity and specificity is therefore required to identify BO patients who are at risk of developing cancer. Candidates will no doubt emerge as a result of the rapid advances in screening for markers using genomic and proteomic approaches.51,52 Such candidate markers should be selected on the basis of what is known about the biology of the disease and be evaluated in appropriate study designs.21,22
Acknowledgments
This study was supported by a grant from Yorkshire Cancer Research L290. The Northern Ireland Barrett’s Oesophagus Register was constructed within the Northern Ireland Cancer Registry, which is supported by the Department of Health, Social Services and Public Safety, Northern Ireland.
REFERENCES
Footnotes
Published online first 8 May 2006
This study was supported by a grant from Yorkshire Cancer Research L290.
Conflict of interest: None declared.