Article Text

Abstract
Complex disease genetics has been revolutionised in recent years by the advent of genome-wide association (GWA) studies. The chronic inflammatory bowel diseases (IBDs), Crohn's disease and ulcerative colitis have seen notable successes culminating in the discovery of 99 published susceptibility loci/genes (71 Crohn's disease; 47 ulcerative colitis) to date. Approximately one-third of loci described confer susceptibility to both Crohn's disease and ulcerative colitis. Amongst these are multiple genes involved in IL23/Th17 signalling (IL23R, IL12B, JAK2, TYK2 and STAT3), IL10, IL1R2, REL, CARD9, NKX2.3, ICOSLG, PRDM1, SMAD3 and ORMDL3. The evolving genetic architecture of IBD has furthered our understanding of disease pathogenesis. For Crohn's disease, defective processing of intracellular bacteria has become a central theme, following gene discoveries in autophagy and innate immunity (associations with NOD2, IRGM, ATG16L1 are specific to Crohn's disease). Genetic evidence has also demonstrated the importance of barrier function to the development of ulcerative colitis (HNF4A, LAMB1, CDH1 and GNA12). However, when the data are analysed in more detail, deeper themes emerge including the shared susceptibility seen with other diseases. Many immune-mediated diseases overlap in this respect, paralleling the reported epidemiological evidence. However, in several cases the reported shared susceptibility appears at odds with the clinical picture. Examples include both type 1 and type 2 diabetes mellitus. In this review we will detail the presently available data on the genetic overlap between IBD and other diseases. The discussion will be informed by the epidemiological data in the published literature and the implications for pathogenesis and therapy will be outlined. This arena will move forwards very quickly in the next few years. Ultimately, we anticipate that these genetic insights will transform the landscape of common complex diseases such as IBD.
- Crohn's disease
- inflammatory bowel disease
- ulcerative colitis
Statistics from Altmetric.com
Introduction
The pace of gene discovery in complex disease genetics has increased rapidly in recent years, catalysed by the advent of genome-wide association (GWA) studies (see box 1 and figure 1). Few complex diseases have seen as much rapid progress as Crohn's disease and ulcerative colitis, thanks in no small part to large collaborations of national and international groups.6 The international inflammatory bowel disease genetics consortium (IIBDGC) presently consists of members from over a dozen countries throughout Europe, North America and Australasia, who together have amassed some 20 000 cases for each of Crohn's disease and ulcerative colitis, along with a similar number of population-based healthy controls (http://www.ibdgenetics.org/). The corresponding statistical power of such large sample sets has proven highly effective in identifying multiple susceptibility loci, even where these confer only modestly increased risk of disease. To date there are 99 IBD susceptibility loci: 71 associated with Crohn's disease, 47 with ulcerative colitis, and 28 with both Crohn's disease and ulcerative colitis (figure 2).
Advances in complex disease genetics
While it has long been established that a wide variety of complex human diseases and traits have an important genetic component (as demonstrated by substantially increased sibling relative risk, twin studies and adoption studies), the specific loci involved have remained largely elusive. Family-based linkage studies, which identified thousands of Mendelian disease genes, had few successes in complex disease (with the notable exception of NOD2 and Crohn's disease).1 2 These studies led to conclusion that complex diseases were not caused by a handful of highly penetrant mutations, but were instead affected by a range of both common and rare mutations that influence disease risk only subtly. The sequencing of the human genome and the generation of public resources of single nucleotide polymorphisms (SNPs),3 allowed the design of powerful population based genome-wide association (GWA) studies.
Within the space of a few years the landscape of complex disease genetics had been transformed by the identification of hundreds of bona fide associations. Studies of inflammatory bowel disease (IBD) led the way both in terms of early results and total number of loci identified to date. These successes underscore several key aspects to study design; large sample sizes (thousands of cases and controls), stringent quality control on the billions of genotypes comprising the typical GWA study, accurate phenotyping, and careful adjustment for potential confounding factors are the hallmarks of successful GWA studies.4
Despite this unprecedented increase in the number of genomic loci associated to complex traits, a number of key questions remain unanswered. Only a fraction of the heritability of most complex traits has been explained by GWAS results,5 and translating association results to biological understanding also requires winnowing down sometimes-long lists of genes in associated regions to implicate functional mutations. The strong correlation between nearby variants (an advantage of the GWA approach which makes the studies more cost efficient) is a barrier to this final step of dissecting associations. This next challenge is now being tackled by both general and disease-tissue specific studies of gene expression, and deep sequencing of associated regions to reveal more completely the allelic spectrum of disease risk.

Genome-wide association studies (GWAS). Thousands of unrelated disease cases and healthy population controls (A) are required for well-powered studies. These are genotyped (B) on up to a million SNPs across the genome, and subsequently called and subjected to rigorous quality control. Case–control association statistics (C) highlight regions of the genome associated with disease. Each of these regions (circled example is IL23R region) must be carefully followed up (D) by replicating the signal in independent samples and studying genes and other functional elements in associated regions. The ultimate outcome of GWAS is a deeper biological understanding of disease (rather than, say, prediction), such as the discovery that a number of genes (E) involved in Th17 cell signalling are all key genetic risk factors for IBD.

Inflammatory bowel disease susceptibility loci. The loci (depicted by lead gene name) attaining genome-wide significance (P<5×10−8) are shown for Crohn's disease (red), ulcerative colitis (blue) and IBD (black where p<5×10-8 in Crohn's disease (CD) and ulcerative colitis (UC); red where p<5×10−8 in Crohn's disease and <5×10−4 in ulcerative colitis; blue where p<5×10−8 in ulcerative colitis and <5×10−4 in Crohn's disease).
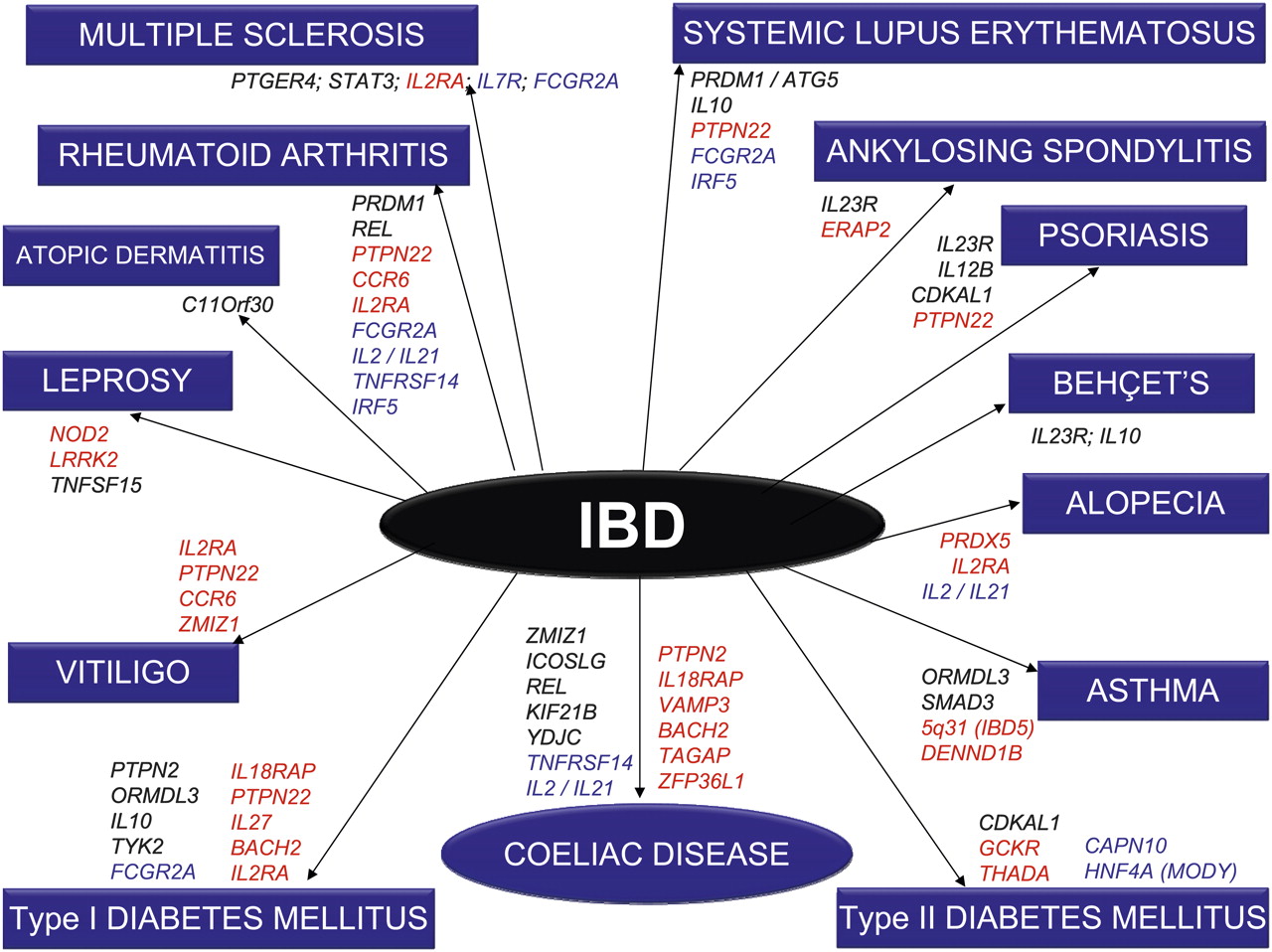
One of the inherent strengths of the new genetic technology is that it is essentially ‘hypothesis-free’. In stark contrast to previous candidate-gene studies, robust statistical analysis precedes biological plausibility, which comes later to interpret signals in multigenic loci. The most obvious benefit from this approach has been the truly novel insights into disease pathogenesis that have arisen: autophagy in Crohn's disease is already a prominent example.7 Among other emerging themes is the realisation that much overlap exists in the genetic contribution to complex diseases.8 9 Much of this might have been anticipated (eg, coeliac disease and ulcerative colitis), but beyond the major histocompatibility complex (MHC), confirmed regions of shared susceptibility were previously lacking. Since the advent of GWA studies, however, some genuinely surprising and novel areas of overlap have become apparent, between both IBD and other immune-mediated diseases (including ankylosing spondylitis, psoriasis, systemic lupus erythematosus (SLE) and type 1 diabetes mellitus (T1DM)) as well as IBD and some non-immune disease (including type 2 diabetes mellitus (T2DM)) (figure 3). Overlap is also seen with other gastrointestinal diseases, including colo-rectal cancer (CRC).

Disease overlap. To ensure clarity of present data as well as presentation, only those genes that have attained genome-wide levels of significance in all diseases are depicted here. IBD loci (Crohn's disease and ulcerative colitis) are depicted in black, Crohn's disease (CD) only loci in red and ulcerative colitis (UC) only loci in blue.
It is hoped that a detailed understanding of these common and, perhaps just as importantly, distinct loci will provide novel insights both into the biology and the treatment of complex genetic disease, including IBD. Clarification of pathogenic pathways specific to Crohn's disease and ulcerative colitis may herald a new era of targeted therapies with less unwanted systemic toxicity than seen with presently available options. Conversely, expensive research and development by the pharmaceutical industry may be offset by opportunities to target multiple phenotypes with the same agent, as has been seen with antibody therapies against tumour necrosis factor (TNF).
Both Crohn's disease and ulcerative colitis are associated with inflammatory disease outside the gut in selected patients. These extra-intestinal manifestations may affect the skin (eg, pyoderma gangrenosum and erythema nodosum), eyes (eg, iritis and uveitis), joints (enteric arthropathy), and liver (eg, primary sclerosing cholangitis/auto-immune hepatitis). Some of these conditions mirror disease activity in the bowel; others are distinct. Precise aetiologies are unknown, but roles for peripheral trafficking of activated lymphocytes and systemic macrophage immunodeficiency, have been proposed. Genetic factors beyond the human leukocyte antigen (HLA) region are probably relevant, but await discovery in large, accurately phenotyped cohorts.
Several recent studies have attempted to document the epidemiological overlap between IBD and various auto-immune diseases.10–13 However, with the exception of the Manitoba population in Canada,10 most of these studies are based on selective disease cohorts and not on population-based data leading to potential ascertainment bias. Overall, these studies point to an increased prevalence of ankylosing spondylitis, psoriasis, asthma, atopic dermatitis, multiple sclerosis (MS) and possibly rheumatoid arthritis (RA), in patients with IBD (table 1).
Epidemiological overlap between inflammatory bowel disease (IBD) and other diseases
In the present review, we first provide a brief overview of the present knowledge of the genetic architecture of IBD (for a detailed review, see Lees and Sansangi6). We then discuss the shared inherited susceptibility between IBD and immune and non-immune mediated disease, before considering the implications for novel therapeutics and future insights. Each disease will be considered in turn, with a brief summary of the epidemiological evidence for clinical overlap preceding discussion of common susceptibility loci. Data were obtained from two main sources: (1) the National Human Genome Research Institute catalogue of published genome-wide association studies (http://www.genome.gov/gwastudies/; last accessed on 1 December 2010)24; and (2) PubMed literature search (“ulcerative colitis OR Crohn's disease OR inflammatory bowel disease AND disease x”). For the sake of clarity we will focus on genes/loci where there is genome-wide evidence for association (p<5×10−8 in combined analysis). Where only nominal evidence for association (p<0.05) has been attained to date this will be clearly described in the text.
Background to IBD genetics
The precise aetiology of Crohn's disease and ulcerative colitis is not presently known. The current working hypothesis is of dysregulated mucosal immune responses to commensal gut flora in genetically susceptible individuals.25 The sibling relative risks (λs) for Crohn's disease and ulcerative colitis have been estimated at 25–42 and 8–15, respectively,26 although these may be over-inflated.27 Hence both genetic and environmental (eg, gut flora, smoking, geography, diet) factors are critical in disease pathogenesis. Gene–environment interactions will almost certainly be central and underlie the complexity of disease phenotype.
At the start of 2010, 60 published IBD susceptibility loci had been discovered and robustly replicated.6 In recent months, meta-analyses of GWA studies in Crohn's disease and ulcerative colitis have been performed by the IIBDGC, increasing the tally to 99.28 29 A much clearer picture of the genetic architecture of Crohn's disease and ulcerative colitis is beginning to emerge, prompting a paradigm shift in our understanding of disease pathogenesis. The majority of confirmed susceptibility loci confer susceptibility to both Crohn's disease and ulcerative colitis (ie, represent general IBD risk loci), but many are specific to either Crohn's disease or ulcerative colitis. The overlap and separations here are in themselves most informative.
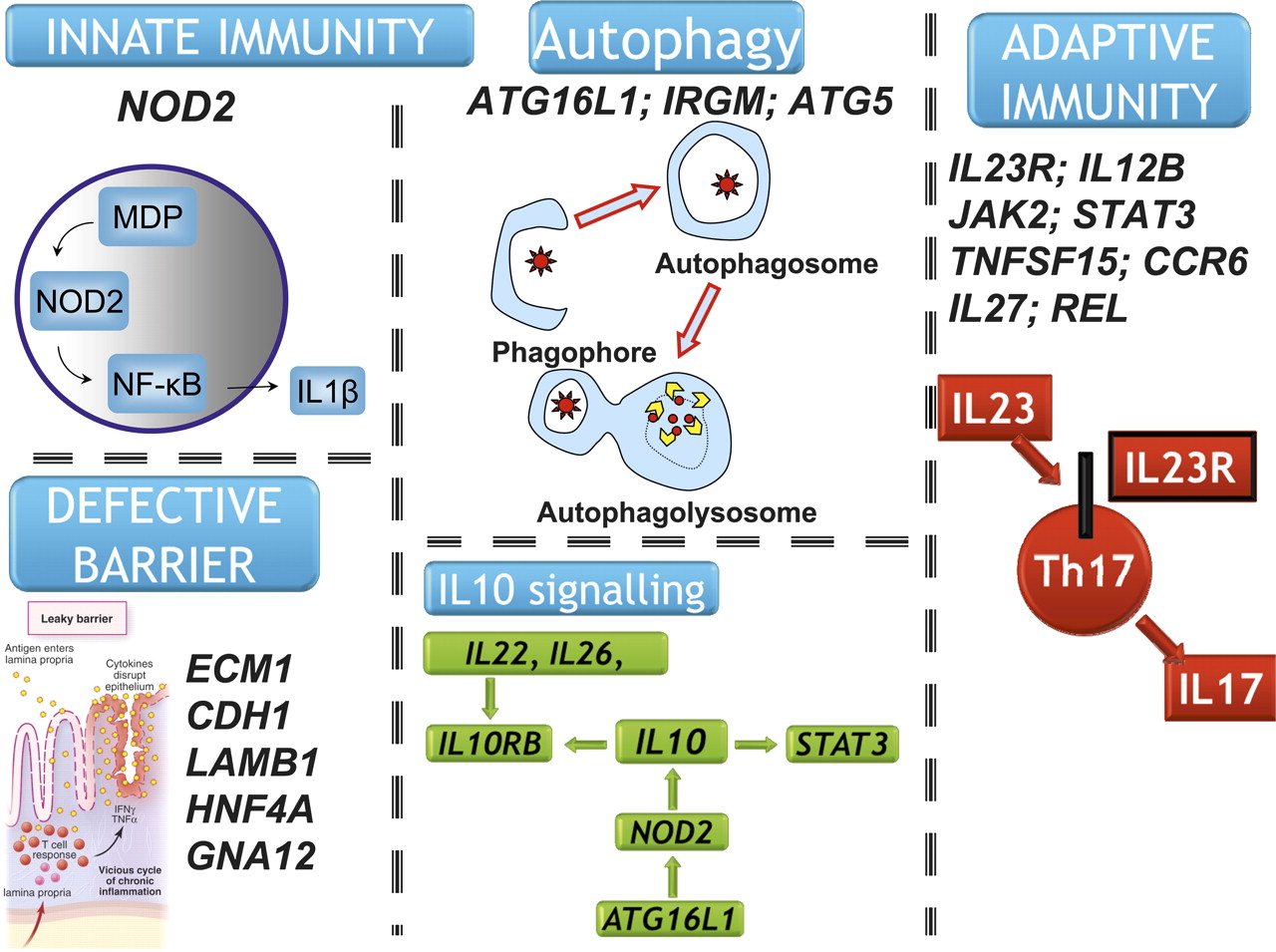
Many components of the IL23 pathway (IL23R, IL12B, STAT3, JAK2 and TYK2) are true IBD susceptibility genes, suggesting a critical role for this pathway in maintaining intestinal immune homeostasis (figure 4).30 Furthermore, IL10 signalling increasingly appears to play a central role in IBD pathogenesis, with germ-line variants associated with ulcerative colitis and Crohn's disease.31 32 This observation is supported by a recently described primary immunodeficiency resulting from autosomal inheritance of functional IL10R1 and IL10R2 mutations and characterised by a severe IBD-like phenotype in the first year of life.33 Other noteworthy IBD susceptibility genes include IL1R2, REL, CARD9, NKX2.3, ICOSLG, PRDM1, SMAD3 and ORMDL3.28

IL23 pathway/Th17 signalling and IBD susceptibility genes. IBD susceptibility genes (confirmed at genome-wide levels of significance) are denoted (*); those that are also associated with other diseases are denoted (**).
A clear emerging theme in Crohn's disease pathogenesis is the central role of defective processing of intracellular bacteria. This critical insight has arisen largely from gene discovery, and the definition of Crohn's disease-specific genes. Among these, NOD2,1 2 ATG16L134 and IRGM35 36 have focused attention on innate immunity generally,37 and more specifically on microbial recognition and autophagy (figure 5). Most recently, key functional links have been elicited between NOD2 and autophagy.38 39 NOD2 initiates autophagy by recruiting ATG16L1 to the cell membrane at the site of bacterial entry, a process that requires RIPK2, ATG5 and ATG7. Dendritic cells from patients with Crohn's disease, with NOD2 or ATG16L1 mutation, are defective in autophagy, bacterial handling and antigen presentation.38 39
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key pathways arising from gene discovery in Crohn's disease and ulcerative colitis.
While NOD2 and autophagy gene variants do not increase risk of ulcerative colitis, recent GWA studies and meta-analysis have shown that a number of epithelial barrier genes (ECM1, CDH1, HNF4A, LAMB1 and GNA12) are specifically associated with ulcerative colitis and not Crohn's disease (figure 5).28 40 41 Other ulcerative colitis specific genes of note include IL7R, IL8RA/IL8RB, DAP, LSP1 and IRF5.28
In Crohn's disease, NOD2 is associated with a severe stricturing/penetrating ileal disease phenotype.42 In ulcerative colitis, the HLA DRB1*0103 is associated with extensive and severe disease.41 Aside from these early observations, genotype–phenotype analysis of IBD genes has seen very little success. A number of associations have been described in single cohorts in the literature, but none of these have replicated consistently. Furthermore, there is to date only one published example of gene–gene interaction (epistasis) among confirmed IBD genes; McGovern and colleagues recently reported epistasis between CARD9 and REL in ulcerative colitis.43
While the biology and relevance to IBD pathogenesis of many genes within confirmed susceptibility loci is rapidly emerging, there remains much work to do in translating these gene discoveries. The increased resolution afforded by fine-mapping studies will further inform this translational effort. There are only a handful of the 99 confirmed susceptibility loci where the causative mutations are known. The three common mutations in the LRR domain of NOD2 are the classic example. However, despite this knowledge, a 5–10 year head-start on other disease loci, multiple animal models and hundreds of published functional studies in vivo and in vitro, there remains debate as to whether they are loss- or gain-of-function mutations.6 Recently, Gallagher and colleagues have provided a potential functional explanation for IL23R mutation (Gallagher G et al, DDW 2010). The rs11209026 mutation (G1142A/p.R31Q) resides in exon 9 which encodes a transmembrane region of IL-23R protein. The protective A allele causes alternative splicing, moves the stop codon and results in removal of the transmembrane domain from the protein. The resulting soluble interleukin 23R (IL-23R) protein (termed delta-9) binds IL-23, taking it out of circulation and decreasing Th17 cell differentiation. Translational efforts are under way to investigate the use of delta-9 as a biomarker and a therapeutic agent in Crohn's disease (http://www.humigen.org/delta9/).
Genetic overlap of IBD with other diseases
Several recent reviews have examined in detail the shared susceptibility genes between different immune-mediated and inflammatory diseases.8 9 Zhernakova analysed data from 28 GWA studies (including six nsSNP scans) and 28 replication meta-analyses in 11 immune-mediated/inflammatory disorders (ankylosing spondylitis, asthma, auto-immune thyroid disease, coeliac disease, MS psoriasis, RA, SLE, T1DM, Crohn's disease and ulcerative colitis).9 Based on data collated at the end of 2008, they identified 23 genes that were shared by two or more diseases. The majority of these were involved in T-cell differentiation, immune cell signalling and the innate immune response. The IMAGEN consortium compared the contribution of the MHC region (1472 tagging SNPs typed across 3.4 Mb) to over 10 000 patients with Crohn's disease, ulcerative colitis, SLE, RA, MS, myasthenia gravis, selective IgA deficiency and healthy controls.44 The data actually reveal surprisingly little overlap, with complex, multilocus effects spanning the entire region contributing to the MHC associations with chronic immune-mediated disease. In contrast, Baranzini's network-based analysis derived from existing GWA studies in Crohn's disease, T1DM, RA, SLE, psoriasis, MS and coeliac disease, demonstrated a cluster of MHC-genes for RA and T1DM, and MS and SLE; there were no MHC associations for Crohn's disease in this analysis.45 While there were no MHC associations for Crohn's disease in this analysis, Crohn's disease was still connected to the network core by virtue of shared genes including IL23, IL12B, ORMDL3, PTPN2, PTPN22, and CDKAL1.45
However, we are in such a rapid phase of data accumulation that the field has moved on significantly. Based on our analysis of the literature and GWA catalogue we now identify approximately 51 IBD genes that overlap with 23 different diseases (figure 3 and box 2). This presently observed overlap includes other gastrointestinal (GI) conditions (coeliac disease, CRC and PBC), autoimmune diseases (MS, RA, psoriasis, atopic dermatitis, SLE, ankylosing spondylitis, T1DM, asthma and vitiligo), mycobacterial infection (leprosy and possibly tuberculosis), and non-immune diseases (T2DM, dyslipidaemia, obesity and osteoporosis). The genes that are associated with the most different diseases are IL2RA, PTPN22 (five each), FCGR2A (four), IRF5, IL10, IL23R, IL2/IL21, and ORMDL3 (three each).
Other themes
Sarcoidosis and Crohn's disease. Sarcoidosis and Crohn's disease are both chronic granulomatous diseases characterised by chronic mucosal inflammation and a similar distribution of extra-intestinal and extra-pulmonary disease (eg, skin, joints, eyes and liver). Coexistent disease is described only in a handful of case reports in the literature.21 A combined analysis of a limited (100 kb) GWA study in Crohn's disease and sarcoidosis identified a common susceptibility locus on 10p12.1 (but not at GWA levels of significance).46 Fine-mapping localised the signal to C10Orf67, a gene of unknown function. Nominal association was also described in ulcerative colitis. BTLN2 is also a possible common susceptibility gene.47 48
Behçet's disease and IBD. Two recent GWAS of Behçet's disease, one from Turkey and the other from Japan, have provided definite evidence of association at HLA, IL23R and IL10.49 50 Such close overlap with IBD is notable given how much these diseases mimic each other. However, the signals at both IL23R and IL10 are distinct to those in Crohn's disease and ulcerative colitis. There is no association in Behçet's disease with the IL23R coding variants associated with Crohn's disease, ulcerative colitis, psoriasis and ankylosing spondylitis. The Behçet's disease-associated IL10 variants, while causing reduced serum IL10 expression, are not associated with ulcerative colitis. Similarly, the rs3024505 variant found with ulcerative colitis, Crohn's disease, T1DM and SLE is not associated with Behçet's disease.
Alopecia areatia and IBD. A recent GWAS in alopecia has identified association at PRDX5, IL2RA (both common with Crohn's disease) and at the IL2/IL21 locus (common with ulcerative colitis).32 51 52
Vitiligo and IBD. Aside from the odd case report, there is no epidemiological evidence for overlap between vitiligo and IBD.13 However, there are common associations with PTPN22, BTLN2, CCR6 and IL2RA in Caucasians,53 54 CCR6 and ZMIZ1 are also associated with vitiligo in Chinese individuals.55
Liver disease and IBD. The clinical overlap between ulcerative colitis and cholestatic liver disease is long established. Only one GWA study in primary sclerosing cholangitis (PSC) has been performed to date.56 The only hit outwith the HLA region that approached GWA significance was at a locus on 13q31 containing GPC6. This has yet to be tested in ulcerative colitis. Nominal association was also seen for 2/15 ulcerative colitis loci tested in the PSC panel: 2q35 (containing ARPC2) and 3p21 (containing MST1). Recent GWA studies in primary biliary cirrhosis have demonstrated association in 3 IBD loci: ORMDL3, IRF5 and MMEL1/ TNFRSF14.57 58
IBD and osteoporosis. There is an increased risk of osteoporosis in patients with IBD, independent of corticosteroid usage. There is common association at TNFSF11 (synonym: RANKL) with Crohn's disease and bone mineral density.59 The most associated SNP in a probable ulcerative colitis locus on 1p36 (just falls short of GWA significance in index study),40 is also the strongest signal in this region for osteoporosis (both hip and spine bone mineral density).59 This locus is a gene desert, with the nearest genes encoding ZBTB40 and WNT4.
E-cadherin and colo-rectal cancer. The link between CRC and chronic colonic inflammation (both ulcerative colitis and colonic Crohn's disease) is well established,60 with routine colonoscopic surveillance recommended for those at greatest risk. The first genetic link between ulcerative colitis and CRC was recently established. A locus containing E-cadherin (CDH1) is associated with CRC,61 ulcerative colitis,40 and possibly Crohn's disease.62
IBD risk loci and miscellaneous malignancies. In addition to CDH1 and CRC, several IBD risk loci have been associated with a variety of solid organ and haematological malignancies. These include prostate cancer (THADA),63 breast cancer (ZNF365, LSP1, and ZMIZ1),64 65 glioma (RTEL1),66 myeloproliferative disorders (JAK2),67 and acute lymphoblastic leukaemia (IKZF1).68 69
It is noteworthy that those diseases that share susceptibility loci with both Crohn's disease and ulcerative colitis typically harbour variants in key immune genes (eg, IL23R, IL10, IL12B, IL27, IL18RAP). In contrast, where variants appear specific to either Crohn's disease or ulcerative colitis, the implicated genes do not, by and large, have such clear roles in adaptive immunity.
While in most cases the direction of effect is the same in all diseases studied (ie, conferring susceptibility or protection to disease), in a few instances it is seen to be clearly divergent. The p.R620W PTPN22 mutation is protective in IBD while conferring susceptibility to SLE, RA, and T1DM.70 The p.H131R mutation in FCGR2A, important for removal of immune complexes, is another example.71 The Arg131 mutation is functionally associated with SLE, MS and T1DM, but it is the His131 variant, that has greater affinity for IgG immune complexes, which is associated with ulcerative colitis.72 For many susceptibility loci, we must await fine-mapping studies to delineate this further.
IBD and ankylosing spondylitis
Genes involved: IL23R, PSMG1 and ERAP1/2
Increased rates of ankylosing spondylitis in IBD have been recognised clinically for many years.15 However, population-based studies have largely been lacking. More recently, data from the IBSEN study reported the prevalence of ankylosing spondylitis in IBD (6 years after diagnosis) to be 3.7% (2.6% in ulcerative colitis; 6% in Crohn's disease), compared to about 1% in the general population.14 Cohen calculated ORs for developed ankylosing spondylitis of 7.8 and 5.8 in two separate North American datasets.11
The contribution of HLA-B27 to ankylosing spondylitis susceptibility was established in the early 1970s. No other susceptibility genes were discovered in the pre-GWA scanning era. Definite association has now been established at IL23R, ERAP1/2, PSMG1 and 2p15,73 74 with nominal evidence at IL1R2, ANTX22, TNFSF15, TNFR1 and TRADD.75 Of these, IL23R, PSMG1, ERAP1/2 and TNFSF15 are also established IBD loci.32 70 76 77
IBD and psoriasis
Genes involved: IL23R, IL12B and CDKAL1
The increased risk of psoriasis in IBD was first documented in a case–control study in 1982, where first-degree relatives of patients with IBD also had increased rates of psoriasis.18 The authors of this study were astute enough to postulate that there might be common genetic factors. More recently, Bernstein and colleagues confirmed these clinical findings in a large population-based study in Manitoba. They demonstrated that patients with Crohn's disease and ulcerative colitis had a significantly increased risk of having psoriasis compared to population controls (greater effect in males vs females).10 Similarly, patients with psoriasis had an increased risk of Crohn's disease and ulcerative colitis. A North Carolina cross-sectional study also reported an increased prevalence of psoriasis in patients with IBD (OR 1.7).13 Moreover, anti-TNF therapy appears to induce psoriatic type lesions in patients with IBD and spondyloarthropathies.78 79 This is most intriguing, given that anti-TNF therapy has established efficacy in psoriasis.
Genetic findings support this clinical overlap. IL23R and IL12B are both confirmed susceptibility genes for Crohn's disease, ulcerative colitis and psoriasis.80 81 There may be an additional shared locus (ulcerative colitis and psoriasis) at 13q13.40 82 CDKAL1, associated with Crohn's disease and T2DM (see below), is also associated with psoriasis but just falls short of genome-wide significance to date.83
IBD and systemic lupus erythematosus
Genes involved: PTPN22, IL10, FCGR2A, PRDM1 and IRF5
True co-existence of IBD and SLE (anti-dsDNA antibody positive) is described in the literature, but is uncommon.20 There may be a slight preponderance of ulcerative colitis cases in the reported series. A definitive diagnosis of both diseases is difficult due to the frequent GI involvement in SLE (occasionally caused by vasculitis of mesenteric vessels) and the systemic features common to both diseases (eg, oral ulceration and arthritis). Furthermore, sulfasalazine, 5-amino salicylic acid (5-ASA) and infliximab therapies for IBD may all result in drug-induced lupus.
The main common genetic factors are between ulcerative colitis and SLE: germline variants in both IL10, FCGR2A and IRF5 are associated with both diseases.31 72 84–86 Of note, the ulcerative colitis-associated nsSNP in FCGR2A is actually protective in SLE. In addition, the p.R620W PTPN22 variant which is protective in Crohn's disease,70 confers susceptibility to SLE.87 The PRDM1/ATG5 locus is common between Crohn's disease, ulcerative colitis and SLE.28 70 86 88 The most associated SNP in the Crohn's disease meta-analysis (rs7746082) is in a small gene desert on 6q21; however, the closest adjacent genes are PRDM1 and ATG5 (112 kb and 197 kb downstream respectively).70 A Caucasian SLE scan demonstrated association within ATG5 itself (rs573775),86 while in a Chinese SLE scan the most strongly associated SNP was within 26 kb of the Crohn's disease SNP.88
IBD and rheumatoid arthritis
Genes involved: PTPN22, CCR6, FCGR2A, REL, PRDM1, IL2/IL21, TNFRSF14, IL2RA and IRF5
An increased prevalence of RA is reported in cross-sectional analyses of IBD patients from three North American health insurance populations (ORs of 1.9 (CI 1.5 to 2.3), 2.7 (CI 2.4 to 3.0) and 2.1 (CI 1.8 to 2.3)).11 13 However, prospective epidemiological studies are required to confirm or refute this observation.
The major genetic contribution to RA susceptibility aside from MHC alleles includes variants affecting nuclear factor kappa B (NF-κB) activation, IL-2 signalling and T cell activation.89 The IL2/IL21, TNFRSF14, and IRF5 loci are common to both RA and ulcerative colitis43 51 90–93; the IL2RA locus and a regulatory variant in CCR6 are associated with RA and Crohn's disease.70 93 94 A variant in PTPN22 confers susceptibility to RA and protection against Crohn's disease.70 95 The REL locus is common to ulcerative colitis, Crohn's disease and RA. GRAIL (Gene Relationships Across Implicated Loci) analysis in RA yielded two further loci previously associated with IBD: the PRDM1/ATG5 locus on 6q21 (at genome-wide significance) and FCGR2A (significant after Bonferroni correction, albeit not at GWA thresholds).72 96 A recent analysis in RA neatly wraps together genotype, serotype and envirotype in complex disease susceptibility. Mahdi's study demonstrated that a specific autoantigen (citrullinated α-enolase) links the environmental (smoking) and genetic (HLA-DRB1 ‘shared epitopes’ and PTPN22) risk factors in RA susceptibility (raising the OR from 2 to 37).97 Similar approaches have not yet been adopted in Crohn's disease or ulcerative colitis.
IBD and asthma
Genes involved: ORMDL3, DENND1, SMAD3 and SLC22A5 (5q31)
IBD is associated with a number of different airways diseases. Typically large airways are involved (bronchiectasis is the single commonest manifestation); patients tend to be in their fifth decade, and ulcerative colitis predominates.21 Only very recently has a robust increase in asthma been documented. In the Manitoba epidemiological study, the prevalence rates of asthma in ulcerative colitis and Crohn's disease were 7.9% and 7.1%, respectively, compared with 4.9% in population controls (using their strict definition of ≥5 healthcare contacts).10 After arthritis, asthma was the most common non-intestinal co-morbidity identified in this cohort. The prevalence of asthma was similarly increased in IBD patients (12.6%) versus matched controls (7.7%) in the North California cross-sectional cohort (OR 1.5).13 In the Manitoba study, ulcerative colitis and Crohn's disease were also more common in patients with asthma compared with population-based controls.10 This latter observation was not reproduced in an English cohort study, where IBD was increased in all patients with airways disease (especially those with a productive cough), except those with asthma.98
Variants in the same region of 17q21 encompassing the ORMDL3 gene have been associated with Crohn's disease,70 ulcerative colitis,43 and childhood-onset asthma.99 Association at this locus has also been described with total white cell count in healthy individuals,100 and with T1DM.101 Of note, these SNPs within ORMDL3 produced the strongest eQTL (expression quantitative trait locus) effect of any of the Crohn's disease genes studied in the Barrett et al meta-analysis.70 Other common associations between Crohn's disease and asthma have been identified at the DENND1B, SMAD3 and SLC22A4/5 (5q31/IBD5) loci.32 102 103
IBD and atopic dermatitis
Gene involved: C11Orf30
There is an increased prevalence of atopic dermatitis (eczema) in both patients with IBD and their first-degree relatives.16 17 Both conditions are characterised by chronic inflammation of the epithelium with defective barrier function and innate immunity resulting in poor bacterial clearance.104
Germline variants in C11Orf30 confer susceptibility to both Crohn's disease and atopic dermatitis.70 105 There is presently no consistent evidence for association at this locus with ulcerative colitis.43 106 Loss of function variants in FLG (Filaggrin) also predispose to atopic dermatitis.107 In a Scottish panel of children with IBD (52% prevalence of coexistent atopy), FLG was associated with eczema (and food allergy) but not with IBD susceptibility.108
IBD and coeliac disease
Genes involved: ICOSLG, ZMIZ1, IL18RAP, IL2/IL21, PTPN2, KIF21B, BACH2, TAGAP and ZFP36L1
The prevalence of coeliac disease in patients with IBD does not appear to be increased compared with the background population (1–2%). Indeed, a large multi-centre Italian study observed lower rates of coeliac disease (0.5%), hinting at a potential protective effect.22 In contrast, following Cottone's description of three Sicilian families with Crohn's disease and coeliac disease,109 there is a suggestion in the literature that IBD (ulcerative colitis > Crohn's disease) may be more common both in patients with coeliac disease,110 111 and in their first degree relatives.112 However, this has not been studied in large population-based cohorts.
Coeliac disease is strongly hereditary (monozygotic concordance 70%, compared with 35% in Crohn's disease). HLA plays a major role; DQ2 carriage is the most important genetic determinant of disease risk (almost 90% of patients are DQ2 positive). There are now 26 definite non-HLA disease genes/loci, with a variety of immunological functions including T cell development, innate immune detection of viral RNA and lymphocyte co-stimulation.113 114 Together with the HLA risk, these loci account for an estimated 40% of the genetic variance in coeliac disease risk.113 Of the confirmed coeliac disease genes, PTPN2, ICOSLG, REL and KIF21B are associated with IBD,28 32 35 70 BACH2, TAGAP, IL18RAP, ZMIZ1, and ZFP36L1 with Crohn's disease,32 70 115 and IL2/IL21 with ulcerative colitis.51
IBD and multiple sclerosis
Genes involved: IL2RA, IL7R, PTGER4, ZMIZ1 and STAT3
It is recognised that there is an increased incidence of MS, demyelination and optic neuritis in patients with IBD (for Crohn's disease, incidence rate ratio (IRR) 2.12, 95% CI 0.94 to 4.50; for ulcerative colitis, IRR 2.63, 95% CI 1.29 to 5.15).12 An increased clustering of MS in IBD patients is also documented in the Kaiser Permanente cohort study (OR 2.3, 95% CI 1.6 to 3.3).13
There is definite common association between Crohn's disease and MS at IL2RA,32 116 and between ulcerative colitis and MS at IL7R.28 117 The association with Crohn's disease on 5p31 lies within a 1.25 Mb gene desert, but eQTL analysis suggests that the adjacent PTGER4 gene is responsible.118 SNPs in linkage disequilibrium with those associated with Crohn's disease are associated with ulcerative colitis,28 and also appear to confer susceptibility to MS, but presently fall just short of genome-wide significance.117 The ZMIZ1 locus in Crohn's disease is also nominally associated with MS.32 115 117 The STAT3 risk allele in Crohn's disease and ulcerative colitis (rs744166-A) confers protection against MS.70 119 This Finnish study was notable for studying a small geographically isolated population with high familial occurrence of MS, with replication in a large heterogenous cohort.119
IBD and type 1 diabetes mellitus
Genes involved: PTPN2, PTPN22, ORMDL3, IL18RAP, IL27, IL10, IL2/IL21, IL2RA, BACH2 and TYK2
There are presently no data to support either an increased or decreased prevalence of T1DM in IBD. This is in contrast to the clear increased incidence of coeliac disease in TIDM (but not T2DM), an observation that led to the identification of seven shared susceptibility loci between the two diseases (including RGS1, IL18RAP, TAGAP, PTPN2, CTLA4 and SH2B3).120 In the Kaiser Permanente Medical Care Program study, T1DM was more common in patients with IBD than in controls (1.0% vs 0.7%), but this did not translate into an increased risk after adjustment for age, gender and smoking.13 No difference in T1DM rates between patients with IBD and controls was observed in another North American health insurance-based study.11 T1DM was not one of the diseases assessed in the Manitoba study.10 A recent Swedish study observed a marginal increase in ulcerative colitis in family members of patients with T1DM (standardised incidence ratio 1.3).121 Finally, a couple of case reports in the literature describe T1DM, ulcerative colitis and PSC affecting the same male individuals (n=4), an association that is very likely related to HLA genotype.122 123
Hence, in the absence of any clinical overlap between T1DM and IBD it is of considerable interest that there are presently 10 loci where definite association is observed in both diseases. Inherited susceptibility to both diseases is in part mediated by germline variants in loci containing PTPN2,35 124 ORMDL3,28 70 101 IL2/IL21,51 101 124 IL2RA,32 101 IL10,31 32 101 IL18RAP,70 120 BACH2,32 101 TYK2,32 125 and IL27.101 115 Overlap is also observed at PTPN22 (protective in Crohn's disease).70 126 Finally, a coding mutation in FCGR2A that is, associated with ulcerative colitis susceptibility in Japanese and Caucasian individuals,43 72 is nominally protective for T1DM in Caucasians.127
Why should such distinct phenotypes as IBD and T1DM apparently result from this shared germline variation, especially given the absence of an epidemiological link between the two diseases? Three possible explanations come to mind: (1) one disease protects from the development of the other; (2) common immune involvement in pathogenesis; and (3) something in the gut (ie, bacterial flora) predisposes to both.
Could the development of IBD or T1DM protect from the development of the other disease? In such a scenario we would expect to see few patients with T1DM in our IBD population; epidemiological data therefore do not support this argument. Do germline mutations that are associated with the development of T1DM confer protection against IBD? The p.R620W PTPN22 variant exerts just such an effect70 126; given its broad association with multiple autoimmune diseases, it may be that protection against Crohn's disease confers some mild selective advantage on this allele.
Common immunological pathways in T1DM and IBD pathogenesis are strongly suggested by the shared genetic susceptibility genes, especially IL2/IL21, IL2RA, IL10, IL27 and IL18RAP. However, very recent in vivo data point to the commensal flora in the gastrointestinal tract as a key player in T1DM pathogenesis.128 It has been recognised for 20 years that alterations in the commensal flora influence the incidence of spontaneous T1DM in non-obese diabetic (NOD) mice. More recently it has been elegantly demonstrated that the innate immune response to gut microbiota is critical to the development of T1DM in these animals. MyD88 deficient NOD mice develop diabetes in germ-free but not in specific pathogen free conditions.128 These animals, with defective signalling via multiple toll-like receptors (except TLR3 and TLR4), are protected from developing T1DM by their commensal flora. This is further demonstrated by the attenuation of the diabetic risk in the germ-free MyD88 NOD mice by early recolonisation of the commensal flora with either altered Schaedler flora (a consortium of bacterial species) or with exposure to specific pathogen free MyD88 negative donors.
IBD and type 2 diabetes mellitus MODY, hyperlipidaemia, coronary artery disease, and obesity
Genes involved: CDKAL1, CAPN10, HNF4A, GCKR, THADA, CPEB4, BTLN2, FADS1 and SMAD3
Apart from an isolated case report,129 no reliable published data are available to address any potential epidemiological interaction between IBD and T2DM. Some slight alterations have been noted in pancreatic endocrine function, with increased insulin resistance noted in Crohn's disease patients.130 Hyperlipidaemia has not been directly studied, but was noted in a single cohort study to be increased in both ulcerative colitis and Crohn's disease compared with healthy controls,23 and there are case reports of statin-induced ulcerative colitis.131 IBD patients have a well-documented increased risk of venous thromboembolic disease.132 There may also be an increased risk of some arterial thrombotic events; Bernstein and Ha both noted an increased risk of cardiac events,23 133 although in the latter study this risk was restricted to females over 40 years.23
The genetic overlap between IBD and T2DM consists of five common loci: HNF4A, CAPN10, CDKAL1, THADA and GCKR. HNF4A and CAPN10 are noteworthy as they are the MODY1 (maturity-onset diabetes of the young) and NIDDM1 genes respectively.134 135 Both HNF4A and CAPN10 are specifically ulcerative colitis genes,40 115 the latter arising from a paediatric gene-discovery cohort.115 MODY is a form of non-ketotic (type 2) DM that presents under the age of 25 years, is characterised by impaired insulin secretion and has an autosomal dominant inheritance with variants in six different genes including HNF4A. A nsSNP in HNF4A is also associated with polygenic dyslipidaemia.136 Variants in the same intronic block of CDKAL1 have been associated with Crohn's disease and T2DM, although the associated alleles are not correlated in the 2 diseases.70 137–140 This gene has to date inconsistent replication in ulcerative colitis.43 106
SMAD3 is associated with IBD and coronary artery disease.28 32 141 FADS1 provides a link between Crohn's disease,32 fasting serum glucose levels,142 hypercholesterolaemia (HDL and LDL cholesterol and triglycerides),136 143 and serum metabolites.144 While the gut flora has not been directly studied in the context of T2DM, we may look to studies in obese individuals for clues here. The CPEB4 locus is associated with Crohn's disease and waist–hip ratio,32 145 while BTLN2 appears common to colonic IBD and body-mass index.47 146 Intriguingly, obesity is associated with reduced microbial diversity in a similar pattern to that seen in IBD.147–150 Signalling through TLR4 potentially links obesity and insulin resistance with innate immunity. Tlr4−/− mice are overweight and have partial protection against diet-induced insulin resistance in association with decreased inflammation in liver and fat.151
Endoplasmic reticulum (ER) stress may be another mechanism that links obesity, insulin resistance, T2DM and IBD. Xbp1−/− mice develop insulin resistance and spontaneous intestinal inflammation.152 153 The XBP1 transcription factor modulates ER stress. ER stress plays an important role in the development of insulin resistance and diabetes by triggering JNK activity.152 PKR (double-stranded RNA-dependent protein kinase) has been identified as a key component of metabolic homeostasis, responding to nutrients and ER stress (marked upregulation with obesity).154 XBP1 has been associated with IBD in a robust candidate gene study, but not at genome-wide significance.153 Xbp1flox/floxVCRE mice (intestinal epithelial cell specific deletion of Xbp1) demonstrate increased ER stress, develop spontaneous mild, patchy mucosal inflammation, and lack Paneth cells.153 ER stress is increased in intestinal epithelial cells in patients with active IBD, and IL-10 inhibits inflammation-induced ER stress in vivo.155 AGR2, another putative IBD gene arising from candidate-gene study,156 also has roles in ER stress, Paneth cell function and intestinal homeostasis.157 Furthermore, aberrant mucin production/assembly results in significant ER stress in ulcerative colitis (even in the absence of inflammation) and spontaneous colitis in mice, suggesting a critical role in the pathogenesis of ulcerative colitis.158 Finally, the IBD and asthma gene ORMDL3 facilitates the unfolded protein response, an endogenous inducer of inflammation, by altering ER-mediated calcium homeostasis.159
Crohn's disease and mycobacterial disease
Genes involved: NOD2, LRRK2 and TNFSF15
A recent Chinese GWA study into leprosy has provided the first definitive evidence for shared susceptibility pathways between mycobacterial disease and Crohn's disease.160 Previously, nominal evidence for association at IRGM had been demonstrated for mycobacterial tuberculosis (albeit only in one subtype),161 and a raft of candidate genes studied, including IFNγ. The context and possible implications of the Chinese study are fascinating. They demonstrated association at the HLA, three confirmed Crohn's disease loci: NOD2, LRRK2 and TNFSF15, and at RIPK2 (an essential downstream mediator of NOD2 signalling).160 This clearly implicates NOD2 signalling in the handling of intracellular mycobacterial leprosy, as defects in this pathway confer susceptibility to disease. It is important to note that the NOD2 variants that predispose to Crohn's disease in Europeans and leprosy in the Chinese are different. Indeed, the Crohn's disease-predisposing NOD2 mutants are not found in the Far East162 163; as such evidence for association between NOD2 and Crohn's disease is lacking in these populations. In contrast, TNFSF15 was initially discovered in a Japanese Crohn's disease population,164 with subsequent replication in Caucasians.70
Do any of the leprosy mutations actually confer protection against Crohn's disease, and therefore result in positive selection? Does leprosy itself develop in the place of Crohn's disease in susceptible individuals? Hence, is the decline of leprosy infection in the developed world somehow linked to the rising incidence of Crohn's disease in the past 50 years or so? Do those individuals with NOD2 mutations have an equal risk of developing leprosy or Crohn's disease depending on whether they are born in China or Scotland (and conditional on other genomic, serotypic, microbiomic factors)? Finally, does this give us any insight into the aetiological microorganisms in Crohn's disease?
This study therefore raises more questions than answers at this stage. We suggest that it is most likely that the defective handling of intracellular bacteria is a common pathogenic pathway in both Crohn's disease and persistent mycobacterial infection. This does not necessarily implicate mycobacteria (of whatever subtype) as aetiological agents in Crohn's disease. Indeed, many groups have now recovered and identified the same strain of adherent–invasive Escherichia coli (AIEC) from the intestines of patients with Crohn's disease (much more consistently than early varied reports of isolation of mycobacteria avarium papatuberculosis subspecies).165 Defects in autophagy (consistent with the disease associated germline variants) have already been shown to increase survival and replication of adherent–invasive E coli in vitro,166 in what is one of the first clear examples of a true gene–environment interaction in Crohn's disease.
Implications for treatment
The application of genetics to IBD therapeutics (pharmacogenetics) may, in future, allow clinicians to predict drug response and drug toxicity prior to initiation of therapy. In fact there is already an established precedent in IBD, with TPMT testing prior to initiation of azathioprine/mercaptopurine now routine clinical practice. This arose directly from the molecular knowledge of azathioprine metabolism and the observation that levels of TPMT, a key enzyme in this pathway, are genetically determined. More recently, researchers have tested whether there are genetic determinants of response to anti-TNF treatment. Two methods have been adopted: (1) examination of existing IBD susceptibility loci, and (2) genome-wide association of treatment response. Adopting the first approach, Jurgens and colleagues described the influence of IL23R genotype status, along with disease activity and ANCA positivity, on infliximab response in ulcerative colitis (n=90).167 Using both approaches (28 IBD susceptibility SNPs and GWA) in a paediatric IBD cohort treated with infliximab (n=94), Dubinsky and colleagues modelled predictors of primary non-response (22/94 in their cohort).168 Their most predictive model incorporated three novel GWA loci, one susceptibility locus (21q22), pANCA status and a diagnosis of ulcerative colitis. Before these data can be applied in the clinic, the pharmacogenetic loci require independent replication and the model subsequently needs validating in a prospective cohort. Indeed, the clinical utility of pharmacogenetics in IBD will be dependent on the generation of robust data, collected prospectively and independently replicated and validated. Further collaborative efforts among members of the IIBDGC will be essential to generate sufficiently powered cohorts.
In addition to the co-existence of other diseases with IBD, there are several related inflammatory diseases where common treatment approaches have been successful. The most recent example of this is the use of anti-TNF therapy in Crohn's disease, ulcerative colitis, RA, ankylosing spondylitis and psoriasis. Other examples range from corticosteroids (many auto-immune/inflammatory phenotypes) to thiopurines (Crohn's disease, ulcerative colitis and auto-immune hepatitis, RA, refractory coeliac disease, SLE) and methotrexate (Crohn's disease and RA, psoriasis) and anti-adhesion therapies (Crohn's disease and MS). Additionally, the 5-amino salicylic acids (5-ASAs) have been shown to work in part through peroxisome proliferator-activated receptor gamma (PPARγ)169; pure agonists (glitazones) have established efficacy in DM and some evidence to support a therapeutic role in IBD (cardiovascular side-effects will likely limit further studies in IBD).170
Industry will be enabled to focus resources on therapies targeting pathways implicated in multiple diseases. Following the success of this approach using anti-TNF antibodies, the IL-23 pathway is being intensively targeted, with potential to treat multiple immune-mediated diseases including Crohn's disease, ulcerative colitis, psoriasis and ankylosing spondylitis (figure 4). The anti-p40 antibody ustekinumab (which blocks IL-12 and IL-23) has demonstrated efficacy superior to anti-TNF agents in psoriasis,171 and shown initial promise in Crohn's disease.172 173 Anti-p19 antibodies may ultimately have a superior efficacy/safety profile, given their specificity for IL-23.
One emerging paradox is the distinction between this broad approach (informed by genetic overlap), and a very specific approach that may finally lead us much closer to truly personalised medicine. This latter line of enquiry will be informed by the elucidation of disease specific defects along with future fine-mapping efforts and functional studies. For example, while antibiotic therapy presently lacks efficacy for luminal Crohn's disease, there may be specific microbial triggers dependent on a specific combination of inherited defects in innate immunity that will require an anti-microbial approach tailored to the individual. It will be of great interest to see how the biotechnology industry responds to this challenge, given the potentially conflicting financial drivers inherent in treating the ‘many’ versus the ‘few’. In all likelihood, some combination of these approaches may ultimately result in a sea-change in the therapy of chronic immune-mediated disease.
Future insights
The recent ‘gold rush’ of gene discovery in complex disease genetics has been phenomenal; the complexity of overlap demonstrated herein between apparently similar and diverse phenotypes is testament to this. How does this translate into meaningful clinical data? In many areas it has highlighted our paucity of accurate epidemiological data. Certainly, the identification of common pathways suitable for therapeutic intervention across multiple phenotypes is one immediately apparent benefit. However, we would suggest that, presently, a significant lack of precision in the genetic detail currently limits the degree to which these observed common susceptibility loci yield substantial pathogenic insights. Throughout mammalian evolution, there have been very high selection pressures on immunity and inflammation associated genes. As such, for most immune-mediated diseases, there remains substantial ‘missing heritability’. In Crohn's disease and ulcerative colitis for example, the missing genetic contribution to disease susceptibility is presently calculated at 77% and 84%, respectively.28 32
We expect that it will be the large international ImmunoChip effort that will really deliver sufficient genetic resolution for substantial insights. At its core, this experiment involves deep replication from Crohn's disease and ulcerative colitis meta-analyses, and fine-mapping of all major immune-mediated diseases by a common, custom-designed chip (∼200k custom-designed SNPs; >100k samples). This level of detail will clarify which genotypes confer risk and protection across multiple diseases. It will also for the first time truly open up the possibility of describing common immune pathways central to complex genetic diseases. Furthermore, it may allow sufficient power for pharmacogenetic experiments to assess drug response and intolerance across multiple phenotypes (although the retrospective data collections required may be prohibitive and clean prospective/inception cohorts may be necessary as part of a future effort here).
When the dust starts to settle on this era of rapid data accumulation, efforts such as to the NHGRI catalogue of published GWA studies will be essential if we are to maximise subsequent data mining. Indeed, there will be a need for more powerful next-generation data cataloguing efforts. This will be true when we start to compare across multiple phenotypes, apply whole-genome sequencing to complex disease genetics,174 and factor in complex variables such as metagenomic level analysis of our microbiome. At any rate, the genomic revolution continues apace, direct application to patient care will follow, and our analysts are going to have plenty of work to do.
Key messages
There are presently 99 published IBD susceptibility loci. The rapid phase of gene discovery is continuing thanks to the successful collaborative efforts of the International IBD Genetics Consortium (IIBDGC).
Key insights from gene discovery include the role of IL-23/Th17 signalling in IBD, defective processing of intracellular bacteria in Crohn's disease and defective barrier function in ulcerative colitis.
There is substantial overlap in susceptibility loci\genes between IBD and other diseases. Presently approximately 51 IBD loci show overlap between up to 23 different diseases.
This overlap includes many disease that are immune-mediated (eg, psoriasis, atopic dermatitis, coeliac disease and type 1 diabetes mellitus) and some that are not (eg, type 2 diabetes mellitus and colo-rectal cancer). This is yielding significant insight into disease pathogenesis.
Many common ‘druggable’ pathways have been identified; of these, the IL-23 pathway is the best characterised. Substantial investment from the pharmaceutical industry should deliver novel therapies arising from gene discovery to the clinic within the next 5 years.
References
Footnotes
Competing interests CL has acted as a consultant to Abbott and Dr Falk, and received honoraria from Shire, Procter and Gamble, Ferring, and Schering-Plough for presentations at academic meetings. JS has acted as a consultant to Schering-Plough, Abbott, UCB, Shire and Ferring, and has on-going research support from Novartis and Genentech. JB and MP have no competing interests.
Provenance and peer review Commissioned; externally peer reviewed.