Article Text

Abstract
Background Little is known about the stem cell organisation of the normal oesophagus or Barrett's metaplastic oesophagus. Using non-pathogenic mitochondrial DNA mutations as clonal markers, the authors reveal the stem cell organisation of the human squamous oesophagus and of Barrett's metaplasia and determine the mechanism of clonal expansion of mutations.
Methods Mutated cells were identified using enzyme histochemistry to detect activity of cytochrome c oxidase (CCO). CCO-deficient cells were laser-captured and mutations confirmed by PCR sequencing. Cell lineages were identified using immunohistochemistry.
Results The normal squamous oesophagus contained CCO-deficient patches varying in size from around 30 μm up to about 1 mm. These patches were clonal as each area within a CCO-deficient patch contained an identical mitochondrial DNA mutation. In Barrett's metaplasia partially CCO-deficient glands indicate that glands are maintained by multiple stem cells. Wholly mutated Barrett's metaplasia glands containing all the expected differentiated cell lineages were seen, demonstrating multilineage differentiation from a clonal population of Barrett's metaplasia stem cells. Patches of clonally mutated Barrett's metaplasia glands were observed, indicating glands can divide to form patches. In one patient, both the regenerating squamous epithelium and the underlying glandular tissue shared a clonal mutation, indicating that they are derived from a common progenitor cell.
Conclusion In normal oesophageal squamous epithelium, a single stem cell clone can populate large areas of epithelium. Barrett's metaplasia glands are clonal units, contain multiple multipotential stem cells and most likely divide by fission. Furthermore, a single cell of origin can give rise to both squamous and glandular epithelium suggesting oesophageal plasticity.
- Oesophagus
- Barrett's metaplasia
- clonality
- mitochondrial DNA
- stem cells
- Barrett's oesophagus
- colorectal adenomas
- mathematical modelling
- Barrett's carcinoma
- microsatellite instability
- carcinogenesis
- colonic neoplasms
- colonic polyps
- colorectal cancer
- histopathology
- liver
- gastrointestinal tract
- molecular carcinogenesis
- gastric adenocarcinoma
- epithelial differentiation
- gastrointestinal pathology
- gastric carcinoma
Statistics from Altmetric.com
- Oesophagus
- Barrett's metaplasia
- clonality
- mitochondrial DNA
- stem cells
- Barrett's oesophagus
- colorectal adenomas
- mathematical modelling
- Barrett's carcinoma
- microsatellite instability
- carcinogenesis
- colonic neoplasms
- colonic polyps
- colorectal cancer
- histopathology
- liver
- gastrointestinal tract
- molecular carcinogenesis
- gastric adenocarcinoma
- epithelial differentiation
- gastrointestinal pathology
- gastric carcinoma
Significance of this study
What is already known on this subject?
-
The identification of rare mutations in gut epithelium allows inference about human gut biology.
-
Specifically, the identification of rare somatic alterations in stem cells allows crypt lineage tracing experiments in humans.
-
Barrett's metaplasia is known to be oligoclonal but the exact mechanism giving rise to this heterogeneity is unknown.
What are the new findings?
-
Clonality of Barrett's metaplasia glands.
-
Multiple stem cells are present in Barrett's metaplasia.
-
Barrett's metaplasia and squamous tissues share a common progenitor.
How might it impact on clinical practice in the foreseeable future?
-
The identification of the alternate clonal patches of Barrett's metaplasia glands means that the efficacy of current biopsy sampling will be scrutinised.
-
The presence of multiple stem cells in Barrett's metaplasia means new therapies to modulate their action could be pursued.
-
The fact that Barrett's metaplasia and squamous tissues share a common progenitor means that therapies could be used to promote squamous growth.
Introduction
The human oesophagus is lined by a non-keratinising, stratified squamous epithelium where superficial layers are desquamated and replaced by an as yet unidentified stem cell.1 The oesophagus has submucosal glands composed of individual mucus producing acini that lead into developed ducts opening onto the surface epithelium.2 The proximal two-thirds of these ducts are lined with columnar epithelium and the distal third by squamous epithelium. Barrett's metaplasia is the replacement of the normal squamous epithelium with a columnar-lined, usually mucus-secreting, epithelium. Barrett's metaplasia develops in association with chronic gastro-oesophageal reflux3 and is the predominant risk factor for oesophageal adenocarcinoma with recent data suggesting a conversion rate of 0.2–0.4% per year.4 ,5
The Barrett's metaplasia stem cell has not been identified and the maintenance of the populations of differentiated cell types within Barrett's metaplasia glands in vivo is unresolved, and this in turn has severely hampered our understanding of the genesis of columnar cells in the oesophagus. Several theories attempt to explain the cellular origins of Barrett's metaplasia: reflux mediated reprogramming of a resident squamous stem cell, stem cell migration from the gastric cardia in order to repopulate the damaged oesophageal mucosa or stem cells within submucosal ducts or glands regenerating the surface epithelium.6 ,7 A recent study gives precedence to the theory that Barrett's metaplasia arises from cells at the squamocolumnar junction (SCJ)8; in this study, p63 knockout mice were shown to develop a Barrett's-like metaplasia in the squamous fore-stomach, and retrograde growth of a population of Car4-expressing cells located at the SCJ was proposed as the origin of the metaplasia although no lineage tracing was performed. However, observations of human Barrett's metaplasia give credence to other hypothesised origins of the metaplasia. Notably, acid suppression and other effective therapies lead to the regrowth of apparently normal neo-squamous epithelium within Barrett's metaplasia lesions and distant from the SCJ. A genetic analysis by Paulson et al 9 showed that the neo-squamous epithelium in 19/20 patients examined did not share the same mutation as the surrounding Barrett's metaplasia, indicating a stem cell source within the Barrett's metaplasia that was distinct from the stem cell population responsible for the routine self-renewal of the metaplastic lesion. Evidence that this stem cell population could be located within the oesophageal ducts or underlying glands was provided by Coad et al who noted that 15/15 neo-squamous islands examined were associated with a submucosal gland and duct.2 Genetic evidence that the ducts could be the source of the Barrett's metaplasia was provided by Leedham et al,10 who found a normal squamous duct that contained the same somatic mutation as the contiguous metaplastic epithelium.
Clonal progression of whole Barrett's metaplasia has been previously studied using flow-purified analysis of a series of whole biopsies and assessing the mutational profile of relevant genes, including CDKN2A (P16) and TP53. Using these methods, it has been shown that an entire Barrett's segment can be clonally derived,11 ,12 with mutations spreading through the epithelium relatively rapidly. However, work by Leedham et al 10 at a crypt-by-crypt level showed that Barrett's metaplasia was were in fact genetically heterogeneous, containing many apparently independent clones. The disparity between these two observations highlights how little is known about the clonal architecture of the normal oesophagus or of individual Barrett's metaplasia glands or the mechanism of mutation accumulation and spread.
By definition, stem cells can maintain their lineage over long periods of time, and produce all differentiated cell types of the tissue in which they reside.13 This means that stem cells, rather than their progeny, are probably the only cell type that live long enough to accrue mutations. While dysplastic or neoplasic mucosa is likely to contain somatic mutations within tumour suppressor genes or oncogenes, these mutations are rare in normal tissues. Consequently, to study the normal and metaplastic oesophagus we have used naturally occurring, non-pathogenic, somatic mutations in mitochondrial DNA (mtDNA). mtDNA is susceptible to mutation as it has poor repair mechanisms, no protective histones and resides in a highly oxidative environment.14 Deficiency of cytochrome c oxidase (CCO), a mitochondrially encoded enzyme which is part of the respiratory chain, is often associated with mtDNA mutations.15 Previous work by our group and colleagues, looking at other regions of the gastrointestinal tract, has shown that CCO-deficient, mtDNA mutant cells increase in number with age, and are rarely found in patients younger than 40 years,16 suggesting mutations accrue only in long-lived stem cells.17–20 Clonal expansion can be visualised by finding patches of adjacent CCO-deficient cells or glands that share the same mtDNA mutation(s).
The aim of this study was to use mtDNA mutations as markers of clonal expansion to investigate the stem cell architecture of the normal oesophagus and the glands in Barrett's metaplasia. We also suggest how Barrett's glands can spread within the epithelium and establish the origins of cell lineages within such glands. Additionally, we investigate the relationship between neo-squamous islands and the columnar epithelium of Barrett's metaplasia.
Materials and methods
Patients
Patients undergoing oesophagectomy or endoscopic mucosal resection for adenocarcinoma or dysplasia at the John Radcliffe Hospital Oxford (n=2), University College Hospital London (n=3) and Leicester Royal Infirmary (n=6) were included in this study (n=11 in total). Tissue was collected from areas of metaplasia or dysplasia as well as areas from morphologically normal epithelium. The mean patient age was 66.7 years (55–75 years). All areas of oesophagus were described histologically by qualified pathologists (NAW, MR-J and RH) experienced in diagnosing Barrett's oesophagus.
Tissue specimens were frozen epithelial surface down on a microscope slide in liquid nitrogen-cooled isopentane (Sigma Aldrich, Poole, UK). All tissue specimens were collected as per UK Home Office regulations under multi-centre ethical approval (07/Q1604/17).
Enzyme histochemistry
Frozen sections were cut at a thickness of 8 μm for routine analysis and 20 μm for laser capture microdissection. Dual CCO and succinate dehydrogenase (SDH, a nuclear-encoded, mitochondrially active enzyme) histochemistry was performed as previously published.17 ,18 ,20 Briefly, air-dried sections were incubated in cytochrome c medium containing 100 mmol/l cytochrome c, 20 μg/ml catalase and 4 mmol/l diaminobenzidine tetrahydrochloride in 0.2 mol/l phosphate buffer, pH 7.0, all sourced from Sigma Aldrich, for a maximum of 50 min at 37°C. Sections then were washed in phosphate-buffered saline (PBS) buffer, pH 7.4 for 3×5 min and then incubated in SDH medium (130 mmol/l sodium succinate, 200 mmol/l phenazine methosulfate, 1 mmol/l sodium azide and 1.5 mmol/l nitroblue tetrazolium in 0.2 mol/l phosphate buffer, pH 7.0) for a maximum of 45 min at 37°C, or until a strong blue stain had developed. Sections again were washed in PBS for 3×5 min and dehydrated in a graded ethanol series (70%, 90%, 100%, 100%), cleared in Histoclear (Lamb Laboratory Supplies, Eastbourne, UK) and mounted with Permount (Fisher Scientific, Fair-Lawn, New Jersey, USA) and allowed to dry overnight. CCO-deficient patches were identified in 7/11 cases from the human oesophagus. DNA from four out of these seven patients was screened and mtDNA mutations were identified within each patient.
Immunohistochemistry
Formalin-fixed paraffin-embedded sections were cut at a thickness of 4 μm. Sections were de-waxed and rehydrated through decreasing alcohol concentrations, and blocked for endogenous peroxidase in 3% methanol/H2O2. Sections were microwaved for 10 min in sodium citrate buffer (pH 6.0) and cooled at room temperature for 30 min.
Detection by horseradish peroxidase
Sections were blocked with rabbit serum at 10% in PBS (Invitrogen, Paisley, UK) for 10 min before primary antibodies were applied for 60 min at room temperature. Antibodies used were: mouse antihuman CCO (Anti-Oxphos complex IV subunit I) (2.5 μg/ml; Invitrogen, UK), mouse antihuman chromogranin A (0.42 μg/ml; Dako, Ely, UK), mouse antihuman Muc-2 (0.66 μg/μl, Novacasta, Milton Keynes, UK), mouse antihuman Muc5AC (1.4 μg/μl, Novacasta, UK) and mouse antihuman cytokeratin 7 (9.2 μg/ml, Dako, UK). All antibodies were diluted in PBS with 10% rabbit serum and a negative immunoglobulin G matched control was used in all experiments. Sections were washed 3×5 min in PBS followed by 40 min incubation with rabbit antimouse secondary antibodies conjugated to biotin. Sections were then incubated with streptavidin peroxidase for 40 min, washed and developed in 4 mmol/l diaminobenzidine and 0.2% hydrogen peroxide. Sections were dehydrated through alcohol, cleared and mounted with DPX.
Detection by immunoflouresence
Sections were blocked in 10% goat serum for 10 min before primary antibodies were applied for 60 min at room temperature. Primary antibodies were mouse antihuman CCO subunit IV (2.5 μg/ml) and rabbit antihuman Porin (8.55 μg/ml, Abcam, Cambridge, UK). Primary antibodies were diluted in 10% goat serum in PBS. Sections were washed 3×5 min in PBS followed by 40 min incubation with secondary antibodies conjugated to a fluorochrome. Secondary antibodies were goat antimouse 555 (Alexafluro, Invitrogen, UK) and goat antirabbit 488 (Alexafluro). Finally, sections were mounted in hardset DAPI (Vector, UK).
Isolation of DNA from cells
Frozen sections (20 μm thick) were cut onto UV-irradiated P.A.L.M. membrane slides (P.A.L.M. Microlaser Biotechnologies, Bernried, Germany). Cells were cut into sterile, UV-irradiated, 0.5 ml tubes with adhesive caps using a P.A.L.M. laser system. Laser capture microdissection was performed on CCO-deficient and CCO-normal areas and the colour boundaries of such cells were easily distinguished. Following centrifugation at 2000 rpm for 2 min, cells were lysed in 14 μl UV-irradiated lysis buffer (Picopure; Arcturus, California, USA) at 65°C overnight and then denatured at 95°C for 10 min.
mtDNA sequencing of individual oesophageal epithelial cells
The entire mitochondrial genome was sequenced in every microdissected cell. The genome was PCR-amplified in overlapping fragments using a series of M13-tailed oligonucleotide primer pairs, as previously described by Greaves et al.17 PCR products were sequenced using BigDye version 3.1 terminator cycle sequencing chemistry on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, California, USA) and compared directly with the revised Cambridge reference sequence by using SEQUENCE ANALYSIS and SEQSCAPE software (Applied Biosystems), as previously described.20 Samples were sequenced with forward and reverse primers and repeated at least twice. Polymorphisms were identified using the MITOMAP database (http://www.mitomap.org) of reported polymorphisms and comparing genotypes with distant CCO normal cells. Any mtDNA mutations identified were confirmed by repeating the PCR and sequencing from the template DNA sample. Furthermore, mutations were observed in multiple distinct areas of tissue that were independently sequenced.
Results
Stratified squamous epithelium and submucosal oesophageal glands and ducts contain stem or progenitor cells that form clonal patches
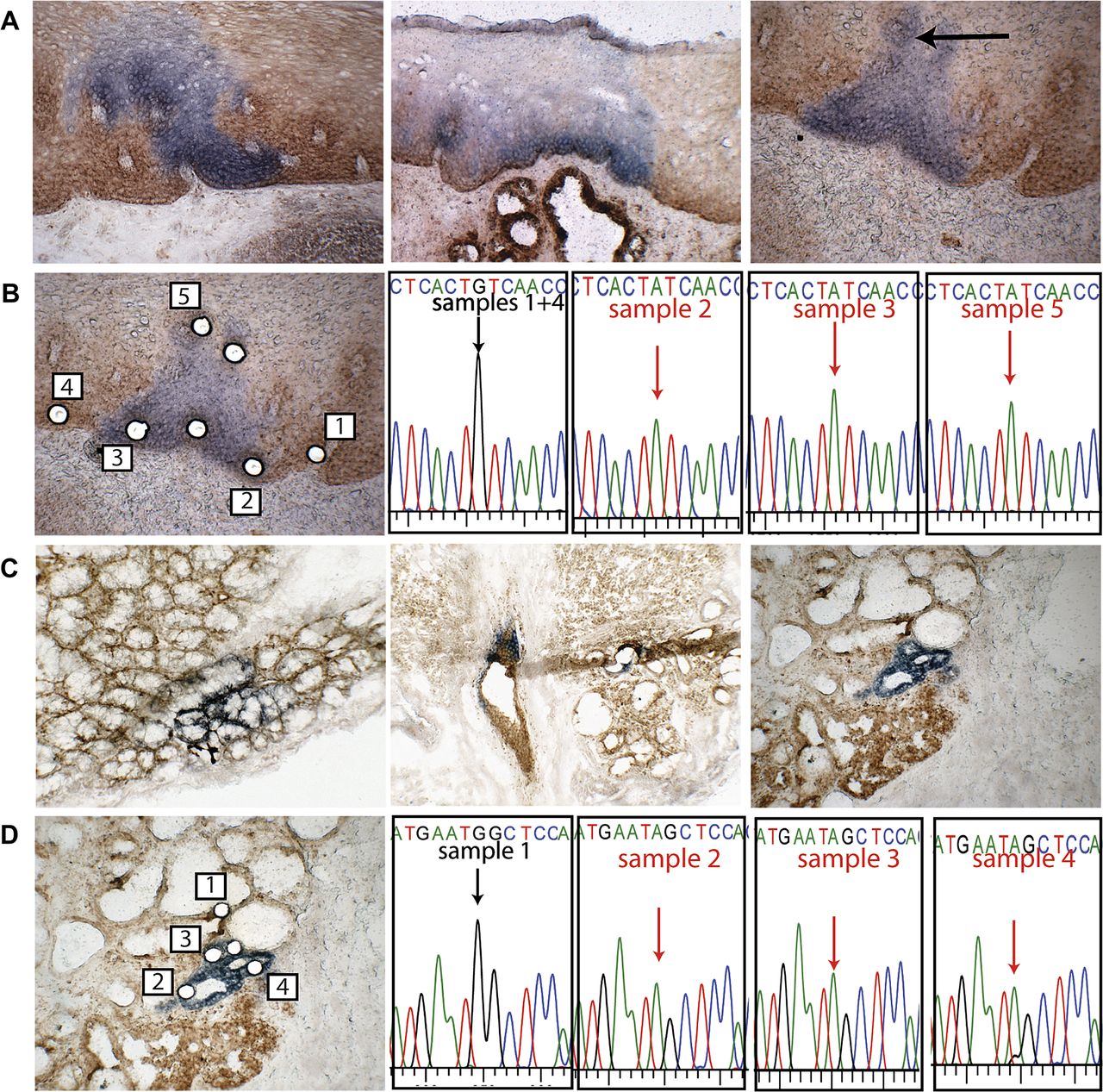
CCO/SDH enzyme histochemistry was performed on fresh frozen oesophageal resection material and patches of CCO-deficient cells were seen within the squamous epithelium of each patient studied (figure 1A). In oesophageal squamous epithelium, CCO-deficient patches varied in size, from around three cells (30 μm) up to about 1 mm in diameter. Laser capture of CCO-deficient cells from one patch (figure 1B, left panel) showed cells within the patch contained a common mtDNA mutation, 2421G→A within the MT-RNR2 gene, which was not present in the adjacent wild-type tissue (figure 1B, right panel) indicating the patch was a clonal proliferation of cells.

Cytochrome c oxidase (CCO)-deficient patches are seen in normal human oesophagus including submucosal oesophageal glands and ducts. (A) Patches of CCO-deficient cells exist in the normal squamous epithelium; a variation of staining is seen which is reflective of the intensity of mitochondria within the section (left panel) (×200). CCO-deficient patches spread from the base to the surface in the epithelium (middle panel) (×100). A CCO-deficient patch pre-laser capture microdissection (LCM) shows cells across the basal layers and covering associated papilli (right panel) (×200). (B) Post-LCM, samples from the CCO-deficient patch as well as areas of normal CCO activity were captured, and two samples (un-numbered) failed PCR (×200). Sequencing showed that CCO-normal cells contain wild-type mtDNA, whereas cells in the CCO-deficient patch contained a 2421G→A mutation within the MT-RNR2 gene. (C) CCO-deficient patches were seen within acini, patches were small and all oesophageal glands seen were partially deficient; a small tear is seen within the section (left panel) (×200). CCO-deficient patches were identified within the gland ducts and ducts were partially CCO-deficient (middle panel) (×200). CCO-deficient acini within a gland, shown here pre-LCM, form a small patch (right panel) (×200). (D) Post-LCM, samples from CCO-deficient areas were captured as well as from areas of normal CCO activity. One un-numbered sample failed PCR. Sequencing showed that CCO-normal cells contained wild-type mitochondrial DNA, whereas CCO-deficient cells all contain a 2635G→A mutation within the MT-RNR2 gene.
Acini within observed oesophageal glands also contained CCO-deficient cells; however, these specific oesophageal glands were partially deficient, also containing a population of CCO-normal cells (figure 1C, left and right panels). A single oesophageal gland duct was also seen to contain CCO-normal and CCO-deficient cells (figure 1C, middle panel). Laser capture was performed on a deficient acinus (figure 1D, left panel) and the mtDNA was analysed. The deficient cells of this acinus contained a clonal mutation, 2635G→A within the MT-RNR2 gene, which was not found in CCO-normal cells (figure 1D, right panel). This suggests that acini within submucosal glands contain clonal units representing the proliferation of a single original (stem) cell; since all glands were partially mutated, they probably contain multiple stem cells. CCO-deficient cells within oesophageal ducts were also found, but did not contain a clonal mutation with nearby submucosal glands.
Barrett's metaplasia glands are clonal units and are maintained by multiple stem cells
Wholly CCO-deficient Barrett's metaplasia glands were observed that could be traced from the surface to the base. Two separate examples are shown in figure 2A. Laser capture of cells from the surface (figure 2B, left panel) towards the base (figure 2B, right panel) showed all cells within this gland contained the same mtDNA mutation, a 7588G→A mutation within the CCOII gene (figure 2C). We also observed partially CCO-deficient Barrett's metaplasia (figure 2D, left and right panels), indicating that Barrett's metaplasia can contain multiple stem cells that respectively produce the CCO-normal and CCO-deficient cell populations.

Barrett's metaplasia glands are seen as both wholly and partially cytochrome c oxidase (CCO) deficient. (A) CCO-deficient glands were seen in Barrett's oesophagus; deficient cells spanned from crypt base to lumen as shown in two serial sections (left panels) (×100). In a different section of Barrett's metaplasia, a CCO-deficient area at the gland surface in a wholly deficient gland is taken onto section 38 through the tissue (middle panel) (×100). A serial section (number 49) shows the gland with CCO-deficient cells at the glands base (right panel) (×100). (B) A serial section (number 36) is shown post-laser capture microdissection (LCM). Cells were captured from within the deficient gland and from areas with normal CCO activity (left panel) (×200). A serial section (number 48) is shown post-LCM of the gland base. Cells were captured from CCO-deficient gland and areas of normal CCO activity (right panel) (×200). (C) Sequencing shows CCO-normal cells contain wild-type mitochondrial DNA whereas all CCO-deficient cells from the base to the surface of the gland contain a 7588G→A mutation within the CCOII gene. (D) Within Barrett's metaplasia partially mutated glands are seen, suggesting that Barrett's metaplasia glands are maintained by more than one stem cell.
All epithelial cell lineages with a Barrett's metaplasia gland derive from a single stem cell
All differentiated lineages were shown to present within single CCO-deficient Barrett's metaplasia glands. Immunohistochemistry showed that a gland lacking CCO expression (CCO subunit-1) (figure 3A, left panel) contained neuroendocrine cells expressing chromogranin A (figure 3B, middle panel) Muc5AC (figure 3B, right panel) expressing foveolar cells and Muc2 expressing goblet cells (figure 3B, left panel). An example of a serial section stained for H&E is shown in figure 3A (centre panel) as well as a negative isotype control (figure 3A, right panel). The presence of both goblet cells and foveolar cells in the same gland indicates a mixed phenotype, a relatively common observation where both gastric and intestinal lineages are present.21 Dual immunohistochemistry for CCO (subunit 1) (figure 3C, left panel) and Porin (a protein expressed on mitochondrial membranes) confirmed that the absence of CCO expression was not a consequence of absence or inactivity of mitochondria within the cells (figure 3C, middle panel).

Cytochrome c oxidase (CCO)-deficient Barrett's metaplasia glands contain all fully differentiated cell types and normal levels of active mitochondria. (A) Two neighbouring CCO-deficient glands within Barrett's metaplasia (left panel) (×100). H&E of a serial section shows the deficient glands (middle panel) (×200). A negative isotype control is shown in the right panel (×100). (B) A serial section stained for Muc-2 to show presence of goblet cells (×400). The middle panel shows a serial section stained for chromogranin A to show the presence of neuroendocrine cells (×400) with a high power insert (×600). The right panel shows a serial section stained for Muc-5AC to show presence of foveolar cells within the glands (×400). (C) Within a different tissue sample, CCO expression shown using Cy3 (red) fluorescence shows a CCO-deficient gland within Barrett's metaplasia (left panel). Porin expression shown using Fitz (green) fluorescence shows the deficient glands containing normal active mitochondria (middle panel) and a DAPI merged image of the glands (right panel) (×200).
Barrett's metaplasia contains large patches of clonally related glands
Patches of adjacent CCO-deficient glands were seen in three patients (figure 4A): here, a patch was defined as at least two neighbouring CCO-deficient glands. Laser capture from each gland within a small patch (three glands) was performed (figure 4B, left panel) and mtDNA sequencing showed they contained the same mtDNA mutation, 9601T→C mutation within the CCOIII gene, which was not seen in wild-type glands (figure 4B, middle and right panel), indicating that this patch was clonal. Large patches, containing approximately 10 CCO-deficient Barrett's metaplasia glands, were also observed (figure 4C, left panel). Each gland within this large patch contained cells with the same mtDNA mutation, a 9715G→A mutation within CCOIII, again not seen in wild-type glands (figure 4C, right panel), indicating that the patch was a clonal proliferation.

Patches of cytochrome c oxidase (CCO)-deficient Barrett's glands contain a clonal mutation. (A) A small patch of around six CCO-deficient glands within Barrett's metaplasia (left panel) (×200). A large patch of around 14 CCO-deficient glands within Barrett's metaplasia is shown in the middle panel (×200). A small patch of CCO-deficient Barrett's metaplasia glands is shown here pre-laser capture microdissection (LCM) (right panel) (×200). (B) Post-LCM, cells from all CCO-deficient glands and neighbouring CCO normal glands were captured. Two samples captured (un-numbered failed PCR). Sequencing shows CCO-normal cells from neighbouring patch contain wild-type mitochondrial DNA (mtDNA), whereas all CCO-deficient glands within the patch contain the same 9601T→C mutation within the CCOIII gene. (C) A large patch of >10 CCO-deficient glands with Barrett's oesophagus pre-LCM. Post-LCM, cells from CCO-deficient glands as well as neighbouring CCO-normal glands were captured and DNA digested prior to PCR. Sequencing shows CCO-normal cells from neighbouring patch contain wild-type mtDNA whereas all CCO-deficient glands within the patch contain the same 9715G→A mutation within the CCOIII gene.
The squamous epithelium of neo-squamous islands and the glandular epithelium of Barrett's metaplasia share a common cell of origin
An oesophageal specimen from a patient subjected to ablative therapy contained glandular epithelium with an overlying neo-squamous island (figure 5A). Serial sections from this specimen showed CCO-deficient areas in glandular tissue and also in the squamous epithelium above (figure 5B). Alcian blue PAS staining confirmed the presence of goblet cells (figure 5C). Cell-like areas were microdissected from each CCO-deficient area (figure 5D). Analysis of the mtDNA showed the same mutation in both squamous and glandular areas within this section, a 6768G→A mutation in the CCOI gene (figure 5D). Thus, the glandular tissue and the overlying squamous epithelium derived from the same precursor cell. Immunohistochemistry for cytokeratin 7 was performed on sections in close to serial (figure 5E), revealing the expected high levels of the CK7 in underlying glands and low expression in the squamous epithelium.22

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Squamous epithelium and underlying glandular epithelium share a clonal mutation suggesting that both cell types contain a common progenitor cell. (A) A sample of Barrett's metaplasia having undergone ablative therapy is stained using H&E. Squamous epithelium (dashed arrow) is seen overlying glandular epithelium (solid arrow) (×100). (B) A serial section stained for cytochrome c oxidase (CCO)/succinate dehydrogenase enzyme histochemistry (×200). CCO-deficient cells are seen with the squamous epithelium (dashed arrow) as well as in the underlying glandular epithelium (solid arrow). (C) Subsequent serial section stained for alcian blue PAS diastase to show presence of mucins from goblet cells (×100). (D) A serial section pre-laser capture microdissection (LCM) shows CCO-deficient cells in both glandular and overlying squamous epithelium (left panel). Post-LCM, cells from CCO-deficient areas as well as CCO normal glands and CCO normal epithelium are captured for analysis (middle panel) (×100). Sequencing shows CCO-normal cells from both glandular and normal squamous epithelium contain wild-type mitochondrial DNA whereas all CCO-deficient cells contain the same 6768G→A mutation within the CCOI gene. (E) Shows CK7 staining of a serial section using alkaline phosphatase substrate. Expression of the cytokeratin is weaker in the overlying squamous epithelium (dashed arrow) than within glands below (solid arrow) (×100). The middle panel shows a further section showing the difference between weak or absent CK7 expression in squamous and stronger in neighbouring glandular tissue (×200). Expression of CK7 in the underlying glandular tissue is shown in the right panel (×600).
Discussion
Our findings are of fundamental importance. We propose that the best explanation for our findings is that Barrett's metaplasia glands are clonal populations. We have shown that individual Barrett's glands contain clonal populations of CCO-deficient cells (figure 2), which contained all the differentiated cell lineages present in Barrett's metaplasia (figure 3), indicating that the gland contained a multipotent stem cell. The identification of partially CCO-deficient glands indicates the presence of multiple stem cells in each gland. We propose that these partially mutated glands are in the process of clonal conversion, whereby the CCO-deficient clone is in the process of replacing the non-mutated cells, in the same way as we have shown for the colonic crypt.23 According to this view, CCO-deficient glands probably form as a result of niche succession, whereby a single stem cell clone, deficient in CCO activity, is able to replace all others leading to a monoclonally converted gland. This process has been previously described in the human gastrointestinal tract.17 ,18 ,20
There is of course the possibility of mutation that mtDNA occurring at the level of committed progenitors could be responsible for partially mutated glands. We would consider this unlikely. In the colon, it has been shown that CCO-deficient cells in partially deficient crypts bear homoplasmic mutations:23 mitochondria have multiple genomes, and cells have hundreds of mitochondria; we would consider it very unlikely that a mutation could become clonal within a mitochondrion, and then clonal within a cell, in the limited life span of such a committed precursor by the accepted mechanism of neutral or random drift.14 ,24
However, immediately adjacent glands may not be derived from the same clone. This explains a previous paradox, which is: despite work showing the expansion of unique genes in the Barrett's metaplasia progression why does the lesion still remain genetically heterogeneous?10 ,11 Furthermore, we have shown shared clonal relationships between contiguous squamous and Barrett's tissue suggesting a common progenitor cell. Once again, this provides a cogent explanation for the observation of neo-squamous epithelium appearing in Barrett's metaplasia as well as the appearance of Barrett's metaplastic islands in squamous oesophagus in the first place.
We have also shown that Barrett's glands can form large clonal patches of several glands, suggesting that Barrett's metaplasia glands can divide by fission. The chance of adjacent glands containing the same mutation by chance alone has been calculated previously as 2.48×109:1,17 and so such patches certainly represent a clonal proliferation; gland fission is the likely mechanism of clone growth. Fission has been established as the canonical mechanism of clone spread elsewhere in the gut.17 ,20 Alternative explanations for the production of these patches are the top–down spread, which we saw no evidence of in non-dysplastic tissue, or migration of cells between glands, which would probably not explain the formation of coherent patches and has no credence in the literature. Taylor et al 15 and Greaves et al 17 have shown that the number of CCO-deficient crypts and mean patch size respectively increase with patient age, suggesting that fission occurs at a basal rate in the gut, although our data are insufficient to demonstrate this phenomenon in the oesophagus.
It seems likely that mutant clones involved in the development of Barrett's adenocarcinoma also clonally expand via gland fission. Genomic DNA mutations in genes such as TP53 or CDKN2A (P16) presumably confer a gland a growth advantage compared with non-mutated neighbours.25 Some mutant clones appear to grow at a rapid rate—spreading through the entire metaplastic lesions in a matter of months—12 indicating that the growth advantage of the tumourigenic mutations could be considerable. The presence of large CCO-deficient patches observed here lends support to the rapid expansion of mutated glands observed by the Reid group.12 An interesting corollary to this point: since Barrett's metaplasia, even those containing pretumour mutant clones, do not appear to increase in length, then gland extinction, either by population pressure or by active competition with those glands with a selective advantage, will be a prominent component of this process. The clonal genetic heterogeneity observed by Leedham et al 10 probably results from multiple rounds of fission of numerous distinct progenitor glands. Notably, given the extensive growth of premalignant clones throughout Barrett's lesions, we cannot discount that the non-dysplastic tissue studied here may have harboured potentially tumourigenic mutations.
It is interesting to consider the time when mtDNA mutated clones arose relative to the development of Barrett's metaplasia: it is likely that this occurs after the formation of the metaplastic glands in most instances. How long before it is difficult to tell: to establish a wholly CCO-deficient crypt takes as many as 40 years in the colon.26 Barrett's glands are larger than colonic crypts, so this clonal stabilisation time may be even longer. Of course, while we do not know how frequently such an event depicted in figure 5 occurs—where an mtDNA mutated bipotential progenitor cell gives rise to glandular and squamous epithelium—it is possible that such an mtDNA mutated Barrett's gland could be derived de novo from such a cell.
We also considered the clonal structure of the histologically normal squamous oesophageal epithelium. Therein, clonal mtDNA mutant CCO-deficient patches were seen to stretch from the basal layer to the luminal surface. Given that the tissue turnover within the squamous epithelium is thought to be around 11 days (J Jankowski, unpublished data), mtDNA mutations occurring in short-lived, presumed non-stem cell, populations would be rapidly lost into the epithelium. Unipotential stem cells, or progenitor cells, are presumed to be located within the basal layer,1 ,27 as cells migrate from here and are eventually shed into the lumen.
The squamous epidermis is thought in some ways to be analogous to the squamous oesophagus. In the epidermis, Clayton et al 28 have shown that clone size can increase with time. If each oesophageal stem cell is responsible for maintaining a characteristic number of epithelial cells, then the identification of varying sizes of clonal patches within the squamous oesophagus is suggestive of lateral replacement of the stem cell pool along the basal layer, and that a process analogous to niche succession occurs in the normal oesophagus. The development of squamous carcinoma of the oesophagus involves mutations of several tumour-suppressor genes, including TP53 and P16, while cyclin D1 and EGFR are frequently amplified.29 ,30 Clones of cells harbouring TP53 mutations have been observed in patients with chronic oesophagitis and we would propose that mutations originate in a progenitor cell and spread in the manner that we have suggested here.
The existence of a stem cell population within oesophageal glands or ducts has previously been proposed2 and our observation of the existence of CCO-deficient patches within the glandular acini and ducts adds new data to this interesting concept. The presence of CCO-deficient and CCO-normal cells within either the same gland or duct indicates at least two stem cell populations within the same structure. Although we were unable to identify a clonal mutation between these structures in these data, previous data suggest that the stem cell populations existing in either ducts or glands are capable of producing both columnar and squamous epithelium.10
The cellular origin of human Barrett's metaplasia remains contentious. Here we have shown one example where both neo-squamous islands and the glandular epithelium of Barrett's metaplastic oesophagus share a common precursor cell. There are several explanations for the source of this unique observation. First, an original squamous stem cell clone exists, and subsequently a proportion of this clone becomes metaplastic leaving two populations of stem cells—respectively producing squamous and glandular epithelium—which share the same clonal origin. This would be in accord with what might be called the ‘classical hypothesis’, whereby the origins of Barrett's metaplasia is from the stratified squamous oesophageal epithelium. Second, due to the acid suppression treatment, an mtDNA mutated Barrett's metaplasia-producing stem cell clone reverted to producing squamous epithelium. This, however, is highly unlikely as neo-squamous epithelium is usually genetic distinct from the surrounding Barrett's metaplasia: Paulson et al 9 showed that the overwhelming majority of neo-squamous islands were wild type and did not share the well-defined mutational burden of the surrounding Barrett's metaplasia, and wild type squamous re-growth can be seen over dysplastic Barrett's that harbours a TP53 mutation (Dr Simon Leedham, University of Oxford, personal communication). A third possibility is that post-treatment, a clone of neo-squamous stem cells with mutated mtDNA transdifferentiated into a clone of mutated multipotential columnar stem cells. This possibly suggests that development of new columnar epithelium continues as the lesion ages. Finally, it is noteworthy that an oesophageal gland duct may underlie both the columnar and the squamous mtDNA mutated areas, and so it is conceivable that this gland could be the source of both lineages.
A recent study using a murine model of Barrett's metaplasia histogenesis speculates that Barrett's metaplasia developed from a gastric derived clone at the SCJ.8 While our data indicate a potential de novo Barrett's metaplasia clone source proximal from the SCJ, our observation of mixed-type metaplasia also provides an interesting facet to this argument for the gastric origins of Barrett's metaplasia. Mixed-type metaplasia is defined by the presence of cells with both gastric and intestinal phenotypes in a single gland.21 Data presented herein show an example of a wholly CCO-deficient gland that contained a mixture of both gastric and intestinal lineages. This suggests that gastric and intestinal cells are derived from a common stem cell, though it should be noted that a genetic clonal analysis could not be performed on this gland. It is interesting to speculate that such mixed glands are in the process of converting from a gastric to intestinal phenotype. As such, conversion to intestinal metaplasia could be heralded from a gastric origin.
This study includes a series of patients who have been selected because they showed mtDNA mutations that were demonstrable by enzyme histochemistry and DNA sequencing: it might be questioned whether this biases the results we have obtained in any way. Naturally, it is rarely possible to observe any system without perturbing it in some way,31 but it has been shown that clonal mutations of this type have only a minor effect upon the crypt population.32 Moreover, sequencing the entire mitochondrial genome of a number of colonic crypt cells has found a wide variety of point mutations in these cells affecting a number of different protein encoding and RNA encoding genes,26 so it is highly likely that these individuals, whose ages range from 55 to 75 years, similar to the many who have developed Barrett's metaplasia, also have multiple mtDNA mutations. We thus have no reason to conclude that these results are not representative, even though an inherent limitation of this study is that the material is derived from individuals who also had dysplasia or carcinoma elsewhere in the specimen. Indeed, rare point mutations arising in stem cell clones can give unprecedented insight into lineage relationships.33
In conclusion, we have shown that: (1) the normal oesophageal squamous epithelium contains clonal patches derived through local expansion of a basally-sited progenitor or stem cell and that lateral replacement of adjacent stem cell populations is possible, strongly suggesting that this is the mechanism for spread of mutated clones; (2) submucosal oesophageal glands and ducts are shown to be maintained by multiple stem cells; (3) Barrett's metaplasia glands contain multiple stem cells and undergo a process of niche succession and monoclonal conversion; (4) Barrett's metaplasia probably spread by fission to clonally expand within the Barrett's metaplasia; and (5) neosquamous epithelium and underlying glandular epithelium can share a common cell of origin that is capable of differentiating into both cell types.
Acknowledgments
We are grateful to Professor Hugh Barr Gloucestershire Royal Hospital, Gloucester, UK, for assisting with the preliminary work for this manuscript.
References
Footnotes
SACM and JAJ contributed equally to this manuscript.
-
Funding C548/A5675—CHOPIN—Chemoprevention of Premalignant Intestinal Neoplasia.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval 07/Q1604/17 Oxfordshire Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statements We are happy to share any data with any interested parties on request.