Article Text

Abstract
Polymorphisms in NOD2, encoding an intracellular pattern recognition receptor, contribute the largest fraction of genetic risk for Crohn's disease among the >40 risk loci identified so far. Autophagy plays a prominent role in the innate immune response towards intracellular bacteria. The discovery of the autophagy genes ATG16L1 and IRGM as risk factors for Crohn's disease turned autophagy into the spotlight in inflammatory bowel disease (IBD). Remarkably, NOD2 has recently been identified as a potent autophagy inducer. A physical interaction of NOD2 and ATG16L1 appears to be required for autophagic clearance of intracellular pathogens. Moreover, Crohn's disease-associated NOD2 and ATG16L1 variants exhibit a defect in the induction of an autophagic response and hence predict autophagy as a key converging mechanism that leads to Crohn's disease. Another pathway that is closely intertwined with autophagy and mutually cross-regulated is the unfolded protein response (UPR), which is induced by endoplasmic reticulum (ER) stress. Genes involved in the UPR (XBP1, ORMDL3) have also been genetically associated with Crohn's disease and ulcerative colitis. Moreover, the intestinal epithelium at the interface between host and microbe appears particularly affected by IBD-associated hypomorphic function of autophagy and the UPR. The functional convergence of main genetic risk factors for IBD on these innate immune pathways has hence important implications for the host's interaction with the microbiota. Moreover, the genetic convergence on these molecular mechanisms may open novel therapeutic options for IBD that deserve further exploration.
- Crohn's disease
Statistics from Altmetric.com
Introduction
Crohn's disease and ulcerative colitis, the main clinical phenotypes of inflammatory bowel disease (IBD), are complex diseases arising from the interplay of genes that regulate immune function with the environment.1 Both diseases exhibit a particularly complex genetic underpinning, and share ∼30–40% of the genetic risk loci, despite their phenotypic differences in presentation.2 3 Apart from the divergent role of smoking in Crohn's disease and ulcerative colitis, little is known about environmental factors influencing IBD pathogenesis. Concordance rates in twin studies suggest a larger environmental contribution in ulcerative colitis compared to Crohn's disease.1
The discovery of an association between polymorphisms in NOD2 and Crohn's disease in 20014 5 (table 1) represented one of the great advances in our understanding of polygenic diseases in general, with important implications beyond IBD. It provided the proof that functional variants of single genes could indeed be identified in a polygenic disease. Due to the advent of high-throughput genotyping strategies, genome-wide association studies (GWAS) became possible and have yielded a dramatic expansion of our insight into the genetic basis of IBD over the last few years, with now ∼50 loci associated with both forms of IBD, Crohn's disease and ulcerative colitis.2 3 6 7 NOD2 was also a ‘bonanza’ in that the genetic variants most strongly associated with Crohn's disease were located in the coding region and led to an altered amino acid sequence, either due to an insertion that resulted in a frame-shift mutation, or through non-synonymous SNPs that resulted in amino acid exchanges (table 1).4 5 8 Crohn's disease-associated genetic variants were all located in the so-called leucine-rich repeat (LRR) region of NOD2, the ligand-binding domain of this intracellular pattern recognition receptor and hence suggested a common functional theme in the relationship between NOD2 and Crohn's disease pathogenesis. Since then, apart from the variants commonly observed in Crohn's disease (termed SNP8 (R702W), SNP12 (G908R), and SNP13 (1007fs) in the initial publication; table 1)4, a number of rare NOD2 variants have been discovered that add to the genetic association of this locus to Crohn's disease, and again almost exclusively localise to the LRR region.9 This deep insight into the ‘functional’ genetic variants of NOD2 contrasts significantly with the level of functional–genetic insights available to date for almost all of the other novel loci discovered through GWAS. While the locus associations based on surveying a limited number (currently 500–1000k) of SNPs are firmly established and reproducible, in many cases these loci harbour multiple genes that appear equally likely as potential causative candidates from a purely genetic point of view.2 Since DNA is inherited in larger chunks of 50–100 kb (haplotypes) across generations, current GWAS technology does not always allow the resolution of associated loci to individual genes or further to causal variants within these genes.10 Moreover, in most instances, the causal variants at a specific locus remain unknown even despite substantial re-sequencing efforts (see below).11 Another major distinction of the NOD2 association from other genetic Crohn's disease (or ulcerative colitis) associations is that its effect size is substantial, reflected in the fact that NOD2 variants alone account for the vast majority of the ∼20% of heritability explained by all genetic loci associated with Crohn's disease thus far.2 6
Summary of genetic variants mentioned in the text
Biology of NOD2
Despite the strong association between Crohn's disease and causative NOD2 variants, a comprehensive picture of how NOD2 contributes to disease pathogenesis has not yet emerged (figure 1).1 3 NOD2 is broadly expressed intracellularly in macrophages, dendritic cells, and at lower levels in intestinal epithelial cells,12 and even in T cells.13 NOD2 is activated by N-acetyl muramyl dipeptide (MDP), a component of bacterial peptidoglycan.14 15 Additional ligands of NOD2 are N-glycolyl muramyl dipeptide from mycobacteria,16 and viral ssRNA.17 NOD2 activated by MDP binds to receptor-interacting serine-threonine kinase 2 (RIP2, RIPK2), which results in TRAF6-mediated ubiquitination of NF-κB essential modulator (NEMO, IKKγ) and hence activation of the NF-κB pathway.1 Activation of NOD2 through ssRNA activates an alternative pathway, triggering interferon-regulatory factor 3 (IRF3) activation through the adapter protein MAVS (mitochondrial antiviral signalling protein) and consequently production of interferon β (IFNβ).17 The common Crohn's disease-associated NOD2 variants abrogate RIP2 binding and consequently decrease NF-κB activation upon MDP stimulation.18 NOD2 is also involved in the modulation of Toll-like receptor (TLR) signalling.19 20 Specifically, Crohn's disease-associated NOD2 variants may be inhibitory with regard to TLR2-induced NF-κB activation,19 especially upon chronic stimulation.21 While these data support a hypomorphic function of Crohn's disease-associated NOD2 variants, the frame-shift NOD23020insC variant (SNP 13) has been suggested to encode a gain-of-function aspect by actively suppressing IL10 transcription via inhibiting the nuclear ribonucleoprotein hnRNP-A1.22 Furthermore, Nod2–/– mice exhibit decreased expression of specific antimicrobial α-defensins in Paneth cells. Along these lines, oral infection with Listeria monocytogenes led to increased translocation of this model pathogen to the liver and spleen in Nod2–/– mice.23 Similarly, patients with Crohn's disease-associated NOD2 variants expressed decreased levels of α-defensins HD5 and HD6.24 Remarkably, neither Nod2–/– mice, nor mice engineered to express the murine homologue of the human NOD23020insC variant (ie, Nod22939insC) instead of wild-type Nod2 develop spontaneous intestinal inflammation under specific pathogen-free (SPF) conditions.23 25 Nod22939insC, but not Nod2–/– mice, exhibited increased severity in the dextran sodium sulfate (DSS)-induced colitis model.23 25 The Nod22939insC model was characterised by increased interleukin 1 β (IL-1β) release from MDP-stimulated macrophages, and blockade of IL-1β signalling alleviated the exaggerated DSS-induced colitis.25

Intracellular NOD2 signalling pathways. NOD2 recognises bacterial muramyl-dipeptide (MDP) and recruits ATG16L1 to the bacterial entry site at the plasma membrane; this results in wrapping of invading bacteria by autophagosomes and consecutive autophagy (1). MDP-stimulated NOD2 signalling limits peptidoglycan/TLR2-dependent activation of NFκB via IKKγ (NEMO) (2). MDP-activated NOD2 recruits receptor-interacting serine-threonine kinase 2 (RIP2) and the adapter molecule TRAF6, which leads to degradation of IKKγ and ultimately activation and nuclear translocation of phosphorylated NFκB (3). Viral ssRNA activates NOD2, which activates the transcription factor interferon regulatory factor 3 (IRF3) via mitochondrial antiviral signalling protein (MAVS) (4). The NOD23020insC variant prevents p38-mediated phosphorylation of the transcription factor nuclear protein hnRNP-A1, which under normal conditions is released in its phosphorylated form from the tripartite complex with NOD2 and p38 to translocate to the nucleus and consecutively activates IL10 transcription (5).
Autophagy: a novel pathway emerges in Crohn's disease
A fundamental cellular pathway that was until quite recently unexplored in IBD is autophagy.26 The discovery of a functional ATG16L1 polymorphism associated with Crohn's disease initially reported by Schreiber and colleagues27 and later independently reported by other groups28 29 brought this pathway into the spotlight in IBD. An association of the regulatory autophagy gene IRGM with Crohn's disease29–31 further highlighted the importance of this pathway for Crohn's disease.32
Autophagy is an evolutionarily conserved self-cannibalistic catabolic lysosomal degradation pathway.26 Basal levels of autophagy occur in essentially all cells as a homeostatic function in the process of protein and organelle turnover. Autophagy is substantially upregulated in response to starvation, when cells need to generate intracellular nutrients and energy (figure 2).26 Autophagy is also upregulated when structural remodelling is required, for example during development, or when cells need to remove damaging cytoplasmic components, for example during intracellular infections, protein aggregate formation, accumulation of misfolded proteins, or oxidative stress.26 In the process of autophagy, cytoplasmic material (eg, organelles, protein aggregates or bacteria) is engulfed into double membrane-coated autophagosomes, which subsequently fuse with endosomes and lysosomes to form autolysosomes, where degradation of its contents occurs.26

Processes inducing autophagy.
Immunity-related GTPase M (IRGM) is required during the initiation phase of autophagy, when it localises to bacteria-containing autophagic vacuoles. IRGM and autophagy are involved in the clearance of intracellular organisms such as Mycobacterium tuberculosis,33 34 and the Crohn's disease-associated IRGM variant is predicted to affect autophagic control of Salmonella typhimurium.31
Insights into ATG16L1 function have been gained through hypofunctional Atg16l1HM and myeloid-specific Atg16l1–/– mice as well as through in vitro transfection experiments (figure 3).35–37 ATG16L1 deficiency disrupts the recruitment of the ATG12-ATG5 conjugate to the isolation membrane, and consequently impairs the engulfment of cellular content destined to autophagic catabolism.35 36 Along these lines, the Crohn's disease-associated ATG16L1T300A risk variant indeed exhibited impaired capture of internalised Salmonella typhimurium within autophagosomes.37

Converging mechanisms of genetically affected pathways in Crohn's disease. Dendritic cells and intestinal epithelial cells (IECs) expressing the Crohn's disease-associated ATG16L1T300A variant or Crohn's disease-associated NOD2 variants exhibit profound defects in autophagy induction (1). Paneth cell degranulation leads to release of a multitude of antimicrobial molecules. Hypomorphic NOD2, ATG16L1 and XBP1 function results in Paneth cell dysfunction with impaired granule biogenesis and decreased α-defensin expression (2). IECs with hypomorphic XBP1 expression exhibit increased inflammatory responsiveness towards microbial (eg, the TLR5 ligand flagellin) and mucosal cytokines (eg, tumour necrosis factor (TNF)) (3), and myeloid cells expressing the Nod22939insC variant (ie, murine homologue of the Crohn's disease-associated NOD23020insC variant), or lacking Atg16l1 exhibit increased inflammasome activation upon stimulation with microbial patterns (eg, the TLR4 ligand LPS). This results in increased caspase-1 activation and consecutive increased IL-1β and IL-18 secretion (4).
Hypomorphic ATG16L1 function leads to structural alterations of Paneth cell granules with a disrupted granule exocytosis pathway.35 Moreover, the transcriptional programme of Paneth cells was profoundly altered.35 Remarkably, these phenotypic and transcriptional changes depend on an environmental contribution, namely infection with a specific type of murine norovirus (MNV, strain CR6).38 Atg16l1HM mice held in an MNV-free enhanced barrier facility exhibit a normal Paneth cell phenotype. Patients homozygous for the Crohn's disease risk-associated ATG16L1 allele (T300A) were also observed to exhibit similar structural and transcriptional alterations in Paneth cells as did Atg16l1HM mice held under conventional SPF conditions (ie, MNV infected).35 While there is no data yet that would support the suggestion that Crohn's disease is a consequence of a MNV infection, the article by Cadwell et al provides an intriguing framework for gene–environment interactions.38
In addition to these alterations in Paneth cells, LPS stimulation of myeloid cells deficient in ATG16L1 overactivates the inflammasome. This leads to IL-1β and IL-18 release through TRIF (Toll/IL-1 receptor domain-containing adaptor inducing IFNβ)-dependent activation of caspase-1.36 Despite these profound alterations in innate immune pathways, spontaneous intestinal inflammation was not observed in MNV-infected or non-infected Atg16l1HM mice, or myeloid-specific Atg16l1–/– mice.35 36 However, MNV-infected, but not uninfected Atg16l1HM mice exhibited increased susceptibility to DSS colitis, which could be abrogated by antibiotic treatment, or administration of anti-TNF or anti-IFNγ antibodies.38 Similarly, myeloid-specific Atg16l1–/– mice (with unknown MNV status) were more susceptible to DSS colitis than wild-type mice, which in this case was alleviated by IL-1β and IL-18 blockade.36
NOD2 regulates autophagy
Remarkably, two intriguing recent papers reported that NOD2 directly intersects with autophagy, and NOD2 might even physically interact with ATG16L1. Autophagy hence appears to be the key converging theme of two of the strongest genetic risk factors of Crohn's disease (figure 3).39 40 Activation of NOD2 by bacterial ligands (MDP) and living bacteria leads to ATG16L1-dependent formation of autophagic vacuoles in dendritic cells and epithelial cells.39 40 Crohn's disease-associated NOD2 variants lack this activity, and MDP-induced autophagy induction is also absent in cells harbouring the Crohn's disease-associated ATG16L1 risk-conferring variant. A NOD2-dependent defect in bacteria-induced autophagy induction and its consequences on intracellular bacterial killing was demonstrated for Salmonella typhimurium,40 enteroadherent invasive Escherichia coli (AIEC; known to be intimately associated with epithelial cells in Crohn's disease mucosa40–42), as well as Shigella flexneri.39 Such a NOD2-related defect in autophagic removal of intracellular bacteria could be overcome by pharmacological induction of autophagy with rapamycin.40 Mechanistically, it appears that normal NOD2, but not Crohn's disease-associated NOD2 variants, recruits ATG16L1 to the plasma membrane at the bacterial entry site, and therefore NOD2 is critical for wrapping invading bacteria by autophagosomes.39 In addition to NOD2-induced autophagic innate immune response towards invading bacteria, NOD2-dependent autophagy in dendritic cells was also required for efficient antigen presentation and consequent induction of CD4+ T cell-dependent immunity towards a bacterial antigen.40 These studies extend the spectrum of bacterial pattern recognition receptors that may induce autophagy from various TLRs (TLR2, TLR3, TLR4, TLR7, TLR8; expressed on the cell surface or in endosomes)43 to the intracellular pattern recognition receptor NOD2 (as well as NOD1).
Endoplasmic reticulum stress
Another pathway that has also recently emerged in IBD pathophysiology is the unfolded protein response (UPR), which is induced by endoplasmic reticulum (ER) stress.44 45 ER stress occurs upon the accumulation of misfolded or unfolded proteins in the ER, which initiates the UPR.46 47 The UPR is orchestrated by three major pathways and their associated transcription factors: PERK-ATF4, ATF6p90-ATF6p50 and IRE1-XBP1 (figure 4).47 ER stress has been genetically associated with both forms of IBD (Crohn's disease and ulcerative colitis) through a candidate gene study of XBP1,48 as well as more recently through the GWAS-based identification of the ORMDL3 locus.6 7 The candidate-gene study of XBP1 was prompted by a mouse model with a partial or complete genetic deletion of Xbp1 specifically in the intestinal epithelium. These mice developed spontaneous small intestinal inflammation with crypt abscesses, neutrophil infiltration, and ulcerations under SPF conditions, and hence closely mimicked the histological features of human IBD.48 Xbp1 deletion in the epithelium resulted in substantial ER stress in the intestinal epithelial cell (IEC) compartment, and a marked pro-inflammatory hyper-reactivity of IECs towards microbial and cytokine stimuli.48 Decreased and absent XBP1 function in IECs resulted in Paneth cell dysfunction and their apoptotic depletion, respectively, with consequent impaired handling of model bacteria.48 A deep sequencing effort in >1000 patients and controls aimed at identifying potentially rare causal variants and indeed discovered 3-fold more rare single nucleotide polymorphisms (SNPs) in patients with IBD compared to healthy controls.48 Functional studies on rare non-synonymous SNPs (ie, SNPs that lead to an amino acid exchange; ‘nsSNPs’) revealed that IBD-only XBP1 nsSNP variants exhibited hypomorphic UPR induction, which was consistent with predictions from the mouse model.48

Proximal effectors of the unfolded protein response (UPR).
The second ER stress-related IBD risk gene product, ORMDL3, localises to the ER membrane,7 and appears to be involved in protein folding and in regulating the extent of UPR activation.7 49 Functional ORMDL3 variants associated with IBD have not yet been identified and hence their biological effect on the UPR remains to be studied.
Remarkably, there appears to be a very old phylogenetic link between innate immunity and the IRE1/XBP1 axis and the UPR in general.50 51 An intriguing study recently revealed that the xbp-1 pathway in Caenorhabditis elegans is required for the survival of the stress associated with an innate immune response towards a microbial infection.50 Along similar lines, TLR3 or TLR4 ligation on macrophages directly engages a UPR-related mechanism that allows inflammatory cells to survive the ER stress associated with their activated state.52 Moreover, only the intestinal epithelium expresses an additional isoform of IRE1 (termed IRE1β), the XBP1-activating endoribonuclease, in addition to ubiquitously expressed IRE1α; hence another phylogenetic hint on the importance of this pathway at the interface with the intestinal microbiome.53 TLR signalling might not only protect cells from ER stress, but may directly activate the IRE1α/XBP1 axis, which in turn is required for optimal pro-inflammatory cytokine secretion by macrophages and hence for the control of infections.54 It is not currently known whether NOD2 may activate IRE1α/XBP1 in a similar way as TLR ligation does.54
Altogether, these studies indicate that the unfolded protein response and autophagy are deeply involved in innate immune mechanisms. This appears particularly relevant for host–microbiota interactions at the epithelial surface of the intestine in the context of IBD.
Inter-relationship of autophagy and ER stress
The unfolded protein response and autophagy are also directly intimately intertwined (figure 5).55–58 ER stress per se is capable of activating autophagy, a function that is conserved from yeast to mammals. Specifically, c-Jun N-terminal kinase (JNK; overactivated in the context of hypomorphic XBP1) downstream of IRE1, and eukaryotic initiation factor 2α (eIF2α) downstream of PERK induce autophagosome formation under conditions of ER stress.59–62 Autophagy thereby complements ER-associated degradation (ERAD) induced during the UPR.62 Moreover, Chop and ATF4 downstream of PERK increase the transcription of essential autophagy genes.63 Another direct link from ER stress to autophagy induction is via Ca2+ release from the ER, which activates 5′-AMP-activated protein kinase (AMPK),64 and protein kinase Cθ (PKCθ),65 both potent inducers of autophagy.

Relationship between endoplasmic reticulum (ER) stress and autophagy. Hypomorphic XBP1 function leads to ER stress and consecutive massive activation of inositol requiring enzyme-1 (IRE1), the kinase end endo-ribonuclease upstream of XBP1, along with activation of other branches of the unfolded protein response (UPR). PERK activates eIF2α and hence halts protein translation, but can also induce autophagy. JNK is phosphorylated due to overactivated IRE1 involving the adaptor TRAF2, which in turn may induce autophagy, but also apoptosis if unrestrained. Furthermore, ER stress induction per se due to hypomorphic XBP1 function may lead to autophagy induction as a compensatory mechanism.
Not only may ER stress induce autophagy, but vice versa, impaired autophagy can also promote ER stress.66 Specifically, suppression of a major protein involved in autophagosome formation, ATG7, in the liver leads to increased ER stress (with its downstream consequences), while restoration of ATG7 expression dampens ER stress.66 Hence, autophagy appears to be an important regulator of organelle—that is ER—function.
Further complicating the mechanistic understanding of the interaction between the UPR and autophagy is that key regulatory components of the class Ia phospho-inositide-3 kinase (PI3K), p85α and p85β, are involved in activation and nuclear shuttling of XBP1s.67 68 At the same time, class I PI3K proteins link receptor tyrosine kinases such as insulin-like or other growth factors to TOR activation and thereby repress autophagy.26 69 Additional central evidence for the close relationship between autophagy and the UPR comes from the fact that loss of tuberous sclerosis complex genes (TSC1 and TSC2), which leads to constitutive activation of mTOR, also leads to ER stress and activation of the UPR.70
IECs appear particularly affected by aberrant autophagic function and impaired UPR signalling as observed in IBD.35 38 48 71 Both the UPR as well as autophagic responses are profoundly regulated in a cell-type and condition-specific way. Studies are currently under way to experimentally investigate the interplay between these disease pathways specifically in IECs. A recent paper on a model of amyotrophic lateral sclerosis (ALS), studying motoneurons in vivo, has provided intriguing insight into the functional interplay of a defective ER stress response and autophagy.72 Genetic deletion of Xbp1 was—contrary to expectations based on the massive induction of ER stress in ALS motoneurons—associated with amelioration of disease.72 This amelioration remarkably resulted from compensatory induction of autophagy secondary to Xbp1 deletion in the nervous system, which led to reduced accumulation of mutant protein aggregates.72 Hence, while it is clear that autophagy and ER stress may intersect at multiple layers, the exact nature of this interaction in IECs (and presumably also myeloid cells like macrophages) and its consequences for the development of IBD remains to be modelled in appropriate experimental systems.
Clinical implications
As outlined above, impaired sensing and handling of (intracellular) bacteria by innate immune cells, in particular IECs, is a central theme in Crohn's disease and related to the interconnected autophagic/UPR pathways. This might have several important implications with regard to the pathogenesis as well as the therapy of this form of IBD.
Impaired handling of the microbiota
Impaired bacterial-induced autophagy as a common theme in Crohn's disease implies that a sizeable proportion of patients with Crohn's disease may harbour a defect in handling of the intestinal microbiota by the host. Some characteristic alterations in the microbial composition at the immediate IEC interface have been described in patients with Crohn's disease.73 High concentrations of bacteria were found attached to the mucosa in Crohn's disease, and those patients with the highest bacterial concentrations had inclusions within individual IECs.73 Moreover, specific strains of E coli, namely adherent-invasive E coli (AIEC), may colonise the intestinal epithelial layer.41 42 AIEC are distinguishable from conventional commensal E coli in that they may adhere, invade and replicate within IECs. Notably, recent in vitro studies indicate that a subpopulation of AIEC resides within autophagosomes, and IRGM- and ATG16L1-deficient cells exhibit increased replication of AIEC.74 Moreover, DCs from patients with Crohn's disease-associated NOD2 variants exhibited impaired intracellular bacterial killing, presumably due to a decrease of AIEC localisation to autophagolysosomes, which was reversible with the autophagy activator rapamycin.40 In addition to these effects, in a specific experimental model system, host genetic alterations may profoundly affect the composition of the intestinal microbiota and in fact lead to ‘reprogramming’ of the intestinal microbiota so that it could be inflammatory (ie, colitis-inducing) in its own right.75 Nod2–/– mice exhibit alterations in the composition of their ileal microbiota, characterised by an overall decreased load of commensal bacteria.76 However, a pathogenic role of these microbial alterations has not yet been reported (Nod2–/– mice do not develop intestinal inflammation). In human IBD, principal component analysis of 16S rRNA sequencing data of Crohn's disease, patients with ulcerative colitis and healthy controls revealed a distinct common ‘IBD subset’ with decreased diversity and which represents ∼1/3 Crohn's disease and ∼¼ of patients with ulcerative colitis.77 At the—presumably—more relevant metagenomic level, IBD patients harboured 25% fewer genes in their microbiome compared to the control population. Principal component analysis of these metagenomic data revealed independent clustering into a healthy control cluster, a Crohn's disease cluster and an ulcerative colitis cluster.78
Host genetic factors might not only alter the composition of the microbiota, the host may also respond improperly towards the microbiota.1 79 In the case of hypomorphic XBP1, NOD2 and ATG16L1 function, IECs and macrophages may become hypersensitive to the products of the bacteria making the process of bacterial mishandling self-replicating.23 35 44 45 48 76 The convergence of the main Crohn's disease-associated genetic risk factors on autophagy could also suggest that the host might be particularly at risk for bacteria that gain access to the cytoplasm of cells such as Mycobacteria sp., Shigella sp., Salmonella sp. or AIEC. Thus, these risk genes could render the host particularly susceptible to enteropathogens or pathobionts that require normal autophagic function for their eradication.
Potential future treatment options
The convergence of major genetic risk factors on autophagy and the inter-related UPR might offer novel avenues to therapeutic intervention. A pre-eminent key proximal regulator of autophagy is the target of rapamycin, mTOR kinase, which provides the major inhibitory signal that shuts off autophagy, when growth factors and nutrients are abundantly present.26 80 81 Pharmacological inhibition of mTOR with specific inhibitors like rapamycin or everolimus releases this inhibitory signal and induces activation of multiple ATG proteins that result in autophagosome formation (figure 6).82 Indeed, rapamycin (sirolimus) treatment in a patient with Crohn's disease who had failed steroid-sparing treatments such as azathioprine, metothrexate and infliximab, has recently been reported in an instructive case report.83 Extending the novel genetic insight on the contribution of ‘hypomorphic’ autophagy to Crohn's disease pathogenesis, rapamycin was administered for 6 months in a dose that yielded serum levels above 5 ng/ml. This was associated with marked clinical and endoscopic improvement. Of note, despite prior failure on infliximab, infliximab was continued during rapamycin therapy on scheduled intervals. Rapamycin is approved for clinical use in conjunction with other immunosuppressive medications in solid transplant recipients to avoid graft rejection,84 but has also been used in preclinical studies for induction of autophagy in a model of Huntington's disease.85 While a double-blind placebo-controlled clinical trial with rapamycin has not yet been performed, results of a trial with everolimus, another mTOR inhibitor, have recently been published.86 Specifically, this study was designed to compare everolimus with azathioprine and placebo to maintain steroid-induced remission, but was terminated early after a negative interim analysis.86 There was no difference between the three treatment arms, hence this study could neither show efficacy of everolimus, nor reproduce the well-established efficacy of azathioprine in this cohort of Crohn's disease subjects.86 Given the latter, a meaningful interpretation of the data is precluded.86 Assuming that the intended mode of action of mTOR inhibitors might be different in Crohn's disease (ie, induction of autophagy in IECs, macrophages, and DC) compared to organ transplantation (ie, T cell activation blockade and growth arrest), issues related to mTOR inhibitor dosing, as well as co-medications require specific scrutiny. For example, corticosteroids, which were used to induce remission in the everolimus trial, are well known to induce autophagic cell death,87 and hence corticosteroids could have precluded any potential benefit of everolimus. In summary, at this point the hypothesis that mTOR inhibitors might provide clinical benefit in Crohn's disease cannot be rejected making additional controlled clinical trials warranted. Since IECs appear to play a particularly important role in the pathogenesis of IBD,1 Crohn's disease and ulcerative colitis might even be amenable to local treatment modalities.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
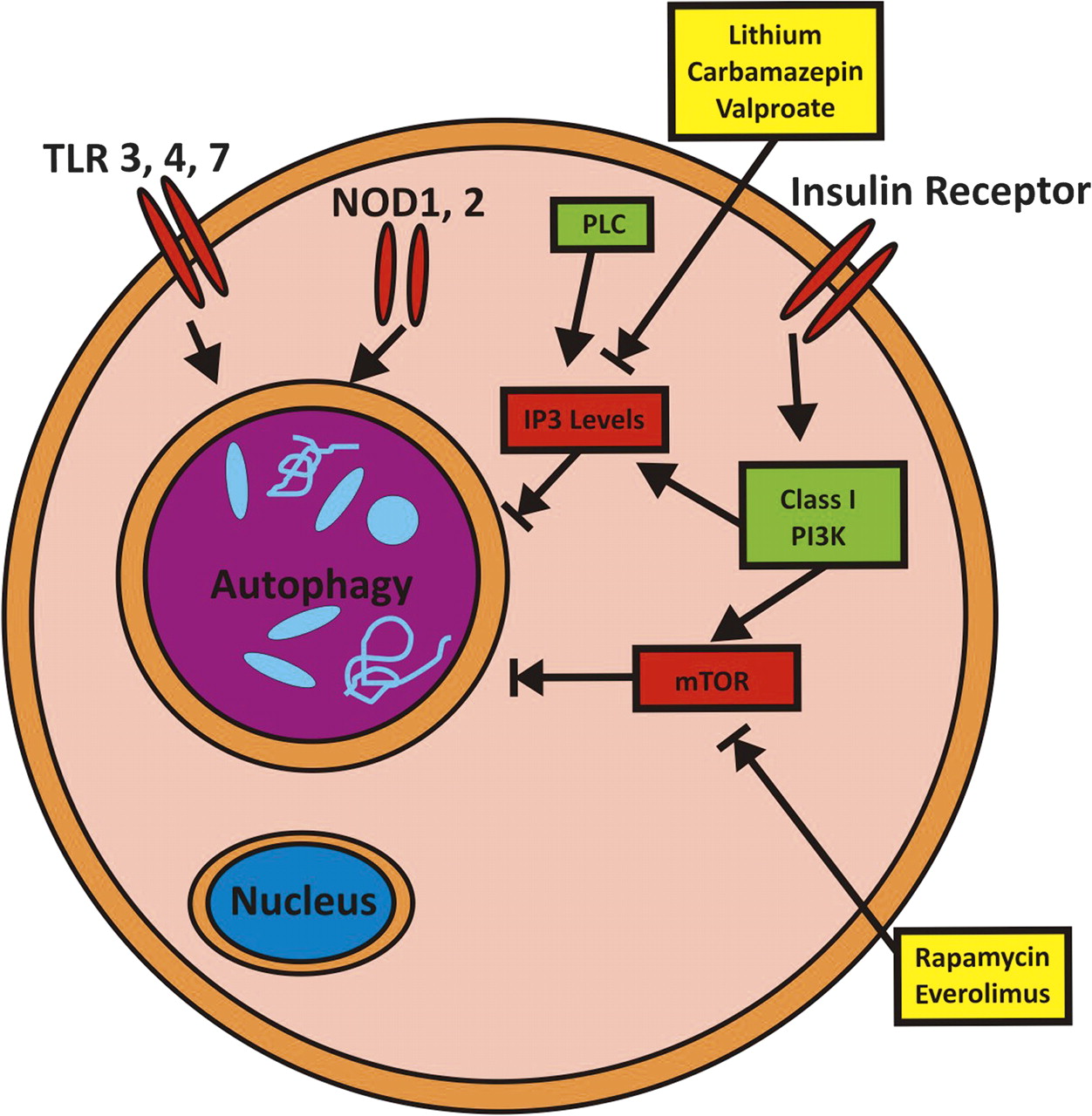
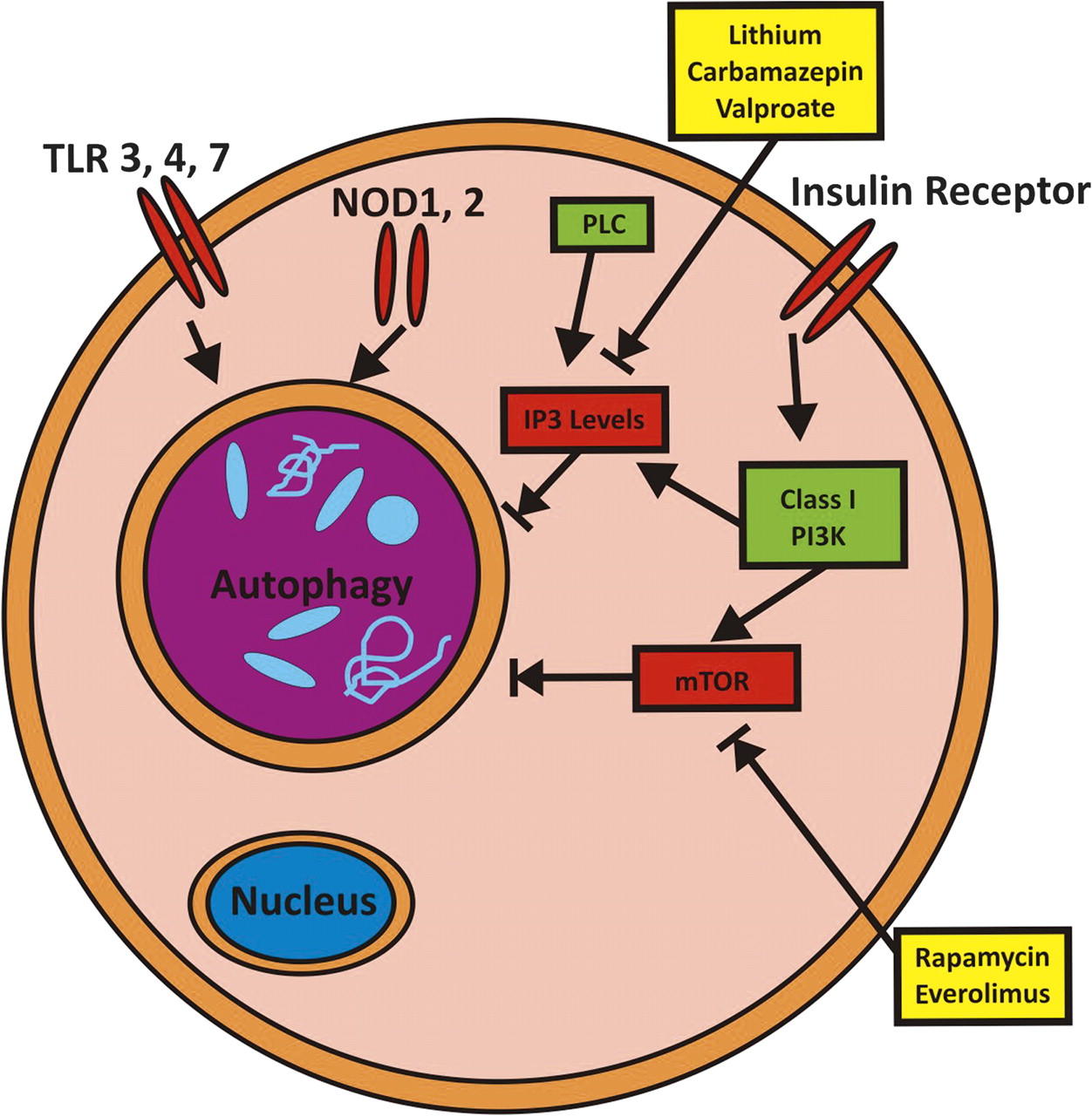
Mechanism of pharmacological autophagy induction. Autophagy is regulated by various signaling pathways, including PI3K-mTOR, IP3, Toll-like receptors (TLRs) and Nod like receptors (NLRs). Examples of pharmacological activators of autophagy are the mTOR inhibitors rapamycin and everolimus; as well as lithium, carbamazepin and valproate by lowering myo-inositol-1,4,5-triphosphate (IP3) levels.
Needless to say, mTOR is not only involved in controlling activation of autophagy, but also affects cell growth and proliferation, cellular metabolism, and other important biological pathways.80 81 Thus, pharmacological induction of autophagy via mTOR inhibition will have numerous additional effects that might or might not be beneficial in the context of Crohn's disease. Autophagy could also be induced by a mTOR-independent route by lowering myo-inositol-1,4,5-triphosphate (IP3) levels82 and via further additional pathways.88 Notably, lithium, carbamazepin and sodium valproate act on inositol metabolism and lead to decreased levels of IP3.89 Among those, carbamazepin has recently been shown to be highly effective in preventing disease via autophagy induction in an in vivo model of α1-antitrypsin (α1AT) deficiency, a disease characterised by hepatocellular aggregation of misfolded α1AT.90 A screen for pharmacological autophagy inducers that do not exert cellular toxicity revealed a further panel of eight autophagy inducers, of which seven represent FDA-approved drugs in use for various indications (fluspirilene, trifluoperazine, pimozide, niguldipine, nicardipine, amiodarone and loperamide).91 In vitro evidence further suggests that combinatorial treatment via mTOR-dependent as well as mTOR-independent induction of autophagy might have synergistic effects and therefore might offer advantages over treatment with maximum doses of single substances.82
ER stress per se may similarly be amenable to pharmacological intervention. Chemical chaperones, like 4-phenyl butyric acid (PBA; an FDA-approved drug for urea-cycle disorders) and taurine-conjugated ursodeoxycholic acid (TUDCA) have been reported to reduce ER stress in vivo with consequent normalisation of insulin sensitivity in murine models of obesity and type 2 diabetes, conditions where increased ER stress plays a central role for impaired glucose homeostasis and inflammation.92 93 Interestingly, oral administration of TUDCA ameliorated inflammation in a rodent model of small intestinal inflammation.94
Another convergent aspect of ATG16L1 and NOD2 function is the marked overproduction of IL-1β from macrophages upon microbe-related stimulation.25 36 As discussed above, hypomorphic NOD2 or ATG16L1 function may lead to an IL-1β-dependent increase in the severity of experimental colitis.25 36 Furthermore, an imbalance of IL-1β expression relative to that of its antagonist (IL-1Ra) in the intestinal lamina propria of patients with Crohn's disease,95 96 as well as efficacy of IL-1 receptor blockade in preclinical models as reported two decades ago are worthy of reconsideration.97 In addition, further preclinical data show that mice genetically deficient in the IL-1β converting enzyme, caspase-1, are protected from experimental colitis.98 It is therefore remarkable that blockade of IL-1β signalling, a quintessential inflammatory pathway, has not yet been adequately investigated in clinical trials in Crohn's disease. It should also be noted that polymorphisms in NLRP3, a critical component of the inflammasome and hence IL-1β activation and secretion, have been associated with Crohn's disease.99 However, counter-intuitively, homozygosity for the NLRP3 risk allele was associated with decreased levels of IL-1β secretion from LPS-stimulated monocytes.99
Conclusions
The observations that genetic loci associated with autophagy (ATG16L1, IRGM and LRRK2), intracellular bacterial sensing (NOD2) and ER stress (XBP1 and ORMDL3) provide risk for the development of Crohn's disease stresses the critical importance of microbial–host interactions in the pathogenesis of Crohn's disease. These insights may also open up new potential considerations for therapy.
Key messages
Intracellular sensing of bacteria and autophagy are regulated by NOD2.
Intracellular bacteria are cleared via autophagy.
Autophagy is a genetically determined (NOD2, ATG16L1 and IRGM), convergent mechanism in Crohn's disease.
Autophagy and endoplasmic reticulum stress are closely intertwined pathways.
Autophagy inducers and ER stress relievers may be potential novel treatment options in Crohn's disease.
References
Footnotes
Div of Gastroenterology and Hepatology, Dept of Medicine, University of Cambridge, Addenbrooke's Hospital, Hills Ave, Cambridge CB2 0QQ, United Kingdom; ak729{at}medschl.cam.ac.uk
Funding Work in the authors' laboratories is supported by the Austrian Science Fund (P21530 and START Y446 to A.K.), Innsbruck Medical University (grant 407 to A.K.), the European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement n 260961 (AK), the National Institutes of Health (RO1s DK088199, DK44319, DK53056 and DK51362 to R.S.B.) and the Harvard Digestive Diseases Center (DK034854 to RSB).
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.