Article Text

Abstract
Background and aims Premature intra-acinar activation of trypsinogen is widely considered key for both the initiation of acute pancreatitis and the development of chronic pancreatitis. However, the biological consequences of intracellular trypsinogen activation have not been directly examined. To do so, a new mouse model was developed.
Methods Mice were engineered to conditionally express an endogenously activated trypsinogen within pancreatic acinar cells (PACE-trypon). Hallmarks of pancreatitis were determined and findings were correlated to the level (zygosity) and extent (temporal and spatial) of conditional PACE-trypon expression. Furthermore, the impact of acinar cell death in PACE-trypon mice was assessed and compared with a model of selective diphtheria toxin (DT)-mediated induction of acinar apoptosis.
Results Initiation of acute pancreatitis was observed with high (homozygous), but not low (heterozygous) levels of PACE-trypon expression. Subtotal (maximal-rapid induction) but not limited (gradual-repetitive induction) conditional PACE-trypon expression was associated with systemic complications and mortality. Rapid caspase-3 activation and apoptosis with delayed necrosis was observed, and loss of acinar cells led to replacement with fatty tissue. Chronic inflammation or fibrosis did not develop. Selective depletion of pancreatic acinar cells by apoptosis using DT evoked similar consequences.
Conclusions Intra-acinar activation of trypsinogen is sufficient to initiate acute pancreatitis. However, the primary response to intracellular trypsin activity is rapid induction of acinar cell death via apoptosis which facilitates resolution of the acute inflammation rather than causing chronic pancreatitis. This novel model provides a powerful tool to improve our understanding of basic mechanisms occurring during pancreatitis.
- Trypsin
- apoptosis
- inflammation
- PACE
- pancreas
- pancreatic disorders
- pancreatic enzymes
- pancreatitis
Statistics from Altmetric.com
Significance of this study
What is already known about the role of trypsin in pancreatitis?
Autodigestion by digestive enzymes was proposed to be the cause of pancreatitis >100 years ago.
Premature activation of intracellular trypsinogen observed both in patients and in animal models of pancreatitis has been considered as the initiation event.
Trypsinogen mutations which lead to increased autoactivation or decreased degradation cause hereditary pancreatitis in humans.
Genetic expression of an endogenously activated form of trypsinogen activation induced cellular apoptosis in vitro.
What are the new findings?
Trypsin activity was directly manipulated for the first time specifically in pancreatic acinar cells in vivo using a genetic approach.
Levels of intracellular trypsin exceeding intrinsic protective mechanisms caused the development of severe acute, but not chronic, pancreatitis.
Acinar cell apoptosis and necrosis were induced by intracellular trypsin.
Excessive levels of acinar cell death initiated acute inflammation of the pancreas.
How might it impact on clinical practice in the foreseeable future?
This study provides a novel model for specifically evaluating the biological effects of intracellular trypsin on pancreatitis.
The animal model described in this study forms an excellent platform for the testing of new treatments aimed at blocking the damaging effects of intracellular trypsin.
This model indicates that chronic pancreatitis is a complex disease and is not fully accounted for by prolonged trypsin activity.
Background and aims
Pancreatitis is the most common disorder of the exocrine pancreas and accounts for significant morbidity and mortality worldwide.1 2 It occurs in acute and chronic variants that share aetiological factors and are mechanistically linked.3 Persistent lack of effective treatments and preventive approaches still pose a challenge for clinicians and reflect our limited understanding of pancreatitis.1–4 Despite intensive investigation, molecular mechanisms that initiate the disease or drive progression to chronic pancreatitis remain poorly understood.
More than a century ago it was proposed that acute pancreatitis results from autodigestion through premature activation of digestive enzymes.5 From that time until now, pancreatic digestive enzymes have remained a major focus of pancreatitis research. Normally these enzymes are produced as inactive forms (zymogens) and only become activated upon secretion into the intestinal lumen. The protease trypsin is the key enzyme for activation of zymogens in the intestine.6 Therefore, it has long been proposed that trypsin plays a fundamental role in their premature activation within pancreatic acinar cells during pancreatitis.7 According to this prevalent hypothesis, activation of trypsinogen to trypsin occurs in response to an initial acinar cell injury and triggers the activation of other zymogens, leading to autodigestion of the pancreatic parenchyma, and initiation of pancreatitis.
Studies on hereditary pancreatitis have provided evidence in favour of this central role for trypsin activity in the disease. Identification of genetic variants of trypsinogen linked the protease to the onset of pancreatitis, and biochemical characterisation proposed an enzymatic gain of function as the initiating mechanism.8–11 However, it is unclear whether genetic variants lead to increased trypsin activity in vivo and some seem not to alter enzymatic properties of the protease at all.12 Mutations of the potent pancreatic serine protease inhibitor Kazal type 1 (SPINK1) also predispose patients to pancreatitis13 putatively by diminished trypsin inhibitory capacity. Yet, the most prevalent of these SPINK1 variants does not show any reduced ability to inhibit or bind active trypsin.14 In summary, although generally favouring a central role for trypsin activity during pancreatitis, none of the hereditary variants provides direct evidence that intracellular trypsin activity alone is sufficient to initiate the disease.
In various animal models of pancreatitis intracellular activation of trypsinogen occurs rapidly and appears to precede other markers of pancreatitis.15–17 However, these models, such as stimulation with supraphysiological doses of caerulein, duct ligation or retrograde injection of bile, are highly artificial and cause severe generalised damage. Recently, the suggested central role of trypsin as the initiator of pancreatitis has been challenged through studies revealing that acute pancreatitis can be initiated independently of trypsinogen activation.18–20 Furthermore, in one report it was suggested that trypsin may play a potential protective role, as prematurely activated trypsin efficiently cleared itself from acinar cells rather than autoactivating its own zymogen.21 Thus, the evidence for premature activation of trypsinogen being sufficient to initiate acute pancreatitis has been correlative and circumstantial.
Here a genetic model has been utilised in which trypsinogen is specifically activated within pancreatic acinar cells without the non-specific trauma of conventional models of acute pancreatitis. Using this novel approach, we found that specific intracellular trypsinogen activation effectively initiates acute pancreatitis if levels are sufficiently high. Surprisingly, intracellular trypsin primarily induced acinar cell apoptosis and caused fatty replacement instead of fibrosis associated with chronic pancreatitis.
Materials and methods
Genetically engineered mice
To target expression of paired basic amino acid-cleaving enzyme (PACE)–trypsinogen22 specifically to pancreatic acinar cells, conditional LGL (loxP-GFP-stop-loxP)–PACE–trypsinogen transgenic mice were bred with full-length elastase promoter-regulated CreErT (tamoxifen (TM)-inducible Cre recombinase) transgenic mice.23 A rat trypsinogen II (PRSS II) was used in this study since this isoform of trypsinogen is the predominant zymogen secreted in pancreatitis and chronic alcoholism.24 To initiate expression of PACE–trypsinogen, Cre recombination was induced with oral administration of TM (5 mg/day) as previously described.25 For maximal-rapid induction, TM was given for five consecutive days (TMmax, Supplementary figure 1A). For gradual-repetitive induction, TM was given every fifth day for a period of 40 days (TMgrad, Supplementary figure 1B). For experimental analyses, sex- and age-matched animals were killed at specified times after the initial TM treatment. Single transgenic littermates received the same treatment and served as controls. To induce apoptosis of pancreatic acinar cells, Cre-inducible diphtheria toxin receptor (iDTR) transgenic mice were utilised.26 After crossing with the CreErT mice, double transgenic mice were treated with TMmax to induce DTR expression in pancreatic acinar cells (DTRon). After an interval of 14 days, DT was applied for three consecutive days (100 ng intraperitoneally, twice daily, Supplementary figure 1C). All experimental procedures and housing of animals followed guidelines of the National Institutes of Health and were approved by the M.D. Anderson Cancer Center Institutional Animal Care and Use Committee.
Supplementary materials and methods
Detailed information regarding histological characterisation, quantification and scoring strategies, western blot analysis and measurement of pancreatitis parameters are described in the online Supplementary Materials and methods.
Results
Active trypsin was conditionally expressed in pancreatic acinar cells of transgenic mice
To evaluate directly the effects of intracellular trypsinogen activation, we developed a modified trypsinogen (PACE–trypsinogen) that can be recognised and activated by PACE in the trans-Golgi network (figure 1A). Functionality and cellular localisation within the secretory pathway of this PACE–trypsinogen was recently described.22 To allow tissue-specific expression, transgenic mice were generated with an LGL cassette under the control of a proximal CAG promoter followed by PACE–trypsinogen (figure 1B). For conditional pancreatic acinar cell-specific expression, these animals were crossed with transgenic mice bearing CreErT expressed from a full-length elastase promoter.23 Activation of Cre recombinase with TM enabled deletion of the floxed GFP (green fluorescent protein)-stop cassette, thereby allowing conditional expression of PACE–trypsinogen (figure 1B). For improved simplicity, these induced double transgenic mice will henceforth be referred to as PACE-trypon. Functionality of the conditional system in vivo was verified by western blot analysis (figure 1C). Measurement of elevated trypsin activity levels in homogenates of pancreatic tissues revealed successful activation of PACE–trypsinogen to proteolytic active trypsin (figure 1D). The primary mutant active trypsinogen utilised in this study had a haemagglutinin (HA) tag associated with the precursor molecule. We previously localised the precursors to the early secretory compartments but were unable to localise the active molecule as the tag is removed upon activation.22 To facilitate the localisation of the active trypsin, we further engineered a construct in which HA was fused in-frame to the C-terminus so that it can be detected after PACE cleavage. Co-localisation of the trypsin–HA and zymogen granule marker amylase indicated that the distribution of these two molecules was completely overlapping, supporting the conclusion that the activated trypsin was sorted to the secretory pathway (figure 1E).

Principle of PACE (paired basic amino acid-cleaving enzyme)-mediated trypsinogen activation in transgenic mice. (A) Wild-type (WT) trypsinogen is activated by enteropeptidase in the duodenum. Insertion of a PACE recognition site allows the genetically modified trypsinogen (PACE–trypsinogen) to be cleaved and activated by PACE within acinar cells. (SP, signal peptide; TAP, trypsinogen activation peptide; HA, haemagglutinin tag). (B) Induction of Cre activity in LGL-PACE-trypsinogen×CreErT double transgenic mice (PACE-trypon) removes the GFP-stop cassette. (CAG, chimeric human CMV-IE enhancer and chicken β-actin promoter; GFP, green fluorescent protein). (C) PACE–trypsinogen was expressed in PACE-trypon mice as shown in this western blot (1 week after induction). (D) Trypsin activity levels in pancreatic homogenates of PACE-trypon mice were significantly elevated 1 week after induction with tamoxifen (TM) (n=4, *p<0.05). (E) PACE–trypsinogen with an HA tag at its C-terminus was expressed in pancreatic acinar cells. The HA tag (red) and amylase (green) were visualised by immunofluorescence using anti-HA and antiamylase antibodies. Co-localization (yellow) of these two proteins indicated that active trypsin was sorted to the secretory pathway (scale bar 10 μm).
Homozygous but not heterozygous PACE-trypon mice developed acute pancreatitis
To determine whether direct activation of PACE–trypsinogen in pancreatic acinar cells would cause the onset of acute pancreatitis, PACE-trypon mice were analysed 5 and 7 days after initiation of Cre recombination with a maximum rapid TM induction strategy (TMmax, Supplementary Figure 1A). We discovered that gene dosage (zygosity), and therefore the level of expression of active trypsin expression, was a primary determinant of the results. Similar to littermate controls (figure 2A,B) we did not observe elevated trypsin activity (data not shown) or evident changes in pancreatic morphology of heterozygous PACE-trypon animals (figure 2C,D). Consistently, oedema and elevation of serum amylase were not detected (figure 2G,H). However, some reduction of zymogen granule density was transiently observed (figure 2D, inset). In contrast, homozygous PACE-trypon animals displayed acinar cell injury and local inflammation as early as 5 days after initiation of TM treatment (figure 2E). By 7 days, widespread deteriorating damage to the pancreatic parenchyma and pronounced local inflammation illustrated the development of severe acute pancreatitis (figure 2F). These findings were supported by significantly increased histopathological scores (figure 2I) and marked increase of both oedema and serum amylase (figure 2G,H). Together these observations illustrate that a sufficient level of active trypsin is able to cause severe acinar cell injury and acute pancreatitis.

Homozygous but not heterozygous PACE-trypon mice rapidly displayed onset of acute pancreatitis. The effects of tamoxifen (TM)-induced recombination (TMmax) on control (A, B) heterozygous PACE-trypon (C, D) and homozygous PACE-trypon (E, F) mice were examined at day 5 (A, C, E) or day 7 (B, D, F) (large panels 100×, insets 600×). Some loss of zymogen granule mass was noted with heterozygotes (D), whereas rapid induction of pancreatitis with obvious damage and oedema was only observed with homozygous PACE-trypon animals (E, F). (G, H) Increased pancreatic water content verified the presence of oedema in homozygous PACE-trypon mice (G) (n=6, p<0.05) and matched elevated levels of serum amylase (H) (n=4, p<0.01). (I) Quantification of histopathological findings confirmed pancreatic injury and acute inflammation in homozygous PACE-trypon mice (control, n=8; PACE-trypon 3–5 days, n=7, 7 days, n=14; p<0.001).
Inflammatory cell infiltration and nuclear factor-κB (NF-κB) nuclear translocation were detected in the pancreas of homozygous PACE-trypon mice
To characterise infiltration of inflammatory cells during acute pancreatitis of PACE-trypon mice we utilised immunohistochemical (IHC) staining. Visualisation of CD45, a universal marker for leucocytes, revealed a dramatic increase of inflammatory cell infiltration over time (figure 3A, upper panel). Gr1-positive neutrophil granulocytes were common in the early acute phase but their numbers declined during progression of the disease (figure 3A, middle panel). In contrast, F4/80-labelled macrophage infiltration occurred relatively later but continued to increase for several days and dominated the local inflammatory cell infiltration after 1 week of induction (figure 3A, lower panel). NF-κB activation plays important roles in the development of pancreatic inflammation.18 In order to study if NF-κB was activated in this model, we examined nuclear translocation of the NF-κB subunit p65 as an indication of NF-κB activation. Nuclear p65 was evident at a late time (7 days) but not at the beginning of active trypsin expression (3 days) (figure 3B). These results were consistent with our previous finding that extracellular but not intracellular trypsin is able to activate this pathway.22

Inflammatory cell infiltration and nuclear factor-κB (NF-κB) activation was evident in homozygous PACE-trypon mice. (A) Immunohistochemical staining with a pan-leucocyte marker CD45 revealed that PACE (paired basic amino acid-cleaving enzyme)–trypsinogen led to dramatically increased inflammatory cell infiltration of the pancreas over time (upper panel). Neutrophil infiltration, as indicated by Gr-1 staining, was abundant in the early phase and declined during progression of the disease (middle panel). Infiltration of mature macrophages (surface marker F4/80) occurred relatively late (lower panel) (all panels 100×). (B) NF-κB activation was detected by monitoring p65 nuclear translocation (brown staining, 400×, inserts are higher magnification of nuclei). PACE-trypon animals were sacrificed at 3 and 7 days after tamoxifen induction and localisation of p65 was conducted using an anti-p65 antibody.
Homozygous PACE-trypon mice displayed both acinar cell apoptosis and necrosis
To detect programmed cell death, we performed TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelling) staining. In contrast to controls, staining was abundant in PACE- trypon mice (figure 4A). In homozygous mice, a 15-fold increase in the number of TUNEL-positive cells was observed (figure 4B). Interestingly, despite the absence of apparent pancreatic injury in heterozygous PACE-trypon mice, a slight increase in TUNEL staining was also detected (figure 4A,B). To investigate if trypsin-induced apoptosis involved caspase activation, IHC staining for cleaved caspase-3 was conducted. Homozygous PACE-trypon mice displayed a dramatic increase of caspase-3-positive cells during the first week (figure 4C) which was 10–30 times more frequent than in controls (figure 4D).

Acinar cell apoptosis was prominent in PACE-trypon mice. (A) Homozygous PACE-trypon mice displayed dramatically increased numbers of TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelling)-positive cells in comparison with heterozygous animals and controls 7 days (7d) after maximal-rapid induction with tamoxifen (TMmax; 200×). (B) Quantification of apoptotic cells revealed a >15-fold increase of apoptotic cells in comparison with control (n=5, p<0.001). (C) Homozygous PACE-trypon mice showed rapid induction of caspase-3 cleavage as revealed by immunohistochemistry (200×). (D) Caspase-3-positive cells increased 10- (3–5 days) to 30-fold (7 days) in homozygous PACE-trypon mice in comparison with control (n=5, p<0.05 and p<0.001).
Despite the predominant early occurrence of apoptosis, at later stages acinar cell necrosis was also observed. Within 1 week, prominent cytosolic translocation of high mobility group protein B1 (HMGB1), a marker of necrosis,27 was observed in acinar cells of homozygous PACE-trypon mice, but not in controls (figure 5A,B). Visualisation of the extent of cellular injury with electron microscopy further supported this finding. Whereas control animals showed normal intact acini, PACE-trypon mice displayed severely damaged acini with flattened cell height and enlarged lumen. Destroyed cells leaking their cytosolic content into the acinar lumen or intercellular surroundings were common (figure 5C).

PACE-trypon mice displayed acinar cell necrosis and increased mortality. (A) Seven days after tamoxifen (TM) treatment, formalin-fixed pancreata were sectioned and stained with anti-high mobility group protein B1 (HMGB1) antibody. Prominent cytosolic translocation of HMGB1, a marker of necrosis, was frequently found in homozygous PACE-trypon mice but not controls (400×). (B) Counting cells with HMGB1 cytosolic translocation allowed quantitative analysis of necrotic cells in the pancreas of TM-treated control and homozygous PACE-trypon mice (7d). (C) Electron microscopy (4000×) demonstrated that control animals had no signs of acinar cell damage with normal endoplasmic reticulum and intact zymogen granules (left panel). In contrast, homozygous PACE-trypon mice (7d) showed abundant intercellular debris and severely damaged acinar cells leaking their content into the enlarged lumen (right panel). (D) Mortality in PACE-trypon mice with maximal-rapid induction with TM (TMmax) was significantly higher than those treated with gradual-repetitive induction with TM (TMgrad; control, n=15; TMgrad, n=12; TMmax, n=15; p<0.05). (E) Prominent neutrophil infiltration in the lung of homozygous PACE-trypon mice (7d) was identified by Gr-1 staining (200×). (F) Examination of lung tissue at a time point where mortality was expected to occur (10 days) revealed significantly elevated histomorphological scores (n=4, p<0.01).
The rate of expression of PACE-trypon determined the severity of pancreatitis
The genetic tool of Cre recombination leads to conditional expression in an on/off fashion. Unlike zygosity, it does not affect cellular expression levels. However, utilisation of an inducible Cre allowed us to modulate the spatial and temporal extent of conditional expression in the pancreas. When active trypsin was expressed simultaneously in the major extent of the pancreas using TMmax, rapid acinar cell death and inflammation throughout the entire exocrine pancreas was observed. This was accompanied by a significant increase in mortality, reaching >50% within the 90 day observation period. Most of the deaths occurred within 30 days of TM treatment (figure 5D). Despite widespread damage to the entire pancreas, islets of Langerhans were mostly intact and glucose homeostasis was not impaired (Supplementary figure 2A,C). The cause of mortality in these animals with acute pancreatitis appeared to be severe systematic inflammation with acute respiratory distress syndrome (ARDS). In order to evaluate pancreatitis-related lung injury, we examined neutrophil infiltration (figure 5E) and analysed the histology score (figure 5F). Whereas controls did not show any lung abnormalities, we found prominent neutrophil infiltrates and lung injury in most of the PACE-trypon mice, as reflected by significantly elevated histomorphological scores (figure 5F).
A single administration of TM leads to Cre recombination in only a fraction of the tissue.25 To determine whether multiple treatments, each causing a limited extent of recombination, would lead to lower mortality, we utilised a gradual-repetitive induction scheme (TMgrad, Supplementary Figure 1B). Indeed, in comparison with the maximal-rapid strategy this approach caused fewer deleterious effects and did not lead to increased mortality (figure 5D). This was despite the fact that TMgrad involved a greater total number of TM injections and would be expected ultimately to cause at least an equal extent of Cre recombination.
Chronic pancreatitis was not observed in homozygous PACE-trypon mice
In PACE-trypon mice that survived maximal induction (TMmax), neither acute nor chronic inflammation was detectable in the long term (>10 weeks). Instead, a dramatic loss of acinar cells replaced by adipocytes was observed (figure 6A). Isolated intact pancreatic acini were found in the midst of large areas of fatty tissue. Similarly, islets of Langerhans and scattered clusters of ductules were intact and completely engulfed by fatty tissue (figure 6A, inset). To determine whether concealed foci of fibrosis were present, we performed histochemical staining for collagen using Sirius red. Staining in PACE-trypon mice (figure 6C) was similar to that of unaffected controls (figure 6B). This was in stark contrast to the abundant staining found in a model of pancreatic fibrosis (figure 6C, inset). Together our observations indicated that acute inflammation receded rapidly after trypsin-induced acute pancreatitis, leaving no persisting foci of pancreatic fibrosis.

PACE-trypon mice did not develop typical chronic pancreatitis. (A) Homozygous PACE-trypon mice that survived maximal-rapid induction with tamoxifen (TMmax) showed no signs of persistent acute damage, chronic inflammation or fibrosis in the long term (>10 weeks). Rather, lost acinar cells were replaced by fatty tissue. Islets of Langerhans (inset) were not affected (large panel 100×, inset 200×). (B and C) Staining for collagen with Sirius red showed little staining in control (B) or PACE-trypon mice (C). In contrast, intense staining was observed in a fibrosis model (C, inset) (all 100×). (D and E) Homozygous PACE-trypon mice that received gradual-repetitive induction with TM (TMgrad) also did not develop fibrosis. Shortly following the last TM episode (+5 days after TMgrad) acute events and ongoing recovery of pancreatic injury were observed (D). In the long term (+30 days after TMgrad), most acute events were resolved with fatty replacement of acinar cells (E) (100×).
To determine whether gradual-repetitive induction of trypsin activation might lead to fibrosis, we examined mice after repetitive administration of single doses of TM (TMgrad) that allowed temporal modulation of PACE–trypsinogen expression and provided recurrent episodes of trypsin-mediated injury and inflammation. Histomorphological findings observed shortly following the last TM treatment (+5 days after TM) included acute events and ongoing recovery of preceding pancreatic injury (figure 6D). Despite the recurring nature of injury in this model, most of the pancreatic damage and inflammation was resolved in the long term (+30 days after TM), leaving no foci of fibrosis. Similarly to maximal induction, we observed that repetitive modulation of trypsin activity also displayed replacement of lost acinar cells with fatty tissue (figure 6E). Limited regeneration of acinar cells was observed with both TM induction strategies, but was restricted to areas of lesser damage (Supplementary figure 3A–C). Therefore, neither TMmax nor TMgrad induction strategies led to development of fibrosis or chronic inflammation.
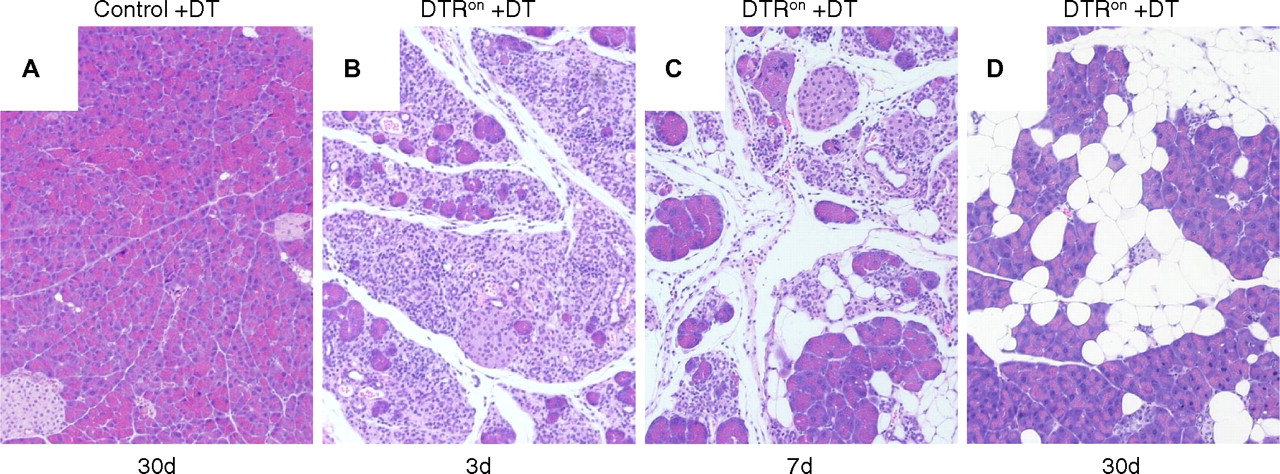
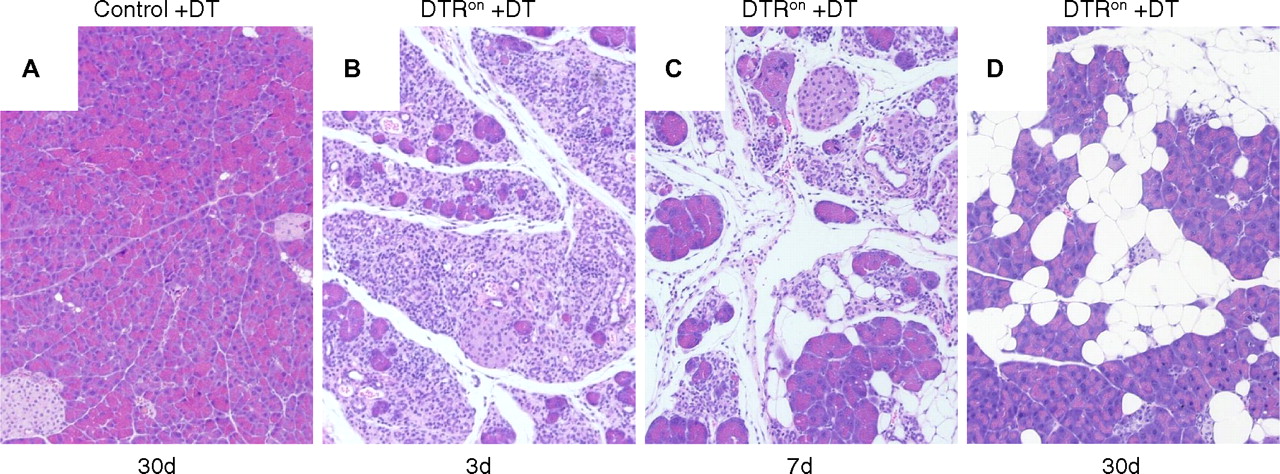
DT-induced acinar cell apoptosis caused acute inflammation and fatty replacement similar to that in PACE-trypon mice
The lack of fibrosis in PACE-trypon mice was unexpected. Because the primary cellular response to intracellular trypsin was apoptosis, we wished to determine whether specific induction of this form of cell death might explain this observation. To this end, iDTR transgenic mice were crossed with acinar cell-specific CreErT mice. Following induction of Cre recombination these animals express DTR on the surface of pancreatic acinar cells (DTRon). Administration of DT to these mice allowed efficient and specific depletion of acinar cells by apoptosis. As expected, control animals were not affected by treatment with DT at any time due to the lack of DTRs in rodents (figure 7A). In contrast, administration of DT caused rapid and efficient ablation of acinar cells in DTRon mice, leading to widespread damage and an acute phase inflammation of the pancreas within 3 days (figure 6B). Over the next several days, DT treatment led to abundant inflammatory infiltrates and loss of acinar cells (figure 6C). In the long term (30 days), large areas of fatty tissue flanked by recovered acinar cells were observed (figure 6D). Notably, no remaining foci of fibrosis or signs of chronic pancreatitis were detected. These results were similar to the observations in PACE-trypon mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diphtheria toxin (DT)-induced acinar cell apoptosis caused acute inflammation and fatty replacement. (A) DT administration did not have any deleterious effects on controls (100×). (B) In contrast, DT caused acinar cell apoptosis and acute inflammation in DTRon mice by 3 days (3d; 100×). (C) Abundant inflammatory cell infiltration and fatty replacement of lost acinar cells were observed at 7 days (7d; 100×). (D) Similar to our long-term findings in PACE-trypon mice, fibrosis did not develop in these mice and lost acinar cells were replaced by adipocytes (30d) (100×).
Discussion
In the current study, we investigated the effects of intracellular trypsin by directly modulating trypsin activity in pancreatic acinar cells of genetically engineered mice. We observed that intracellular trypsin activity was capable of initiating pancreatitis. The outcome was dependent on both the level of active trypsin expression in acinar cells and the initial extent to which the exocrine pancreas was affected. However, intracellular activation of trypsinogen in the absence of other factors was not sufficient to drive the development of chronic pancreatitis.
In agreement with the general paradigm proposing a key role for trypsin during initiation of acute pancreatitis, we observed that trypsin activity levels exceeding a threshold were able to precipitate typical acute pancreatitis with widespread damage to the pancreatic parenchyma, oedema and hyperamylasaemia, and infiltration of inflammatory cells. This genetic model provides significant advantages over conventional animal models of acute pancreatitis in elucidating the role of intracellular trypsin during pancreatitis. Most of the common models, including the caerulein hyperstimulation or bile salt injection models, not only result in activation of trypsinogen but also simultaneously stimulate multiple injury-related signalling pathways.28 29 Therefore, these models are not suitable to specifically dissect the role of trypsin activity during the onset of the disease. The novel genetic approach described in this study using endogenous PACE-mediated activation of trypsinogen allows direct evaluation of the biological effects of intracellular trypsin activity on the exocrine pancreas without the use of artificial stimuli. This model may be especially useful for preclinical evaluation of therapeutics aimed to control trypsin activity.
The trypsin activity observed in this model has a unique time course compared with that of other models. For example, the supraphysiological doses of caerulein led to a rapid and transient increase in trypsin activity.30 In contrast, in this model, trypsin activity is gradual but sustained. The peak value of caerulein-induced trypsin activity is very high and appears to be higher than that observed in the PACE-trypon mice. However, the duration of elevated trypsin is much longer in the genetic model. Likewise, the onset of acute pancreatitis is delayed in this genetic model. Evidence that the pancreatitis observed in this model is due to the expression of active trypsin includes: (1) overexpression of exogenous proteins including GFP from the same promoter does not induce pancreatitis; (2) levels of expression in heterozygotic animals which were not sufficient to induce measurable trypsin activity had no effects; and (3) in our previous in vitro study, we showed that pancreatic acinar cell damage was caused only by expression of active trypsin but not a variant of trypsin that lacked enzyme activity.22
Onset of typical acute pancreatitis was only observed in mice homozygous for the PACE–trypsinogen allele. Since trypsin is capable of activating trypsinogen and other zymogens, it may have seemed likely that even small amounts of trypsin activity would have caused deleterious effects to the pancreas. However, expression at lower levels in heterozygous mice caused only a transient loss of zymogen granules and the pancreas quickly recovered with no overt pathology despite the continued expression of active trypsin. This suggests that the intrinsic antiprotease potential of the pancreas is able to control increases of trypsin activity up to a certain amount, making it surprisingly resistant to trypsin-mediated injury.
The compartment in which trypsin activity occurs is likely to be of great importance. Currently, it remains unclear where trypsin activity arises during pancreatitis in patients. From studies with animal models, using artificial means to induce pancreatitis, several different compartments have been proposed to play a role in the initial activation of trypsinogen, including zymogen granules,31 lysosome-fused zymogen granules32 and specialised endosomes.33 Furthermore, it is likely, but unproven, that trypsin must escape from membrane-bound compartments in order to have pathological effects.17 In patients with hereditary pancreatitis, ‘gain-of-function’ effects of genetic variants were proposed to occur within the secretory pathway.31 Several of these genetic trypsinogen variants caused cell death when expressed in cells, and lysosomal cathepsins were not involved in their activation.34 35 In the current study, PACE–trypsinogen was expressed and activated within the secretory pathway and induced acinar cell death and acute pancreatitis. These findings emphasise the validity of this approach. However, it should be noted that this model is aimed to study the biological effecs of active trypsin which cannot be elucidated by non-specific injury-based models. Thus, like other artificial animal models currently in use for investigations of acute pancreatitis, this model may also not fully recapitulate the pathological process of human conditions. Nonetheless, this new model provides an excellent tool for investigation of the influence of intracellular active trypsin on pancreatitis in the absence of the confounding features of other models.
Besides the level of trypsin activity, the initial extent of PACE–trypsinogen activation was an important determinant of the outcome of pancreatitis. When maximum coordinated recombination was achieved, trypsin-mediated acinar cell injury caused severe acute pancreatitis with considerable mortality. Our observations suggest that death was due to systemic complications arising from the massive level of pancreatic damage, including necrotic cell death. This finding was similar to severe necrotising pancreatitis in patients, where mortality can be >30%.36 In contrast, repetitive but limited Cre recombination led to conditional activation in only a fraction of the pancreas at a given time, and no severe systemic complications or mortality were observed using this approach. These results suggest that normal defences can only cope with a limited level of pancreatic damage at a time. Injuries beyond that limit generate systemic effects, leading to life-threatening complications.
PACE–trypsinogen-mediated acute pancreatitis was accompanied by high levels of acinar cell death. The major form of cell death was apoptosis, although acinar cell necrosis was also identified. Apoptosis was observed earlier and some was present even in the absence of obvious deleterious effects in heterozygous animals. Thus, it appears that apoptosis is the major pathway of cell death induced by increased intracellular trypsin activity in this model. The observation of necrosis was probably of secondary nature, as late occurrence of necrosis was also observed in a model using a known apoptosis inducer, DT. Induction of apoptosis has been suggested to improve outcome of acute pancreatitis, while necrosis is considered to propagate inflammation and increase mortality.4 37–40 Therefore, the response of acinar cells to intracellular trypsin is apparently less injury provoking than might have been expected.
A surprising observation was that intracellular activation of trypsinogen did not lead to fibrosis and chronic pancreatitis. Clinical and experimental data suggest that chronic pancreatitis may develop by progression from acute pancreatitis and that trypsin activity may be a mechanistic link.3 41–43 In our model, activation of PACE–trypsinogen caused acute pancreatic inflammation and acinar cell death but no fibrosis or chronic pancreatitis. Rather, in areas of abundant acinar cell loss fatty replacement occurred. Limited regeneration was evident in areas with less dramatic damage. This suggests that when an excessive number of acinar cells are eliminated there may simply be too few healthy acinar cells left to regenerate lost exocrine pancreas. Fatty replacement is also observed in human pancreas, especially after injury.44 However, how this process is orchestrated and the origins of the adipocytes remain to be determined.
Our results suggest that trypsin-mediated injury in the absence of other factors may not be sufficient to drive pancreatic fibrosis. Even with repetitive induction of trypsin activity no smouldering inflammation or foci of fibrosis were observed. These results are in disagreement with the observation of chronic inflammation and fibrosis after repetitive treatments with caerulein.45 This suggests that caerulein treatment involves additional mechanisms apart from activating intracellular trypsin to drive fibrosis. Recently, chronic pancreatitis was reported in one46 but not another47 transgenic model of hereditary pancreatitis expressing R122H-trypsinogen. However, the R122H variant was not shown to increase intracellular trypsin activity in vivo in either study. Therefore, it remains unclear whether the biological effects observed with this mutant trypsin are related to trypsin activity. The current observations support the concept that fibrogenesis is an active process requiring other factors beyond trypsin activity and not simply the default programme following pancreatic injury.48
Our findings suggested that trypsin-induced acinar cell apoptosis might account for the majority of the biological consequences observed in PACE-trypon mice. To test this hypothesis, we employed a DT-based model to induce acinar apoptosis. DT is an exotoxin that binds to a surface receptor, the DTR, which is found in humans but not rodents. DTR internalises the toxin and causes apoptotic cell death. DTR expression has been widely utilised specifically to ablate cells in vivo26 and was adapted for pancreatic acinar cells in this study. We observed that DT-mediated apoptotic ablation of pancreatic acinar cells in the short term caused an acute phase reaction similar to acute pancreatitis. In the long term, we observed fatty replacement of lost acinar cells and partial recovery of the remaining tissue. This outcome was similar to that of mice with expression of PACE–trypsinogen and strongly supports the hypothesis that the major effect of acinar cells in response to trypsin activity is apoptosis.
In summary, these findings have major implications for our understanding of the role of trypsin activity during initiation of acute pancreatitis and its progression to chronic pancreatitis. This novel model will provide a unique tool to improve the understanding of basic mechanisms of pancreatitis and allow the testing of new treatments and preventive measures to reduce trypsin-induced acinar cell injury.
Acknowledgments
We thank Dr Georg Halder from the M.D. Anderson Cancer Center for critical review of the manuscript and valuable discussions during the progress of the project. We also thank Kenneth Dunner Jr of the M.D. Anderson High Resolution Electron Microscopy Facility for his skilful electron microscopy analysis of pancreatic samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
- Data supplement 5 - Online figure 4
Footnotes
See Commentary, p 1305
Linked article 241703.
CDL and BJ contributed equally to this work.
Funding This research was supported by funds from DK068414 (to BJ), DK052067 (to CDL), Cancer Center Support Core grant CA16672 (to M.D. Anderson Cancer Center), Pancreatic Specialized Programs of Research Excellence (SPORE) grant P20 CA101936 (to M.D. Anderson Cancer Center) and by the Lockton Endowment (to CDL).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.